
Molecular Foundations of Neuroplasticity in Brain Tumours: From Microscopic Adaptation to Functional Reorganisation



Abstract
1. Introduction
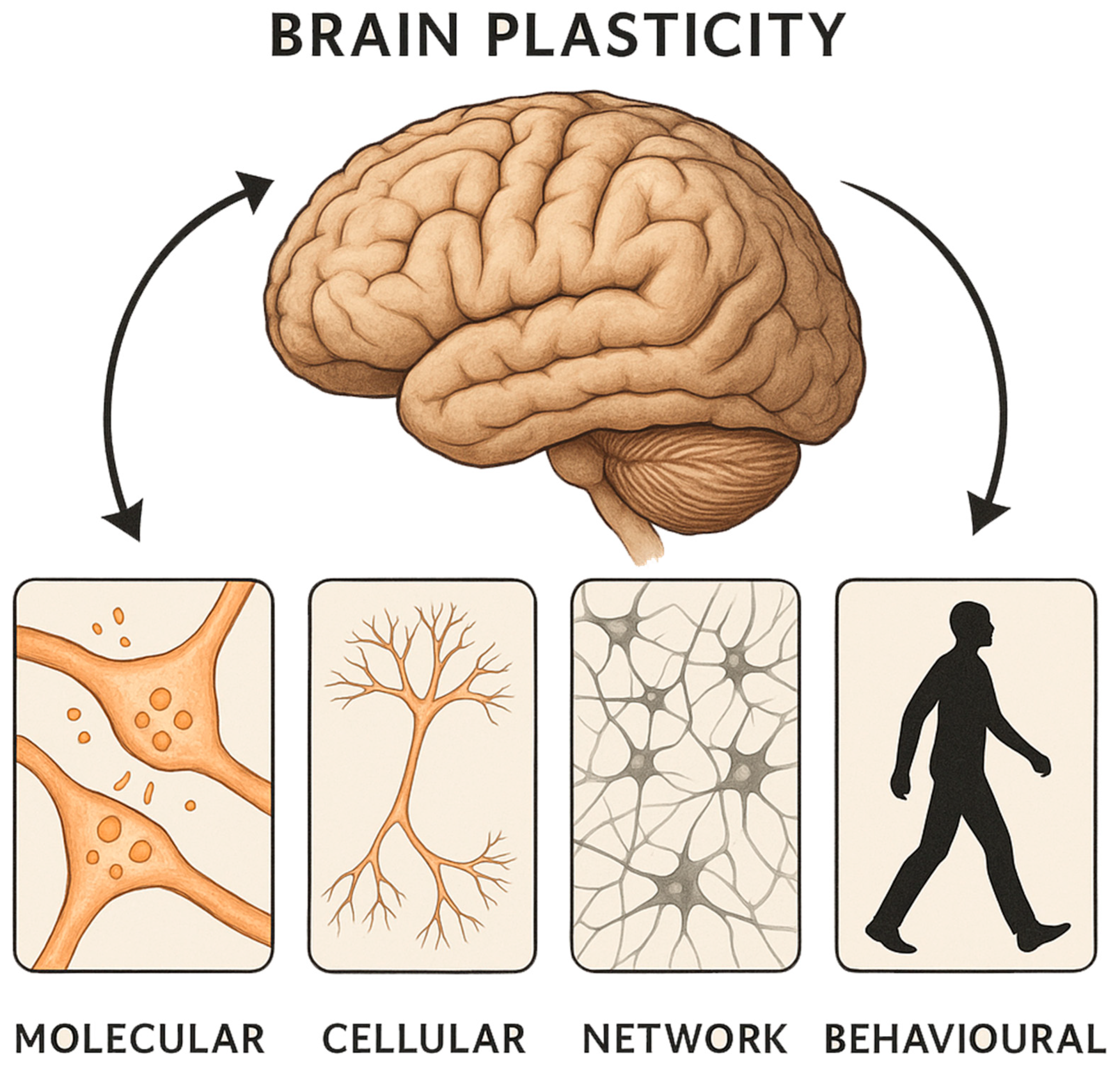
2. Plasticity Events at the Micromolecular Level
2.1. Genetic Changes
2.2. Trophic Factors and Protein Changes
2.3. Synaptic Changes
2.4. Neuronal Morphological/Structural Changes
3. Bridging Molecular and Macroscopic Plasticity
4. Plasticity Events at the Macroscopic Level
4.1. Macroscopic Plasticity Depending on the Spatial Location of the Tumour
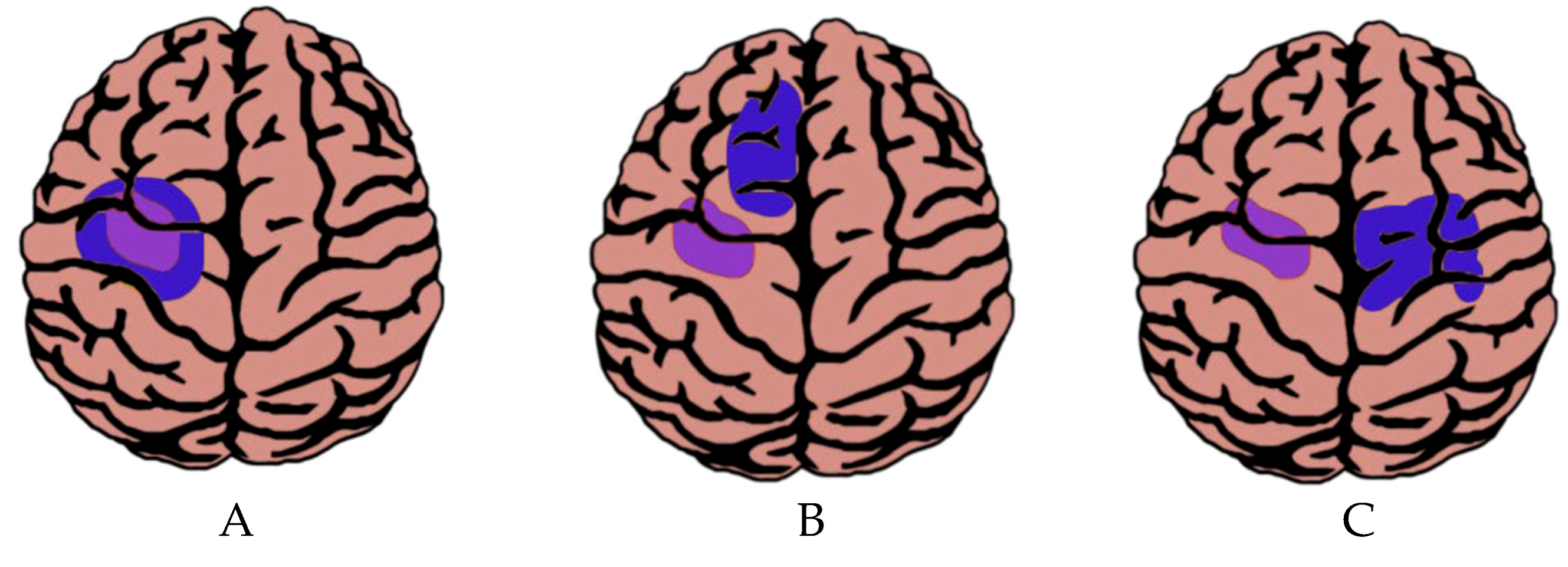
4.1.1. Cortical Plasticity
4.1.2. Subcortical Plasticity
4.2. Macroscopic Plasticity Depending on Temporal Aspects
4.3. Macroscopic Plasticity Depending on the Size of the Tumour
5. Clinical Relevance of Neuroplasticity in Gliomas
6. Limitations and Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Keyvani, K.; Schallert, T. Plasticity-Associated Molecular and Structural Events in the Injured Brain. J. Neuropathol. Exp. Neurol. 2002, 61, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Duffau, H. Brain Plasticity and Tumors. In Advances and Technical Standards in Neurosurgery; Pickard, J.D., Akalan, N., Di Rocco, C., Dolenc, V.V., Antunes, J.L., Mooij, J.J.A., Schramm, J., Sindou, M., Eds.; Springer: Vienna, Austria, 2008; pp. 3–33. ISBN 978-3-211-72283-1. [Google Scholar]
- Duffau, H. Lessons from Brain Mapping in Surgery for Low-Grade Glioma: Insights into Associations between Tumour and Brain Plasticity. Lancet Neurol. 2005, 4, 476–486. [Google Scholar] [CrossRef]
- Pascual-Leone, A.; Amedi, A.; Fregni, F.; Merabet, L.B. The Plastic Human Brain Cortex. Annu. Rev. Neurosci. 2005, 28, 377–401. [Google Scholar] [CrossRef] [PubMed]
- Duffau, H.; Capelle, L.; Denvil, D.; Sichez, N.; Gatignol, P.; Lopes, M.; Mitchell, M.C.; Sichez, J.P.; Effenterre, R.V. Functional Recovery after Surgical Resection of Low Grade Gliomas in Eloquent Brain: Hypothesis of Brain Compensation. J. Neurol. Neurosurg. Psychiatry 2003, 74, 901–907. [Google Scholar] [CrossRef]
- Fields, R.D. White Matter in Learning, Cognition and Psychiatric Disorders. Trends Neurosci. 2008, 31, 361–370. [Google Scholar] [CrossRef]
- Bonafina, A.; Paratcha, G.; Ledda, F. Deciphering New Players in the Neurogenic Adult Hippocampal Niche. Front. Cell Dev. Biol. 2020, 8, 548. [Google Scholar] [CrossRef]
- Carrera, E.; Tononi, G. Diaschisis: Past, Present, Future. Brain J. Neurol. 2014, 137, 2408–2422. [Google Scholar] [CrossRef] [PubMed]
- Duffau, H. Does Post-Lesional Subcortical Plasticity Exist in the Human Brain? Neurosci. Res. 2009, 65, 131–135. [Google Scholar] [CrossRef]
- Herbet, G.; Maheu, M.; Costi, E.; Lafargue, G.; Duffau, H. Mapping Neuroplastic Potential in Brain-Damaged Patients. Brain J. Neurol. 2016, 139, 829–844. [Google Scholar] [CrossRef]
- Desmurget, M.; Bonnetblanc, F.; Duffau, H. Contrasting Acute and Slow-Growing Lesions: A New Door to Brain Plasticity. Brain 2007, 130, 898–914. [Google Scholar] [CrossRef]
- Karns, C.M.; Dow, M.W.; Neville, H.J. Altered Cross-Modal Processing in the Primary Auditory Cortex of Congenitally Deaf Adults: A Visual-Somatosensory fMRI Study with a Double-Flash Illusion. J. Neurosci. Off. J. Soc. Neurosci. 2012, 32, 9626–9638. [Google Scholar] [CrossRef]
- Carmichael, S.T. Plasticity of Cortical Projections after Stroke. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2003, 9, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Sydnor, V.J.; Satterthwaite, T.D. Neuroimaging of Plasticity Mechanisms in the Human Brain: From Critical Periods to Psychiatric Conditions. Neuropsychopharmacology 2023, 48, 219–220. [Google Scholar] [CrossRef] [PubMed]
- Leon-Rojas, J.; Cornell, I.; Rojas-Garcia, A.; D’Arco, F.; Panovska-Griffiths, J.; Cross, H.; Bisdas, S. The Role of Preoperative Diffusion Tensor Imaging in Predicting and Improving Functional Outcome in Pediatric Patients Undergoing Epilepsy Surgery: A Systematic Review. BJR Open 2021, 3, 20200002. [Google Scholar] [CrossRef]
- Alfonso, J.; Fernández, M.E.; Cooper, B.; Flugge, G.; Frasch, A.C. The Stress-Regulated Protein M6a Is a Key Modulator for Neurite Outgrowth and Filopodium/Spine Formation. Proc. Natl. Acad. Sci. USA 2005, 102, 17196–17201. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Chopp, M.; Weiland, B.; Zhang, X.; Tepley, N.; Jiang, F.; Schallert, T. Sensorimotor Deficits Associated with Brain Tumor Progression and Tumor-Induced Brain Plasticity Mechanisms. Exp. Neurol. 2007, 207, 357–367. [Google Scholar] [CrossRef]
- Goel, S.; Wharton, S.B.; Brett, L.P.; Whittle, I.R. Morphological Changes and Stress Responses in Neurons in Cerebral Cortex Infiltrated by Diffuse Astrocytoma. Neuropathology 2003, 23, 262–270. [Google Scholar] [CrossRef]
- Cicvaric, A.; Yang, J.; Krieger, S.; Khan, D.; Eun-Jung, K.; Dominguez-Rodriguez, M.; Cabatic, M.; Molz, B.; Acevedo Aguilar, J.P.; Milicevic, R.; et al. The Brain-Tumor Related Protein Podoplanin Regulates Synaptic Plasticity and Hippocampus-Dependent Learning and Memory. Ann. Med. 2016, 48, 652–668. [Google Scholar] [CrossRef]
- Inácio, R.F.; Zanon, R.G.; de Castro, M.V.; de Souza, H.M.; Bajgelman, M.C.; Verinaud, L.; de Oliveira, A.L.R. Astroglioma Conditioned Medium Increases Synaptic Elimination and Correlates with Major Histocompatibility Complex of Class I (MHC I) Upregulation in PC12Cells. Neurosci. Lett. 2016, 634, 160–167. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, S.; Yang, Z.; Zhang, T. Impaired Hippocampal Synaptic Plasticity in C6 Glioma-Bearing Rats. J. Neurooncol. 2011, 103, 469–477. [Google Scholar] [CrossRef]
- Becker, D.; Deller, T.; Vlachos, A. Tumor Necrosis Factor (TNF)-Receptor 1 and 2 Mediate Homeostatic Synaptic Plasticity of Denervated Mouse Dentate Granule Cells. Sci. Rep. 2015, 5, 12726. [Google Scholar] [CrossRef]
- Lonjon, M.; Quentien, M.H.; Risso, J.J.; Michiels, J.F.; Carre, E.; Rostain, J.C.; Darbin, O. Alteration of Striatal Dopaminergic Function Induced by Glioma Development: A Microdialysis and Immunohistological Study in the Rat Striatum. Neurosci. Lett. 2004, 354, 131–134. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Peng, D.; Huang, Y.; Cao, Y.; Li, H.; Zhang, X. Podoplanin: Its Roles and Functions in Neurological Diseases and Brain Cancers. Front. Pharmacol. 2022, 13, 964973. [Google Scholar] [CrossRef]
- Eng, L.F.; Ghirnikar, R.S.; Lee, Y.L. Glial Fibrillary Acidic Protein: GFAP-Thirty-One Years (1969–2000). Neurochem. Res. 2000, 25, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rheinstein, P.H.; Green, S.; Rosenzweig, K.E. Von Willebrand Factor Gene Expression in Primary Lower Grade Glioma: Mutually Co-Occurring Mutations in von Willebrand Factor, ATRX, and TP53. Brain Tumor Res. Treat. 2019, 7, 33–38. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic Synaptic Input to Glioma Cells Drives Brain Tumour Progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell 2015, 161, 803–816. [Google Scholar] [CrossRef]
- Duffau, H. The Huge Plastic Potential of Adult Brain and the Role of Connectomics: New Insights Provided by Serial Mappings in Glioma Surgery. Cortex 2014, 58, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Johung, T.; Monje, M. Neuronal Activity in the Glioma Microenvironment. Curr. Opin. Neurobiol. 2017, 47, 156–161. [Google Scholar] [CrossRef]
- Brandao, M.; Simon, T.; Critchley, G.; Giamas, G. Astrocytes, the Rising Stars of the Glioblastoma Microenvironment. Glia 2019, 67, 779–790. [Google Scholar] [CrossRef]
- Munno, D.W.; Syed, N.I. Synaptogenesis in the CNS: An Odyssey from Wiring Together to Firing Together. J. Physiol. 2003, 552, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Kong, N.W.; Gibb, W.R.; Tate, M.C. Neuroplasticity: Insights from Patients Harboring Gliomas. Neural. Plast. 2016, 2016, 2365063. [Google Scholar] [CrossRef]
- Yuan, T.; Ying, J.; Zuo, Z.; Gui, S.; Gao, Z.; Li, G.; Zhang, Y.; Li, C. Structural Plasticity of the Bilateral Hippocampus in Glioma Patients. Aging 2020, 12, 10259–10274. [Google Scholar] [CrossRef]
- Otten, M.L.; Mikell, C.B.; Youngerman, B.E.; Liston, C.; Sisti, M.B.; Bruce, J.N.; Small, S.A.; McKhann, G.M. Motor Deficits Correlate with Resting State Motor Network Connectivity in Patients with Brain Tumours. Brain J. Neurol. 2012, 135, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jin, Y.; Wu, D.; Zhang, L. A Depression Network Caused by Brain Tumours. Brain Struct. Funct. 2022, 227, 2787–2795. [Google Scholar] [CrossRef] [PubMed]
- Cao, T.; Pang, J.C.; Segal, A.; Chen, Y.-C.; Aquino, K.M.; Breakspear, M.; Fornito, A. Mode-Based Morphometry: A Multiscale Approach to Mapping Human Neuroanatomy. Hum. Brain Mapp. 2024, 45, e26640. [Google Scholar] [CrossRef]
- Almairac, F.; Duffau, H.; Herbet, G. Contralesional Macrostructural Plasticity of the Insular Cortex in Patients with Glioma. Neurology 2018, 91, e1902–e1908. [Google Scholar] [CrossRef]
- Ballato, M.; Germanà, E.; Ricciardi, G.; Giordano, W.G.; Tralongo, P.; Buccarelli, M.; Castellani, G.; Ricci-Vitiani, L.; D’Alessandris, Q.G.; Giuffrè, G.; et al. Understanding Neovascularization in Glioblastoma: Insights from the Current Literature. Int. J. Mol. Sci. 2025, 26, 2763. [Google Scholar] [CrossRef]
- Bourdillon, P.; Apra, C.; Guénot, M.; Duffau, H. Similarities and Differences in Neuroplasticity Mechanisms between Brain Gliomas and Nonlesional Epilepsy. Epilepsia 2017, 58, 2038–2047. [Google Scholar] [CrossRef]
- Cargnelutti, E.; Ius, T.; Skrap, M.; Tomasino, B. What Do We Know about Pre- and Postoperative Plasticity in Patients with Glioma? A Review of Neuroimaging and Intraoperative Mapping Studies. NeuroImage: Clin. 2020, 28, 102435. [Google Scholar] [CrossRef]
- Fisicaro, R.A.; Jost, E.; Shaw, K.; Brennan, N.P.; Peck, K.K.; Holodny, A.I. Cortical Plasticity in the Setting of Brain Tumors. Top. Magn. Reson. Imaging 2016, 25, 25. [Google Scholar] [CrossRef] [PubMed]
- Kunesch, E.; Classen, J.; Bettag, M.; Kahn, T.; Ulrich, F.; Bock, W.J.; Freund, H.J.; Seitz, R.J. Representational Cortical Plasticity Associated with Brain Tumours: Evidence from Laser-Induced Interstitial Thermotherapy. Acta Neurol. Scand. 2003, 108, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Charras, P.; Herbet, G.; Deverdun, J.; de Champfleur, N.M.; Duffau, H.; Bartolomeo, P.; Bonnetblanc, F. Functional Reorganization of the Attentional Networks in Low-Grade Glioma Patients: A Longitudinal Study. Cortex 2015, 63, 27–41. [Google Scholar] [CrossRef]
- Cho, N.S.; Peck, K.K.; Zhang, Z.; Holodny, A.I. Paradoxical Activation in the Cerebellum During Language fMRI in Patients with Brain Tumors: Possible Explanations Based on Neurovascular Uncoupling and Functional Reorganization. Cerebellum 2018, 17, 286–293. [Google Scholar] [CrossRef]
- Meyer, P.T.; Sturz, L.; Schreckenberger, M.; Spetzger, U.; Meyer, G.F.; Setani, K.S.; Sabri, O.; Buell, U. Preoperative Mapping of Cortical Language Areas in Adult Brain Tumour Patients Using PET and Individual Non-Normalised SPM Analyses. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 951–960. [Google Scholar] [CrossRef]
- Fandino, J.; Kollias, S.S.; Wieser, H.G.; Valavanis, A.; Yonekawa, Y. Intraoperative Validation of Functional Magnetic Resonance Imaging and Cortical Reorganization Patterns in Patients with Brain Tumors Involving the Primary Motor Cortex. J. Neurosurg. 1999, 91, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Parisot, S.; Darlix, A.; Baumann, C.; Zouaoui, S.; Yordanova, Y.; Blonski, M.; Rigau, V.; Chemouny, S.; Taillandier, L.; Bauchet, L.; et al. A Probabilistic Atlas of Diffuse WHO Grade II Glioma Locations in the Brain. PLoS ONE 2016, 11, e0144200. [Google Scholar] [CrossRef]
- Chieffo, D.P.R.; Lino, F.; Ferrarese, D.; Belella, D.; Della Pepa, G.M.; Doglietto, F. Brain Tumor at Diagnosis: From Cognition and Behavior to Quality of Life. Diagnostics 2023, 13, 541. [Google Scholar] [CrossRef]
- Zilioli, A.; Misirocchi, F.; Mutti, C.; Pancaldi, B.; Mannini, E.; Spallazzi, M.; Parrino, L.; Cerasti, D.; Michiara, M.; Florindo, I. Volumetric Hippocampal Changes in Glioblastoma: A Biomarker for Neuroplasticity? J. Neurooncol. 2023, 163, 261–267. [Google Scholar] [CrossRef]
- Duffau, H. Stimulation Mapping of White Matter Tracts to Study Brain Functional Connectivity. Nat. Rev. Neurol. 2015, 11, 255–265. [Google Scholar] [CrossRef]
- Saur, D.; Ronneberger, O.; Kümmerer, D.; Mader, I.; Weiller, C.; Klöppel, S. Early Functional Magnetic Resonance Imaging Activations Predict Language Outcome after Stroke. Brain 2010, 133, 1252–1264. [Google Scholar] [CrossRef]
- Duffau, H. A Two-Level Model of Interindividual Anatomo-Functional Variability of the Brain and Its Implications for Neurosurgery. ortex J. Devoted Study Nerv. Syst. Behav. 2017, 86, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Nakada, M.; Kinoshita, M.; Hamada, J. Functional Reorganization in the Patient with Progressing Glioma of the Pure Primary Motor Cortex: A Case Report with Special Reference to the Topographic Central Sulcus Defined by Somatosensory-Evoked Potential. World Neurosurg. 2014, 82, 536.e1–536.e4. [Google Scholar] [CrossRef] [PubMed]
- Ius, T.; Isola, M.; Budai, R.; Pauletto, G.; Tomasino, B.; Fadiga, L.; Skrap, M. Low-Grade Glioma Surgery in Eloquent Areas: Volumetric Analysis of Extent of Resection and Its Impact on Overall Survival. A Single-Institution Experience in 190 Patients: Clinical Article. J. Neurosurg. 2012, 117, 1039–1052. [Google Scholar] [CrossRef]
- Mandonnet, E.; Capelle, L.; Duffau, H. Extension of Paralimbic Low Grade Gliomas: Toward an Anatomical Classification Based on White Matter Invasion Patterns. J. Neurooncol. 2006, 78, 179–185. [Google Scholar] [CrossRef]
- Leon-Rojas, J.E.; Ekert, J.O.; Kirkman, M.A.; Sewell, D.; Bisdas, S.; Samandouras, G. Experience with Awake throughout Craniotomy in Tumour Surgery: Technique and Outcomes of a Prospective, Consecutive Case Series with Patient Perception Data. Acta Neurochir. 2020, 162, 3055–3065. [Google Scholar] [CrossRef]
- Duffau, H. The Challenge to Remove Diffuse Low-Grade Gliomas While Preserving Brain Functions. Acta Neurochir. 2012, 154, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Kesler, S.R.; Harrison, R.A.; Rao, V.; Dyson, H.; Petersen, M.; Prinsloo, S. Predicting Overall Survival in Diffuse Glioma from the Presurgical Connectome. Sci. Rep. 2022, 12, 18783. [Google Scholar] [CrossRef]
- Kiran, S.; Thompson, C.K. Neuroplasticity of Language Networks in Aphasia: Advances, Updates, and Future Challenges. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef]
- Ali, J.I.; Viczko, J.; Smart, C.M. Efficacy of Neurofeedback Interventions for Cognitive Rehabilitation Following Brain Injury: Systematic Review and Recommendations for Future Research. J. Int. Neuropsychol. Soc. JINS 2020, 26, 31–46. [Google Scholar] [CrossRef]
- De Luca, R.; Calabrò, R.S.; Bramanti, P. Cognitive Rehabilitation after Severe Acquired Brain Injury: Current Evidence and Future Directions. Neuropsychol. Rehabil. 2018, 28, 879–898. [Google Scholar] [CrossRef] [PubMed]
- Weyer-Jamora, C.; Brie, M.S.; Luks, T.L.; Smith, E.M.; Hervey-Jumper, S.L.; Taylor, J.W. Postacute Cognitive Rehabilitation for Adult Brain Tumor Patients. Neurosurgery 2021, 89, 945–953. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Molecule/ Protein | Setting * | Category ¶ | Regulation | Plasticity Change | Promotes (+) Reduces (−) Plasticity | Reference |
|---|---|---|---|---|---|---|
| M6a | stress | 5 | ↑ | neurite and filopodium growth + synapse formation | (+) | [16] |
| MAP2 | gliosarcoma | 5 | ↑ | neuronal migration and neurite outgrowth | (+) | [17,18] |
| GFAP | gliosarcoma | 6 | ↑ | synaptic plasticity | (+) | [17] |
| vWF | gliosarcoma | 7 | ↑ | angiogenesis | (+) | [17] |
| Synaptophysin | astroglioma medium | 6 | ↑ | synaptogenesis | (+) | [16,20] |
| MHC I | astroglioma medium | 6 | ↑ | negative influence on synaptic stability | (−) | [20] |
| Glutamate | glioma | 6 | ↑ | impairment of excitatory and inhibitory synaptic transmission | (−) | [21] |
| Glu/GABA | glioma | 6 | ↑ | impairment of excitatory and inhibitory synaptic transmission | (−) | [21] |
| Neurofilament | glioblastoma | 6 | ↓ | staining showed gradual reduction within tumour centre | (−) | [23] |
| Tyrosine hydroxylase | glioblastoma | 6 | ↓ | staining showed gradual reduction within tumour centre | (−) | [23] |
| Dopamine transporter | glioblastoma | 6 | ↓ | decrease in density of dopaminergic endings | (−) | [23] |
| GAP-43 | diffuse astrocytoma | 5 | No change | no change in tumour model vs. control | No change | [18] |
| Podoplanin | overexpression experiments | 7 | ↑ | brain neuronal outgrowth, synaptic plasticity | (+) | [19] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vinueza, L.; Pineda, S.; Leon-Rojas, J.E. Molecular Foundations of Neuroplasticity in Brain Tumours: From Microscopic Adaptation to Functional Reorganisation. Int. J. Mol. Sci. 2025, 26, 7049. https://doi.org/10.3390/ijms26157049
Vinueza L, Pineda S, Leon-Rojas JE. Molecular Foundations of Neuroplasticity in Brain Tumours: From Microscopic Adaptation to Functional Reorganisation. International Journal of Molecular Sciences. 2025; 26(15):7049. https://doi.org/10.3390/ijms26157049
Chicago/Turabian StyleVinueza, Lizeth, Salvador Pineda, and Jose E. Leon-Rojas. 2025. "Molecular Foundations of Neuroplasticity in Brain Tumours: From Microscopic Adaptation to Functional Reorganisation" International Journal of Molecular Sciences 26, no. 15: 7049. https://doi.org/10.3390/ijms26157049
APA StyleVinueza, L., Pineda, S., & Leon-Rojas, J. E. (2025). Molecular Foundations of Neuroplasticity in Brain Tumours: From Microscopic Adaptation to Functional Reorganisation. International Journal of Molecular Sciences, 26(15), 7049. https://doi.org/10.3390/ijms26157049