
Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Fabry Disease: Phenotypic Variability
Diagnosis
3. Inflammation in Lysosomal Storage Disease
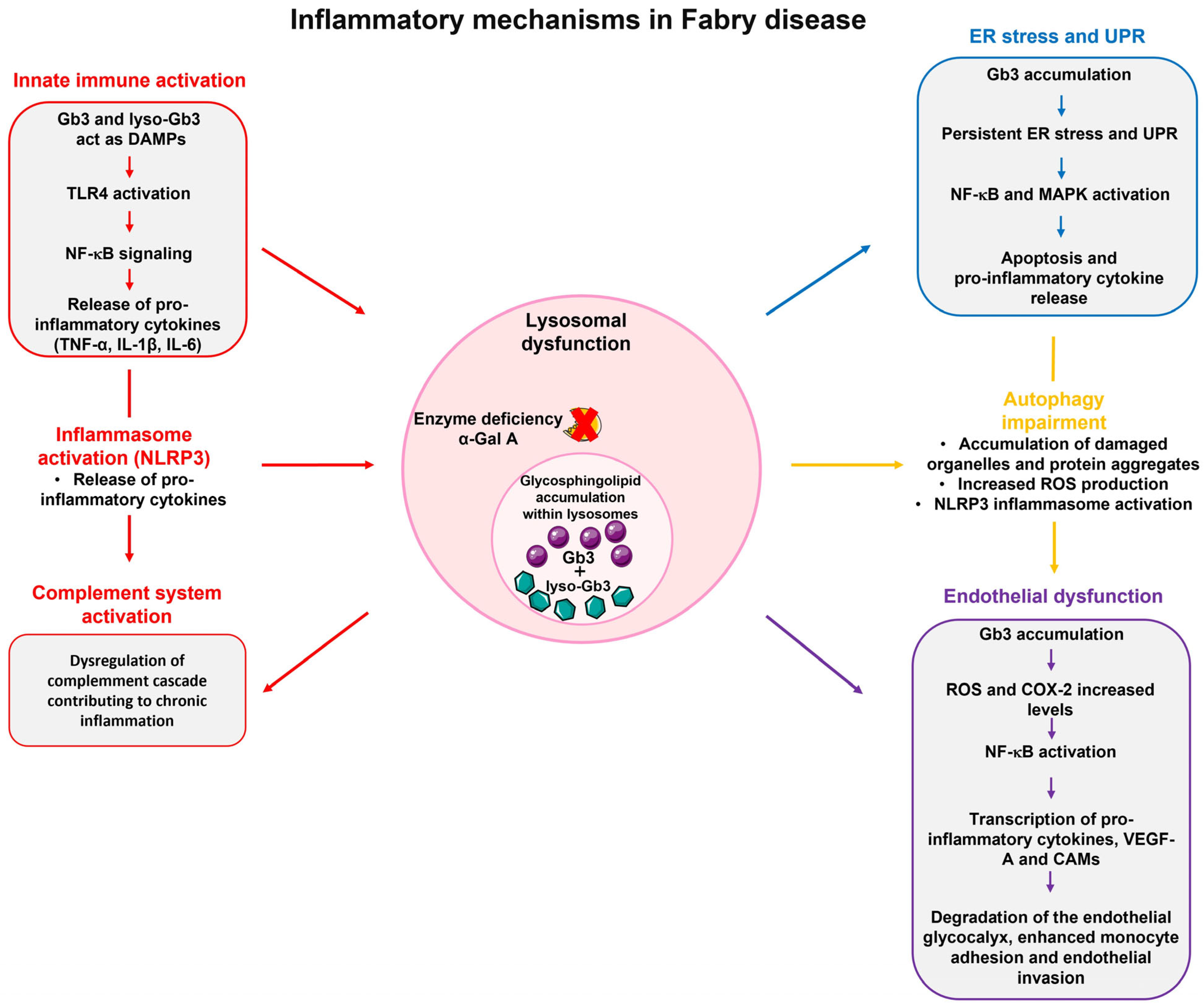
3.1. Inflammation in Fabry Disease and Molecular Mechanisms
3.2. Immune System Activation in Fabry Disease
3.3. Endoplasmic Reticulum Stress and the Unfolded Protein Response
3.4. Impairment of Autophagic Processes
3.5. Endothelial Dysfunction
4. Clinical Implications of Chronic Inflammation in Fabry Disease
4.1. Chronic Inflammation and Fibrosis in Fabry Nephropathy
4.2. Cardiac Involvement and Inflammation in Fabry Disease
4.3. Central and Peripheral Nervous System in Fabry Disease: The Role of Inflammation
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sun, A. Lysosomal storage disease overview. Ann. Transl. Med. 2018, 6, 476. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Ribeiro, B.; Silva, H.G.; Sampaio-Marques, B.; Fraga, A.G.; Azevedo, O.; Pedrosa, J.; Ludovico, P. Inflammation and Exosomes in Fabry Disease Pathogenesis. Cells 2024, 13, 654. [Google Scholar] [CrossRef]
- Thompson, S.E.; Roy, A.; Geberhiwot, T.; Gehmlich, K.; Steeds, R.P. Fabry Disease: Insights into Pathophysiology and Novel Therapeutic Strategies. Biomedicines 2025, 13, 624. [Google Scholar] [CrossRef]
- Schiffmann, R. Chapter 17—Fabry disease. In Handbook of Clinical Neurology; Islam, M.P., Roach, E.S., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Volume 132, pp. 231–248. [Google Scholar]
- Marques, A.R.A.; Saftig, P. Lysosomal storage disorders—Challenges, concepts and avenues for therapy: Beyond rare diseases. J. Cell Sci. 2019, 132, jcs221739. [Google Scholar] [CrossRef]
- Michaud, M.; Mauhin, W.; Belmatoug, N.; Garnotel, R.; Bedreddine, N.; Catros, F.; Ancellin, S.; Lidove, O.; Gaches, F. When and How to Diagnose Fabry Disease in Clinical Pratice. Am. J. Med. Sci. 2020, 360, 641–649. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Simonetta, I.; Riolo, R.; Todaro, F.; Di Chiara, T.; Miceli, S.; Pinto, A. Pathogenesis and Molecular Mechanisms of Anderson-Fabry Disease and Possible New Molecular Addressed Therapeutic Strategies. Int. J. Mol. Sci. 2021, 22, 10088. [Google Scholar] [CrossRef]
- Simonetta, I.; Tuttolomondo, A.; Daidone, M.; Pinto, A. Biomarkers in Anderson–Fabry Disease. Int. J. Mol. Sci. 2020, 21, 8080. [Google Scholar] [CrossRef]
- Ferreira, C.R.; Gahl, W.A. Lysosomal storage diseases. Transl. Sci. Rare Dis. 2017, 2, 27. [Google Scholar] [CrossRef]
- Amodio, F.; Caiazza, M.; Monda, E.; Rubino, M.; Capodicasa, L.; Chiosi, F.; Simonelli, V.; Dongiglio, F.; Fimiani, F.; Pepe, N.; et al. An Overview of Molecular Mechanisms in Fabry Disease. Biomolecules 2022, 12, 1460. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry Disease: The Current Treatment Landscape. Drugs 2021, 81, 635–645. [Google Scholar] [CrossRef] [PubMed]
- Lenders, M.; Nowak, A.; Cybulla, M.; Kaufeld, J.; Köhn, A.F.; Muschol, N.M.; Kurschat, C.; Brand, E. Impact of enzyme replacement therapy and migalastat on disease progression in females with fabry disease. Orphanet J. Rare Dis. 2025, 20, 79. [Google Scholar] [CrossRef]
- Iacobucci, I.; Hay Mele, B.; Cozzolino, F.; Monaco, V.; Cimmaruta, C.; Monti, M.; Andreotti, G.; Monticelli, M. Enzyme Replacement Therapy for FABRY Disease: Possible Strategies to Improve Its Efficacy. Int. J. Mol. Sci. 2023, 24, 4548. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The lysosome: From waste bag to potential therapeutic target. J. Mol. Cell Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed]
- Vitner, E.B.; Platt, F.M.; Futerman, A.H. Common and uncommon pathogenic cascades in lysosomal storage diseases. J. Biol. Chem. 2010, 285, 20423–20427. [Google Scholar] [CrossRef] [PubMed]
- Schmid, D.; Münz, C. Immune surveillance of intracellular pathogens via autophagy. Cell Death Differ. 2005, 12, 1519–1527. [Google Scholar] [CrossRef]
- Schmid, D.; Dengjel, J.; Schoor, O.; Stevanovic, S.; Münz, C. Autophagy in innate and adaptive immunity against intracellular pathogens. J. Mol. Med. 2006, 84, 194–202. [Google Scholar] [CrossRef]
- Hsing, L.C.; Rudensky, A.Y. The lysosomal cysteine proteases in MHC class II antigen presentation. Immunol. Rev. 2005, 207, 229–241. [Google Scholar] [CrossRef]
- Castaneda, J.A.; Lim, M.J.; Cooper, J.D.; Pearce, D.A. Immune system irregularities in lysosomal storage disorders. Acta Neuropathol. 2008, 115, 159–174. [Google Scholar] [CrossRef]
- Scerra, G.; De Pasquale, V.; Scarcella, M.; Caporaso, M.G.; Pavone, L.M.; D’Agostino, M. Lysosomal positioning diseases: Beyond substrate storage. Open Biol. 2022, 12, 220155. [Google Scholar] [CrossRef]
- Ebner, M.; Fröhlich, F.; Haucke, V. Mechanisms and functions of lysosomal lipid homeostasis. Cell Chem. Biol. 2025, 32, 392–407. [Google Scholar] [CrossRef]
- Tanaka, H.; Tsuji, D.; Watanabe, R.; Ohnishi, Y.; Kitaguchi, S.; Nakae, R.; Teramoto, H.; Tsukimoto, J.; Horii, Y.; Itoh, K. Aberrant autophagy in lysosomal storage disorders marked by a lysosomal SNARE protein shortage due to suppression of endocytosis. J. Inherit. Metab. Dis. 2022, 45, 1191–1202. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Medina, D.L.; Ballabio, A. The rapidly evolving view of lysosomal storage diseases. EMBO Mol. Med. 2021, 13, e12836. [Google Scholar] [CrossRef] [PubMed]
- Kurdi, H.; Lavalle, L.; Moon, J.C.C.; Hughes, D. Inflammation in Fabry disease: Stages, molecular pathways, and therapeutic implications. Front. Cardiovasc. Med. 2024, 11, 1420067. [Google Scholar] [CrossRef]
- Rozenfeld, P.; Feriozzi, S. Contribution of inflammatory pathways to Fabry disease pathogenesis. Mol. Genet. Metab. 2017, 122, 19–27. [Google Scholar] [CrossRef]
- Del Pinto, R.; Ferri, C. The role of Immunity in Fabry Disease and Hypertension: A Review of a Novel Common Pathway. High Blood Press. Cardiovasc. Prev. 2020, 27, 539–546. [Google Scholar] [CrossRef]
- Üçeyler, N.; Urlaub, D.; Mayer, C.; Uehlein, S.; Held, M.; Sommer, C. Tumor necrosis factor-α links heat and inflammation with Fabry pain. Mol. Genet. Metab. 2019, 127, 200–206. [Google Scholar] [CrossRef]
- Chen, K.H.; Chien, Y.; Wang, K.L.; Leu, H.B.; Hsiao, C.Y.; Lai, Y.H.; Wang, C.Y.; Chang, Y.L.; Lin, S.J.; Niu, D.M.; et al. Evaluation of Proinflammatory Prognostic Biomarkers for Fabry Cardiomyopathy with Enzyme Replacement Therapy. Can. J. Cardiol. 2016, 32, e1221. [Google Scholar] [CrossRef]
- Yogasundaram, H.; Nikhanj, A.; Putko, B.N.; Boutin, M.; Jain-Ghai, S.; Khan, A.; Auray-Blais, C.; West, M.L.; Oudit, G.Y. Elevated Inflammatory Plasma Biomarkers in Patients with Fabry Disease: A Critical Link to Heart Failure with Preserved Ejection Fraction. J. Am. Heart Assoc. 2018, 7, e009098. [Google Scholar] [CrossRef]
- Weissman, D.; Dudek, J.; Sequeira, V.; Maack, C. Fabry Disease: Cardiac Implications and Molecular Mechanisms. Curr. Heart Fail. Rep. 2024, 21, 81–100. [Google Scholar] [CrossRef]
- Biddeci, G.; Spinelli, G.; Colomba, P.; Duro, G.; Anania, M.; Francofonte, D.; Di Blasi, F. Fabry Disease and Inflammation: Potential Role of p65 iso5, an Isoform of the NF-κB Complex. Cells 2025, 14, 230. [Google Scholar] [CrossRef]
- Spinelli, G.; Biddeci, G.; Artale, A.; Valentino, F.; Tarantino, G.; Gallo, G.; Gianguzza, F.; Conaldi, P.G.; Corrao, S.; Gervasi, F.; et al. A new p65 isoform that bind the glucocorticoid hormone and is expressed in inflammation liver diseases and COVID-19. Sci. Rep. 2021, 11, 22913. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhao, Y.; Li, F.; Ling, C.; Wu, Y.; Ma, W.; Wang, Z.; Yuan, Y.; Hao, H.; Zhang, W. Inflammatory cytokine expression in Fabry disease: Impact of disease phenotype and alterations under enzyme replacement therapy. Front. Immunol. 2024, 15, 1367252. [Google Scholar] [CrossRef] [PubMed]
- Rosa, N.S.; Bento, J.C.d.B.; Caparbo, V.d.F.; Pereira, R.M.R. Increased Serum Interleukin-6 and Tumor Necrosis Factor Alpha Levels in Fabry Disease: Correlation with Disease Burden. Clinics 2021, 76, e2643. [Google Scholar] [CrossRef] [PubMed]
- De Francesco, P.N.; Mucci, J.M.; Ceci, R.; Fossati, C.A.; Rozenfeld, P.A. Fabry disease peripheral blood immune cells release inflammatory cytokines: Role of globotriaosylceramide. Mol. Genet. Metab. 2013, 109, 93–99. [Google Scholar] [CrossRef]
- Feriozzi, S.; Rozenfeld, P. The inflammatory pathogenetic pathways of Fabry nephropathy. Rare Dis. Orphan Drugs J. 2024, 3, 11. [Google Scholar] [CrossRef]
- Lerario, S.; Monti, L.; Ambrosetti, I.; Luglio, A.; Pietra, A.; Aiello, V.; Montanari, F.; Bellasi, A.; Zaza, G.; Galante, A.; et al. Fabry disease: A rare disorder calling for personalized medicine. Int. Urol. Nephrol. 2024, 56, 3161–3172. [Google Scholar] [CrossRef]
- Mehta, A.; Ricci, R.; Widmer, U.; Dehout, F.; Garcia de Lorenzo, A.; Kampmann, C.; Linhart, A.; Sunder-Plassmann, G.; Ries, M.; Beck, M. Fabry disease defined: Baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur. J. Clin. Investig. 2004, 34, 236–242. [Google Scholar] [CrossRef]
- Germain, D.P. Fabry disease. Orphanet J. Rare Dis. 2010, 5, 30. [Google Scholar] [CrossRef]
- Arends, M.; Wanner, C.; Hughes, D.; Mehta, A.; Oder, D.; Watkinson, O.T.; Elliott, P.M.; Linthorst, G.E.; Wijburg, F.A.; Biegstraaten, M.; et al. Characterization of Classical and Nonclassical Fabry Disease: A Multicenter Study. J. Am. Soc. Nephrol. JASN 2017, 28, 1631–1641. [Google Scholar] [CrossRef]
- Waldek, S.; Patel, M.R.; Banikazemi, M.; Lemay, R.; Lee, P. Life expectancy and cause of death in males and females with Fabry disease: Findings from the Fabry Registry. Genet. Med. 2009, 11, 790–796. [Google Scholar] [CrossRef]
- Azevedo, O.; Gago, M.F.; Miltenberger-Miltenyi, G.; Sousa, N.; Cunha, D. Fabry Disease Therapy: State-of-the-Art and Current Challenges. Int. J. Mol. Sci. 2020, 22, 206. [Google Scholar] [CrossRef]
- Capelli, I.; Aiello, V.; Gasperoni, L.; Comai, G.; Corradetti, V.; Ravaioli, M.; Biagini, E.; Graziano, C.; La Manna, G. Kidney Transplant in Fabry Disease: A Revision of the Literature. Medicina 2020, 56, 284. [Google Scholar] [CrossRef]
- Barba-Romero, M.-Á.; Sánchez-Martínez, R. Fabry disease in women: Beyond the role of “carriers”. Rare Dis. Orphan Drugs J. 2025, 4, 2. [Google Scholar] [CrossRef]
- Echevarria, L.; Benistan, K.; Toussaint, A.; Dubourg, O.; Hagege, A.A.; Eladari, D.; Jabbour, F.; Beldjord, C.; De Mazancourt, P.; Germain, D.P. X-chromosome inactivation in female patients with Fabry disease. Clin. Genet. 2016, 89, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Juchniewicz, P.; Piotrowska, E.; Kloska, A.; Podlacha, M.; Mantej, J.; Węgrzyn, G.; Tukaj, S.; Jakóbkiewicz-Banecka, J. Dosage Compensation in Females with X-Linked Metabolic Disorders. Int. J. Mol. Sci. 2021, 22, 4514. [Google Scholar] [CrossRef] [PubMed]
- Cammarata, G.; Fatuzzo, P.; Rodolico, M.S.; Colomba, P.; Sicurella, L.; Iemolo, F.; Zizzo, C.; Alessandro, R.; Bartolotta, C.; Duro, G.; et al. High variability of Fabry disease manifestations in an extended Italian family. BioMed Res. Int. 2015, 2015, 504784. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Simonetta, I.; Duro, G.; Pecoraro, R.; Miceli, S.; Colomba, P.; Zizzo, C.; Nucera, A.; Daidone, M.; Di Chiara, T.; et al. Inter-familial and intra-familial phenotypic variability in three Sicilian families with Anderson-Fabry disease. Oncotarget 2017, 8, 61415–61424. [Google Scholar] [CrossRef]
- Oliveira, J.P.; Ferreira, S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. Appl. Clin. Genet. 2019, 12, 35–50. [Google Scholar] [CrossRef]
- Ezgu, F.; Alpsoy, E.; Bicik Bahcebasi, Z.; Kasapcopur, O.; Palamar, M.; Onay, H.; Ozdemir, B.H.; Topcuoglu, M.A.; Tufekcioglu, O. Expert opinion on the recognition, diagnosis and management of children and adults with Fabry disease: A multidisciplinary Turkey perspective. Orphanet J. Rare Dis. 2022, 17, 90. [Google Scholar] [CrossRef]
- Schiffmann, R.; Fuller, M.; Clarke, L.A.; Aerts, J.M.F.G. Is it Fabry disease? Genet. Med. 2016, 18, 1181–1185. [Google Scholar] [CrossRef]
- van der Tol, L.; Smid, B.E.; Poorthuis, B.J.; Biegstraaten, M.; Deprez, R.H.; Linthorst, G.E.; Hollak, C.E. A systematic review on screening for Fabry disease: Prevalence of individuals with genetic variants of unknown significance. J. Med. Genet. 2014, 51, 1–9. [Google Scholar] [CrossRef]
- Smid, B.E.; van der Tol, L.; Cecchi, F.; Elliott, P.M.; Hughes, D.A.; Linthorst, G.E.; Timmermans, J.; Weidemann, F.; West, M.L.; Biegstraaten, M.; et al. Uncertain diagnosis of Fabry disease: Consensus recommendation on diagnosis in adults with left ventricular hypertrophy and genetic variants of unknown significance. Int. J. Cardiol. 2014, 177, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Laney, D.A.; Bennett, R.L.; Clarke, V.; Fox, A.; Hopkin, R.J.; Johnson, J.; O’Rourke, E.; Sims, K.; Walter, G. Fabry disease practice guidelines: Recommendations of the National Society of Genetic Counselors. J. Genet. Couns. 2013, 22, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Jehn, U.; Bayraktar, S.; Pollmann, S.; Van Marck, V.; Weide, T.; Pavenstädt, H.; Brand, E.; Lenders, M. α-Galactosidase a Deficiency in Fabry Disease Leads to Extensive Dysregulated Cellular Signaling Pathways in Human Podocytes. Int. J. Mol. Sci. 2021, 22, 11339. [Google Scholar] [CrossRef] [PubMed]
- Daitx, V.V.; Mezzalira, J.; Goldim, M.P.d.S.; Coelho, J.C. Comparison between alpha-galactosidase A activity in blood samples collected on filter paper, leukocytes and plasma. Clin. Biochem. 2012, 45, 1233–1238. [Google Scholar] [CrossRef]
- Scalia, S. DBS assay in the diagnosis of Fabry disease. G. Di Tec. Nefrol. E Dial. 2017, 29, S5–S6. [Google Scholar] [CrossRef]
- Froissart, R.; Guffon, N.; Vanier, M.T.; Desnick, R.J.; Maire, I. Fabry disease: D313Y is an α-galactosidase A sequence variant that causes pseudodeficient activity in plasma. Mol. Genet. Metab. 2003, 80, 307–314. [Google Scholar] [CrossRef]
- Izhar, R.; Borriello, M.; La Russa, A.; Di Paola, R.; De, A.; Capasso, G.; Ingrosso, D.; Perna, A.F.; Simeoni, M. Fabry Disease in Women: Genetic Basis, Available Biomarkers, and Clinical Manifestations. Genes 2024, 15, 37. [Google Scholar] [CrossRef]
- Duro, G.; Anania, M.; Zizzo, C.; Francofonte, D.; Giacalone, I.; D’Errico, A.; Marsana, E.M.; Colomba, P. Diagnosis of Fabry Disease Using Alpha-Galactosidase A Activity or LysoGb3 in Blood Fails to Identify Up to Two Thirds of Female Patients. Int. J. Mol. Sci. 2024, 25, 5158. [Google Scholar] [CrossRef]
- Taguchi, A.; Ishii, S.; Mikame, M.; Maruyama, H. Distinctive accumulation of globotriaosylceramide and globotriaosylsphingosine in a mouse model of classic Fabry disease. Mol. Genet. Metab. Rep. 2023, 34, 100952. [Google Scholar] [CrossRef]
- Shin, J.-h.; Kim, S.H. Screening and diagnosis of Fabry disease in chronic kidney disease: The important role of globotriaosylsphingosine. Kidney Res. Clin. Pract. 2024, 43, 2. [Google Scholar] [CrossRef] [PubMed]
- Sueoka, H.; Ichihara, J.; Tsukimura, T.; Togawa, T.; Sakuraba, H. Nano-LC-MS/MS for Quantification of Lyso-Gb3 and Its Analogues Reveals a Useful Biomarker for Fabry Disease. PLoS ONE 2015, 10, e0127048. [Google Scholar] [CrossRef] [PubMed]
- Dupont, F.O.; Gagnon, R.; Boutin, M.; Auray-Blais, C. A metabolomic study reveals novel plasma lyso-Gb3 analogs as Fabry disease biomarkers. Curr. Med. Chem. 2013, 20, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Changsila, E.; Iaonou, C.; Goker-Alpan, O. Impaired autophagic and mitochondrial functions are partially restored by ERT in Gaucher and Fabry diseases. PLoS ONE 2019, 14, e0210617. [Google Scholar] [CrossRef]
- Maalouf, K.; Jia, J.; Rizk, S.; Brogden, G.; Keiser, M.; Das, A.; Naim, H.Y. A modified lipid composition in Fabry disease leads to an intracellular block of the detergent-resistant membrane-associated dipeptidyl peptidase IV. J. Inherit. Metab. Dis. 2010, 33, 445–449. [Google Scholar] [CrossRef]
- Medzhitov, R. Inflammation 2010: New adventures of an old flame. Cell 2010, 140, 771–776. [Google Scholar] [CrossRef]
- Oronsky, B.; Caroen, S.; Reid, T. What Exactly Is Inflammation (and What Is It Not?). Int. J. Mol. Sci. 2022, 23, 14905. [Google Scholar] [CrossRef]
- Lawrence, T. The nuclear factor NF-kappaB pathway in inflammation. Cold Spring Harb. Perspect. Biol. 2009, 1, a001651. [Google Scholar] [CrossRef]
- Libby, P. Inflammatory Mechanisms: The Molecular Basis of Inflammation and Disease. Nutr. Rev. 2007, 65, S140–S146. [Google Scholar] [CrossRef]
- Kiss, A.L. Inflammation in Focus: The Beginning and the End. Pathol. Oncol. Res. 2022, 27, 1610136. [Google Scholar] [CrossRef]
- Fiorenza, M.T.; Moro, E.; Erickson, R.P. The pathogenesis of lysosomal storage disorders: Beyond the engorgement of lysosomes to abnormal development and neuroinflammation. Hum. Mol. Genet. 2018, 27, R119–R129. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.S.; Sohn, D.H. Damage-Associated Molecular Patterns in Inflammatory Diseases. Immune Netw. 2018, 18, e27. [Google Scholar] [CrossRef] [PubMed]
- Bi, J.; Sun, Y.; Guo, M.; Sun, X.; Sun, J.; Jiang, R.; Wang, N.; Huang, G. Lysosomes: Guardians and healers within cells- multifaceted perspective and outlook from injury repair to disease treatment. Cancer Cell Int. 2025, 25, 136. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.Y.; Yao, R.Q.; Li, Y.X.; Zhao, P.Y.; Ren, C.; Du, X.H.; Yao, Y.M. Lysosomal quality control of cell fate: A novel therapeutic target for human diseases. Cell Death Dis. 2020, 11, 817. [Google Scholar] [CrossRef]
- Di Mambro, T.; Pellielo, G.; Agyapong, E.D.; Carinci, M.; Chianese, D.; Giorgi, C.; Morciano, G.; Patergnani, S.; Pinton, P.; Rimessi, A. The Tricky Connection between Extracellular Vesicles and Mitochondria in Inflammatory-Related Diseases. Int. J. Mol. Sci. 2023, 24, 8181. [Google Scholar] [CrossRef]
- Anders, H.J.; Banas, B.; Schlöndorff, D. Signaling danger: Toll-like receptors and their potential roles in kidney disease. J. Am. Soc. Nephrol. JASN 2004, 15, 854–867. [Google Scholar] [CrossRef]
- Wicherska-Pawłowska, K.; Wróbel, T.; Rybka, J. Toll-Like Receptors (TLRs), NOD-Like Receptors (NLRs), and RIG-I-Like Receptors (RLRs) in Innate Immunity. TLRs, NLRs, and RLRs Ligands as Immunotherapeutic Agents for Hematopoietic Diseases. Int. J. Mol. Sci. 2021, 22, 13397. [Google Scholar] [CrossRef]
- Kawai, T.; Ikegawa, M.; Ori, D.; Akira, S. Decoding Toll-like receptors: Recent insights and perspectives in innate immunity. Immunity 2024, 57, 649–673. [Google Scholar] [CrossRef]
- Kim, J.W.; Kim, H.W.; Nam, S.A.; Lee, J.Y.; Cho, H.J.; Kim, T.-M.; Kim, Y.K. Human kidney organoids reveal the role of glutathione in Fabry disease. Exp. Mol. Med. 2021, 53, 1580–1591. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Carpio, D.; Sanz, A.B.; Ruiz-Ortega, M.; Mezzano, S.; Ortiz, A. Lyso-Gb3 activates Notch1 in human podocytes. Hum. Mol. Genet. 2015, 24, 5720–5732. [Google Scholar] [CrossRef]
- Laffer, B.; Lenders, M.; Ehlers-Jeske, E.; Heidenreich, K.; Brand, E.; Köhl, J. Complement activation and cellular inflammation in Fabry disease patients despite enzyme replacement therapy. Front. Immunol. 2024, 15, 1307558. [Google Scholar] [CrossRef]
- Pandey, M.K. Exploring Pro-Inflammatory Immunological Mediators: Unraveling the Mechanisms of Neuroinflammation in Lysosomal Storage Diseases. Biomedicines 2023, 11, 1067. [Google Scholar] [CrossRef] [PubMed]
- Feriozzi, S.; Rozenfeld, P. Pathology and pathogenic pathways in fabry nephropathy. Clin. Exp. Nephrol. 2021, 25, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Hanawa, H.; Jiao, S.; Hasegawa, G.; Ohno, Y.; Yoshida, K.; Suzuki, T.; Kashimura, T.; Obata, H.; Tanaka, K.; et al. Elevated Endomyocardial Biopsy Macrophage-Related Markers in Intractable Myocardial Diseases. Inflammation 2015, 38, 2288–2299. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, M.M.; Dao, J.; Friedman, A.; Kasaci, N.; Goker-Alpan, O. Sex Differences in Circulating Inflammatory, Immune, and Tissue Growth Markers Associated with Fabry Disease-Related Cardiomyopathy. Cells 2025, 14, 322. [Google Scholar] [CrossRef]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Dupuis, M.L.; Maselli, A.; Pagano, M.T.; Pierdominici, M.; Ortona, E. Immune response and autoimmune diseases: A matter of sex. Ital. J. Gender-Specif. Med. 2019, 5, 11–20. [Google Scholar] [CrossRef]
- Hughes, D.A.; Barba Romero, M.; Hollak, C.E.; Giugliani, R.; Deegan, P.B. Response of women with Fabry disease to enzyme replacement therapy: Comparison with men, using data from FOS--the Fabry Outcome Survey. Mol. Genet. Metab. 2011, 103, 207–214. [Google Scholar] [CrossRef]
- Lenders, M.; Nowak, A.; Cybulla, M.; Kaufeld, J.; Köhn, A.F.; Muschol, N.M.; Kurschat, C.; Brand, E. Impact of enzyme replacement therapy on clinical manifestations in females with Fabry disease. Orphanet J. Rare Dis. 2024, 19, 490. [Google Scholar] [CrossRef]
- Deegan, P.B. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J. Inherit. Metab. Dis. 2012, 35, 227–243. [Google Scholar] [CrossRef]
- Byun, J.H.; Lebeau, P.F.; Trink, J.; Uppal, N.; Lanktree, M.B.; Krepinsky, J.C.; Austin, R.C. Endoplasmic reticulum stress as a driver and therapeutic target for kidney disease. Nat. Rev. Nephrol. 2025, 21, 299–313. [Google Scholar] [CrossRef]
- Lemmer, I.L.; Willemsen, N.; Hilal, N.; Bartelt, A. A guide to understanding endoplasmic reticulum stress in metabolic disorders. Mol. Metab. 2021, 47, 101169. [Google Scholar] [CrossRef]
- Consolato, F.; De Fusco, M.; Schaeffer, C.; Pieruzzi, F.; Scolari, F.; Gallieni, M.; Lanzani, C.; Feriozzi, S.; Rampoldi, L. α-Gal A missense variants associated with Fabry disease can lead to ER stress and induction of the unfolded protein response. Mol. Genet. Metab. Rep. 2022, 33, 100926. [Google Scholar] [CrossRef]
- Riillo, C.; Bonapace, G.; Moricca, M.T.; Sestito, S.; Salatino, A.; Concolino, D. c.376A>G, (p.Ser126Gly) Alpha-Galactosidase A mutation induces ER stress, unfolded protein response and reduced enzyme trafficking to lysosome: Possible relevance in the pathogenesis of late-onset forms of Fabry Disease. Mol. Genet. Metab. 2023, 140, 107700. [Google Scholar] [CrossRef] [PubMed]
- Nikolaenko, V.; Warnock, D.G.; Mills, K.; Heywood, W.E. Elucidating the toxic effect and disease mechanisms associated with Lyso-Gb3 in Fabry disease. Hum. Mol. Genet. 2023, 32, 2464–2472. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Shen, X.; Wu, J.; Sakaki, K.; Saunders, T.; Rutkowski, D.T.; Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress Activates Cleavage of CREBH to Induce a Systemic Inflammatory Response. Cell 2006, 124, 587–599. [Google Scholar] [CrossRef]
- Satoh, K. Globotriaosylceramide induces endothelial dysfunction in fabry disease. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2–4. [Google Scholar] [CrossRef]
- Stepien, K.M.; Roncaroli, F.; Turton, N.; Hendriksz, C.J.; Roberts, M.; Heaton, R.A.; Hargreaves, I. Mechanisms of Mitochondrial Dysfunction in Lysosomal Storage Disorders: A Review. J. Clin. Med. 2020, 9, 2596. [Google Scholar] [CrossRef]
- Plotegher, N.; Duchen, M.R. Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders. Trends Mol. Med. 2017, 23, 116–134. [Google Scholar] [CrossRef]
- Hamzeh, O.; Rabiei, F.; Shakeri, M.; Parsian, H.; Saadat, P.; Rostami-Mansoor, S. Mitochondrial dysfunction and inflammasome activation in neurodegenerative diseases: Mechanisms and therapeutic implications. Mitochondrion 2023, 73, 72–83. [Google Scholar] [CrossRef]
- Zhang, G.; Wei, H.; Zhao, A.; Yan, X.; Zhang, X.; Gan, J.; Guo, M.; Wang, J.; Zhang, F.; Jiang, Y.; et al. Mitochondrial DNA leakage: Underlying mechanisms and therapeutic implications in neurological disorders. J. Neuroinflamm. 2025, 22, 34. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, X.; Escames, G.; Lei, W.; Zhang, X.; Li, M.; Jing, T.; Yao, Y.; Qiu, Z.; Wang, Z.; et al. The NLRP3 inflammasome: Contributions to inflammation-related diseases. Cell. Mol. Biol. Lett. 2023, 28, 51. [Google Scholar] [CrossRef]
- Schumann, A.; Schaller, K.; Belche, V.; Cybulla, M.; Grünert, S.C.; Moers, N.; Sass, J.O.; Kaech, A.; Hannibal, L.; Spiekerkoetter, U. Defective lysosomal storage in Fabry disease modifies mitochondrial structure, metabolism and turnover in renal epithelial cells. J. Inherit. Metab. Dis. 2021, 44, 1039–1050. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.S.; Meng, X.L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Zampetti, A.; Gnarra, M.; Borsini, W.; Giurdanella, F.; Antuzzi, D.; Piras, A.; Smaldone, C.; Pieroni, M.; Cadeddu, C.; de Waure, C.; et al. Vascular Endothelial Growth Factor (VEGF-a) in Fabry disease: Association with cutaneous and systemic manifestations with vascular involvement. Cytokine 2013, 61, 933–939. [Google Scholar] [CrossRef] [PubMed]
- Aerts, J.M.; Groener, J.E.; Kuiper, S.; Donker-Koopman, W.E.; Strijland, A.; Ottenhoff, R.; van Roomen, C.; Mirzaian, M.; Wijburg, F.A.; Linthorst, G.E.; et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc. Natl. Acad. Sci. USA 2008, 105, 2812–2817. [Google Scholar] [CrossRef]
- Namdar, M.; Gebhard, C.; Studiger, R.; Shi, Y.; Mocharla, P.; Schmied, C.; Brugada, P.; Lüscher, T.F.; Camici, G.G. Globotriaosylsphingosine accumulation and not alpha-galactosidase-A deficiency causes endothelial dysfunction in Fabry disease. PLoS ONE 2012, 7, e36373. [Google Scholar] [CrossRef]
- Rombach, S.M.; Twickler, T.B.; Aerts, J.M.; Linthorst, G.E.; Wijburg, F.A.; Hollak, C.E. Vasculopathy in patients with Fabry disease: Current controversies and research directions. Mol. Genet. Metab. 2010, 99, 99–108. [Google Scholar] [CrossRef]
- Collins, T.; Read, M.A.; Neish, A.S.; Whitley, M.Z.; Thanos, D.; Maniatis, T. Transcriptional regulation of endothelial cell adhesion molecules: NF-kappa B and cytokine-inducible enhancers. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1995, 9, 899–909. [Google Scholar] [CrossRef]
- Pollmann, S.; Scharnetzki, D.; Manikowski, D.; Lenders, M.; Brand, E. Endothelial Dysfunction in Fabry Disease Is Related to Glycocalyx Degradation. Front. Immunol. 2021, 12, 789142. [Google Scholar] [CrossRef]
- Thurberg, B.L.; Rennke, H.; Colvin, R.B.; Dikman, S.; Gordon, R.E.; Collins, A.B.; Desnick, R.J.; O’Callaghan, M. Globotriaosylceramide accumulation in the Fabry kidney is cleared from multiple cell types after enzyme replacement therapy. Kidney Int. 2002, 62, 1933–1946. [Google Scholar] [CrossRef]
- Pisani, A.; Pieruzzi, F.; Cirami, C.L.; Riccio, E.; Mignani, R. Interpretation of GFR slope in untreated and treated adult Fabry patients. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2023, 39, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Mignani, R.; Biagini, E.; Cianci, V.; Pieruzzi, F.; Pisani, A.; Tuttolomondo, A.; Pieroni, M. Effects of Current Therapies on Disease Progression in Fabry Disease: A Narrative Review for Better Patient Management in Clinical Practice. Adv. Ther. 2025, 42, 597–635. [Google Scholar] [CrossRef] [PubMed]
- Riccio, E.; Sabbatini, M.; Bruzzese, D.; Annicchiarico Petruzzelli, L.; Pellegrino, A.; Spinelli, L.; Esposito, R.; Imbriaco, M.; Feriozzi, S.; Pisani, A. Glomerular Hyperfiltration: An Early Marker of Nephropathy in Fabry Disease. Nephron 2019, 141, 10–17. [Google Scholar] [CrossRef]
- Perretta, F.J.; Jaurretche, S.; Antongiovanni, N. Sun-008 Glomerular Hyperfiltration in Fabry Disease. Kidney Int. Rep. 2019, 4, S155. [Google Scholar] [CrossRef]
- Pisani, A.; Petruzzelli Annicchiarico, L.; Pellegrino, A.; Bruzzese, D.; Feriozzi, S.; Imbriaco, M.; Tedeschi, E.; Cocozza, S.; De Rosa, D.; Mignani, R.; et al. Parapelvic cysts, a distinguishing feature of renal Fabry disease. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2018, 33, 318–323. [Google Scholar] [CrossRef]
- Capuano, I.; Buonanno, P.; Riccio, E.; Crocetto, F.; Pisani, A. Parapelvic Cysts: An Imaging Marker of Kidney Disease Potentially Leading to the Diagnosis of Treatable Rare Genetic Disorders? A Narrative Review of the Literature. J. Nephrol. 2022, 35, 2035–2046. [Google Scholar] [CrossRef]
- Tøndel, C.; Bostad, L.; Hirth, A.; Svarstad, E. Renal Biopsy Findings in Children and Adolescents with Fabry Disease and Minimal Albuminuria. Am. J. Kidney Dis. 2008, 51, 767–776. [Google Scholar] [CrossRef]
- Najafian, B.; Tøndel, C.; Svarstad, E.; Gubler, M.C.; Oliveira, J.P.; Mauer, M. Accumulation of Globotriaosylceramide in Podocytes in Fabry Nephropathy Is Associated with Progressive Podocyte Loss. J. Am. Soc. Nephrol. JASN 2020, 31, 865–875. [Google Scholar] [CrossRef]
- Trimarchi, H. Mechanisms of Podocyte Detachment, Podocyturia, and Risk of Progression of Glomerulopathies. Kidney Dis. 2020, 6, 324–329. [Google Scholar] [CrossRef]
- Levstek, T.; Vujkovac, B.; Trebusak Podkrajsek, K. Biomarkers of Fabry Nephropathy: Review and Future Perspective. Genes 2020, 11, 1091. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Visciano, B.; Imbriaco, M.; Di Nuzzi, A.; Mancini, A.; Marchetiello, C.; Riccio, E. The kidney in Fabry’s disease. Clin. Genet. 2014, 86, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Schiffmann, R.; Warnock, D.G.; Banikazemi, M.; Bultas, J.; Linthorst, G.E.; Packman, S.; Sorensen, S.A.; Wilcox, W.R.; Desnick, R.J. Fabry disease: Progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2009, 24, 2102–2111. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef]
- Humphreys, B.D. Mechanisms of Renal Fibrosis. Annu. Rev. Physiol. 2018, 80, 309–326. [Google Scholar] [CrossRef]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Patón, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar] [CrossRef]
- Rozenfeld, P.A.; de Los Angeles Bolla, M.; Quieto, P.; Pisani, A.; Feriozzi, S.; Neuman, P.; Bondar, C. Pathogenesis of Fabry nephropathy: The pathways leading to fibrosis. Mol. Genet. Metab. 2020, 129, 132–141. [Google Scholar] [CrossRef]
- Zhou, D.; Liu, Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat. Rev. Nephrol. 2016, 12, 68–70. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Jung, N.; Park, J.W.; Park, H.Y.; Jung, S.C. Epithelial-Mesenchymal Transition in Kidney Tubular Epithelial Cells Induced by Globotriaosylsphingosine and Globotriaosylceramide. PLoS ONE 2015, 10, e0136442. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef]
- Pieroni, M.; Moon, J.C.; Arbustini, E.; Barriales-Villa, R.; Camporeale, A.; Vujkovac, A.C.; Elliott, P.M.; Hagege, A.; Kuusisto, J.; Linhart, A.; et al. Cardiac Involvement in Fabry Disease: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2021, 77, 922–936. [Google Scholar] [CrossRef]
- Havranek, S.; Linhart, A.; Urbanova, Z.; Ramaswami, U. Early cardiac changes in children with anderson-fabry disease. JIMD Rep. 2013, 11, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Mauhin, W.; Lidove, O.; Masat, E.; Mingozzi, F.; Mariampillai, K.; Ziza, J.M.; Benveniste, O. Innate and Adaptive Immune Response in Fabry Disease. In JIMD Reports; Springer: Berlin/Heidelberg, Germany, 2015; Volume 22, pp. 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Sanz, A.B.; Carrasco, S.; Saleem, M.A.; Mathieson, P.W.; Valdivielso, J.M.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Globotriaosylsphingosine actions on human glomerular podocytes: Implications for Fabry nephropathy. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2011, 26, 1797–1802. [Google Scholar] [CrossRef] [PubMed]
- Nordin, S.; Kozor, R.; Medina-Menacho, K.; Abdel-Gadir, A.; Baig, S.; Sado, D.M.; Lobascio, I.; Murphy, E.; Lachmann, R.H.; Mehta, A.; et al. Proposed Stages of Myocardial Phenotype Development in Fabry Disease. JACC Cardiovasc. Imaging 2019, 12, 1673–1683. [Google Scholar] [CrossRef]
- Augusto, J.B.; Nordin, S.; Vijapurapu, R.; Baig, S.; Bulluck, H.; Castelletti, S.; Alfarih, M.; Knott, K.; Captur, G.; Kotecha, T.; et al. Myocardial Edema, Myocyte Injury, and Disease Severity in Fabry Disease. Circulation. Cardiovasc. Imaging 2020, 13, e010171. [Google Scholar] [CrossRef]
- Camporeale, A.; Pieroni, M.; Pieruzzi, F.; Lusardi, P.; Pica, S.; Spada, M.; Mignani, R.; Burlina, A.; Bandera, F.; Guazzi, M.; et al. Predictors of Clinical Evolution in Prehypertrophic Fabry Disease. Circ. Cardiovasc. Imaging 2019, 12, e008424. [Google Scholar] [CrossRef]
- Gatterer, C.; Beitzke, D.; Sunder-Plassmann, G.; Friedl, M.; Hohensinner, P.; Mann, C.; Ponleitner, M.; Graf, S.; Lenz, M. NT-proBNP Reflects Left Ventricular Hypertrophy Rather than Left Ventricular Dilatation or Systolic Dysfunction in Patients with Fabry Disease. J. Clin. Med. 2024, 13, 5953. [Google Scholar] [CrossRef]
- Chimenti, C.; Scopelliti, F.; Vulpis, E.; Tafani, M.; Villanova, L.; Verardo, R.; De Paulis, R.; Russo, M.A.; Frustaci, A. Increased oxidative stress contributes to cardiomyocyte dysfunction and death in patients with Fabry disease cardiomyopathy. Hum. Pathol. 2015, 46, 1760–1768. [Google Scholar] [CrossRef]
- Sheppard, M.N.; Cane, P.; Florio, R.; Kavantzas, N.; Close, L.; Shah, J.; Lee, P.; Elliott, P. A detailed pathologic examination of heart tissue from three older patients with Anderson–Fabry disease on enzyme replacement therapy. Cardiovasc. Pathol. 2010, 19, 293–301. [Google Scholar] [CrossRef]
- Bosch, M.E.; Kielian, T. Neuroinflammatory paradigms in lysosomal storage diseases. Front. Neurosci. 2015, 9, 417. [Google Scholar] [CrossRef]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef]
- Pará, C.; Bose, P.; Pshezhetsky, A.V. Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J. Clin. Med. 2020, 9, 616. [Google Scholar] [CrossRef] [PubMed]
- Carnicer-Cáceres, C.; Arranz-Amo, J.A.; Cea-Arestin, C.; Camprodon-Gomez, M.; Moreno-Martinez, D.; Lucas-Del-Pozo, S.; Moltó-Abad, M.; Tigri-Santiña, A.; Agraz-Pamplona, I.; Rodriguez-Palomares, J.F.; et al. Biomarkers in Fabry Disease. Implications for Clinical Diagnosis and Follow-up. J. Clin. Med. 2021, 10, 1664. [Google Scholar] [CrossRef] [PubMed]
- Moore, D.F.; Kaneski, C.R.; Askari, H.; Schiffmann, R. The cerebral vasculopathy of Fabry disease. J. Neurol. Sci. 2007, 257, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Tuttolomondo, A.; Baglio, I.; Riolo, R.; Todaro, F.; Parrinello, G.; Miceli, S.; Simonetta, I. Molecular Pathogenesis of Central and Peripheral Nervous System Complications in Anderson–Fabry Disease. Int. J. Mol. Sci. 2024, 25, 61. [Google Scholar] [CrossRef]
- Doran, A.C.; Meller, N.; McNamara, C.A. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 812–819. [Google Scholar] [CrossRef]
- Rock, R.B.; Gekker, G.; Hu, S.; Sheng, W.S.; Cheeran, M.; Lokensgard, J.R.; Peterson, P.K. Role of microglia in central nervous system infections. Clin. Microbiol. Rev. 2004, 17, 942–964. [Google Scholar] [CrossRef]
- Thundyil, J.; Lim, K.L. DAMPs and neurodegeneration. Ageing Res. Rev. 2015, 24, 17–28. [Google Scholar] [CrossRef]
- Amor, S.; Peferoen, L.A.; Vogel, D.Y.; Breur, M.; van der Valk, P.; Baker, D.; van Noort, J.M. Inflammation in neurodegenerative diseases—An update. Immunology 2014, 142, 151–166. [Google Scholar] [CrossRef] [PubMed]
- de Leeuw, F.-E.; de Groot, J.C.; Achten, E.; Oudkerk, M.; Ramos, L.M.P.; Heijboer, R.; Hofman, A.; Jolles, J.; van Gijn, J.; Breteler, M.M.B. Prevalence of cerebral white matter lesions in elderly people: A population based magnetic resonance imaging study. The Rotterdam Scan Study. J. Neurol. Neurosurg. Psychiatry 2001, 70, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Medala, V.K.; Üçeyler, N. Neuropathy and pain in Fabry disease. Rare Dis. Orphan Drugs J. 2024, 3, 20. [Google Scholar] [CrossRef]
- Biegstraaten, M.; Hollak, C.E.M.; Bakkers, M.; Faber, C.G.; Aerts, J.M.F.G.; van Schaik, I.N. Small fiber neuropathy in Fabry disease. Mol. Genet. Metab. 2012, 106, 135–141. [Google Scholar] [CrossRef]
- Lenders, M.; Brand, E. Fabry disease—A multisystemic disease with gastrointestinal manifestations. Gut Microbes 2022, 14, 2027852. [Google Scholar] [CrossRef]
- Hung, A.L.; Lim, M.; Doshi, T.L. Targeting cytokines for treatment of neuropathic pain. Scand. J. Pain 2017, 17, 287–293. [Google Scholar] [CrossRef]
- Basbaum, A.I.; Bautista, D.M.; Scherrer, G.; Julius, D. Cellular and molecular mechanisms of pain. Cell 2009, 139, 267–284. [Google Scholar] [CrossRef]
- Milligan, E.D.; Watkins, L.R. Pathological and protective roles of glia in chronic pain. Nat. Rev. Neurosci. 2009, 10, 23–36. [Google Scholar] [CrossRef]
- Grace, P.M.; Hutchinson, M.R.; Maier, S.F.; Watkins, L.R. Pathological pain and the neuroimmune interface. Nat. Rev. Immunol. 2014, 14, 217–231. [Google Scholar] [CrossRef]
- Vanderwall, A.G.; Milligan, E.D. Cytokines in Pain: Harnessing Endogenous Anti-Inflammatory Signaling for Improved Pain Management. Front. Immunol. 2019, 10, 3009. [Google Scholar] [CrossRef]
- Dhulkifle, H.; Diab, M.I.; Algonaiah, M.; Korashy, H.M.; Maayah, Z.H. Apabetalone (RVX-208): A Potential Epigenetic Therapy for the Treatment of Cardiovascular, Renal, Neurological, Viral, and Cancer Disorders. ACS Pharmacol. Transl. Sci. 2024, 7, 546–559. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ray, K.K.; Johansson, J.O.; Gordon, A.; Sweeney, M.; Halliday, C.; Kulikowski, E.; Wong, N.; Kim, S.W.; Schwartz, G.G. Selective BET Protein Inhibition with Apabetalone and Cardiovascular Events: A Pooled Analysis of Trials in Patients with Coronary Artery Disease. Am. J. Cardiovasc. Drugs Drugs Devices Other Interv. 2018, 18, 109–115. [Google Scholar] [CrossRef]
- Wiesinger, A.-M.; Bigger, B.; Giugliani, R.; Scarpa, M.; Moser, T.; Lampe, C.; Kampmann, C.; Lagler, F.B. The Inflammation in the Cytopathology of Patients with Mucopolysaccharidoses- Immunomodulatory Drugs as an Approach to Therapy. Front. Pharmacol. 2022, 13, 863667. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biddeci, G.; Spinelli, G.; Colomba, P.; Duro, G.; Giacalone, I.; Di Blasi, F. Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression. Int. J. Mol. Sci. 2025, 26, 7054. https://doi.org/10.3390/ijms26157054
Biddeci G, Spinelli G, Colomba P, Duro G, Giacalone I, Di Blasi F. Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression. International Journal of Molecular Sciences. 2025; 26(15):7054. https://doi.org/10.3390/ijms26157054
Chicago/Turabian StyleBiddeci, Giuseppa, Gaetano Spinelli, Paolo Colomba, Giovanni Duro, Irene Giacalone, and Francesco Di Blasi. 2025. "Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression" International Journal of Molecular Sciences 26, no. 15: 7054. https://doi.org/10.3390/ijms26157054
APA StyleBiddeci, G., Spinelli, G., Colomba, P., Duro, G., Giacalone, I., & Di Blasi, F. (2025). Fabry Disease Beyond Storage: The Role of Inflammation in Disease Progression. International Journal of Molecular Sciences, 26(15), 7054. https://doi.org/10.3390/ijms26157054