
Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms


Abstract
1. Introduction
2. Results
2.1. Identification and Evaluation of Rust Resistance in 24 Leymus chinensis Germplasms Under Laboratory Conditions
2.2. Transcriptomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Fungus Infection
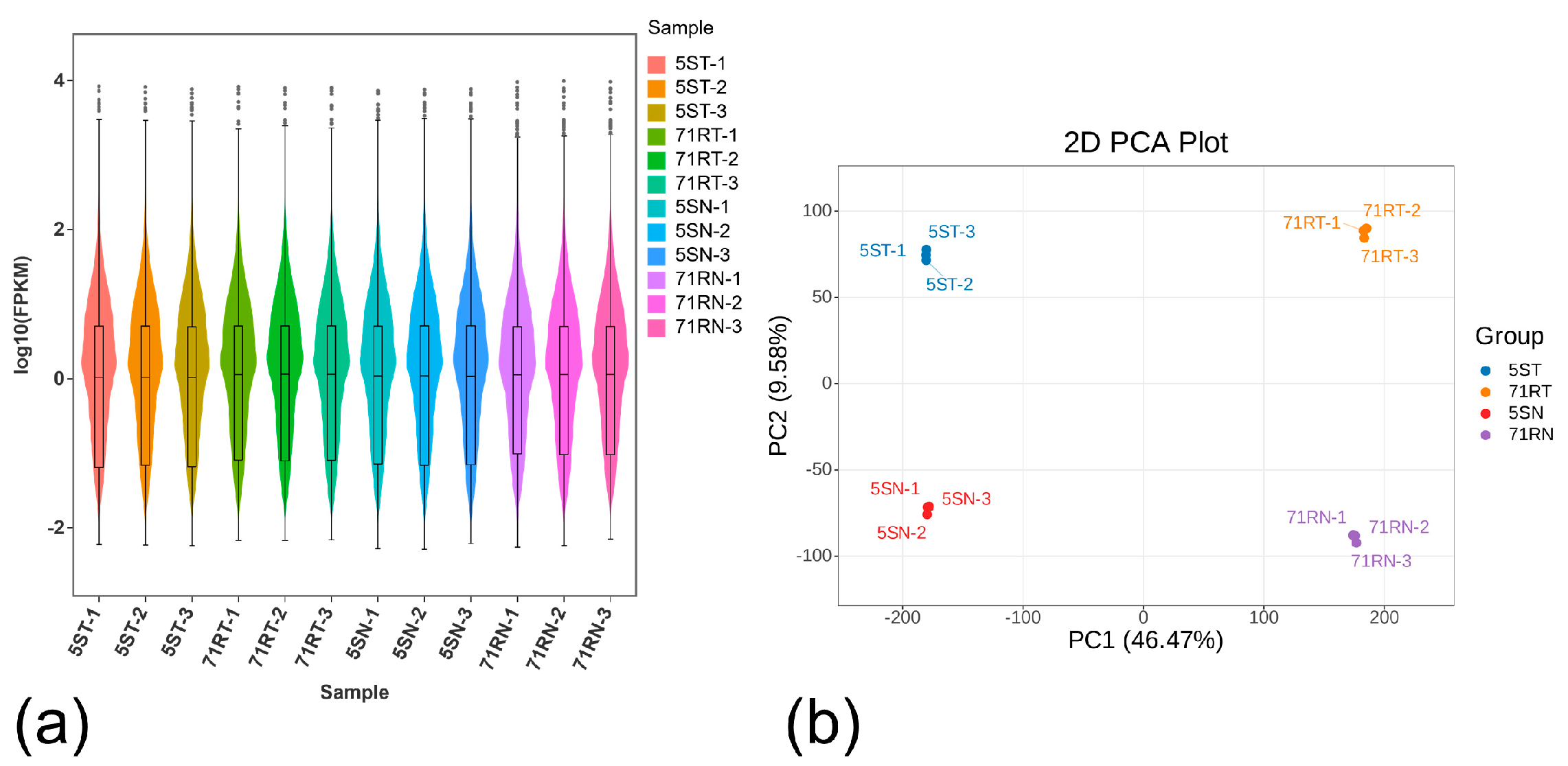
2.2.1. RNA Sequencing and Quality Control of Samples
2.2.2. Analysis of Gene Expression Levels and Principal Component Analysis (PCA) Between Samples
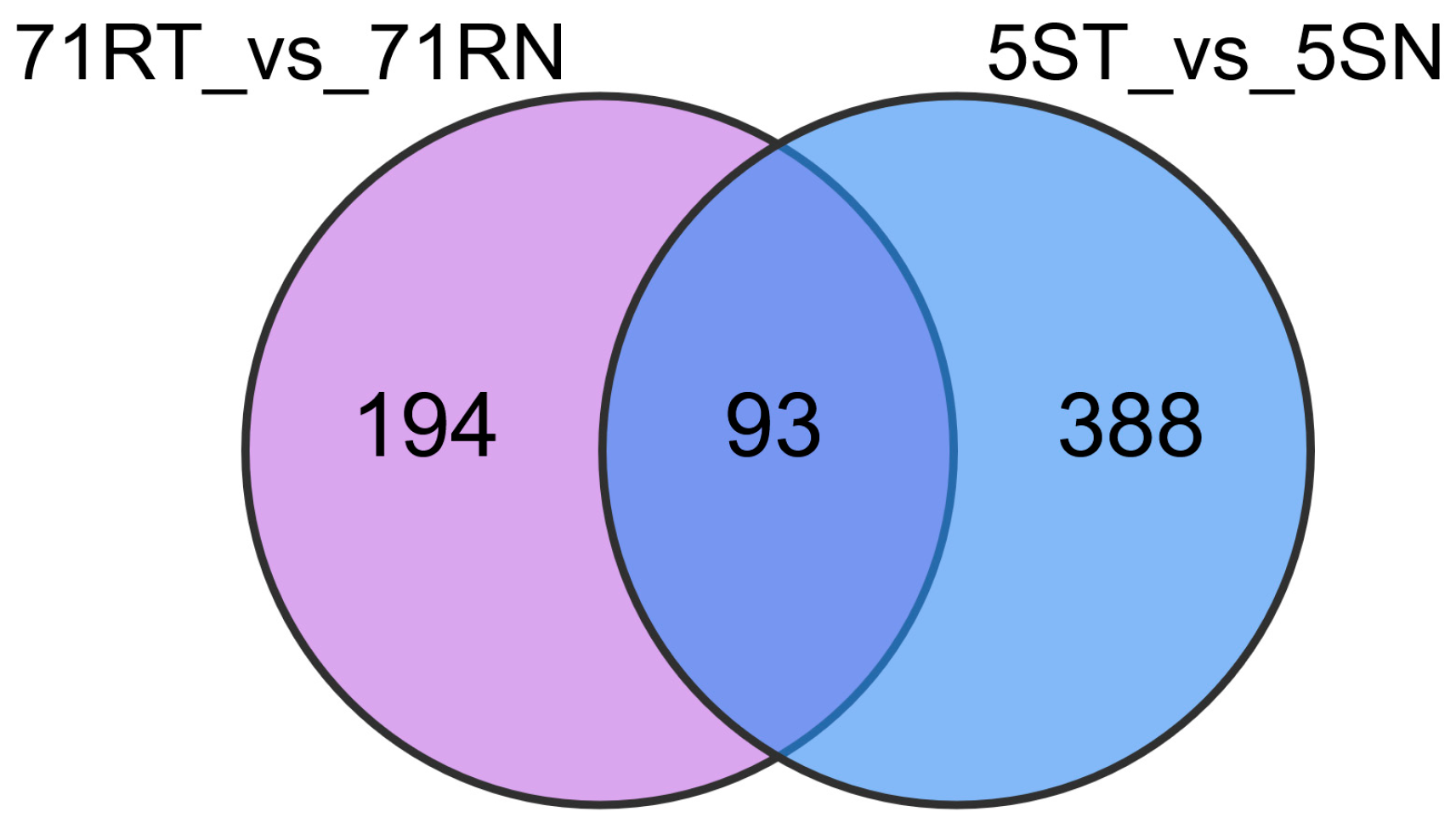
2.2.3. Screening of Differentially Expressed Genes (DEGs)
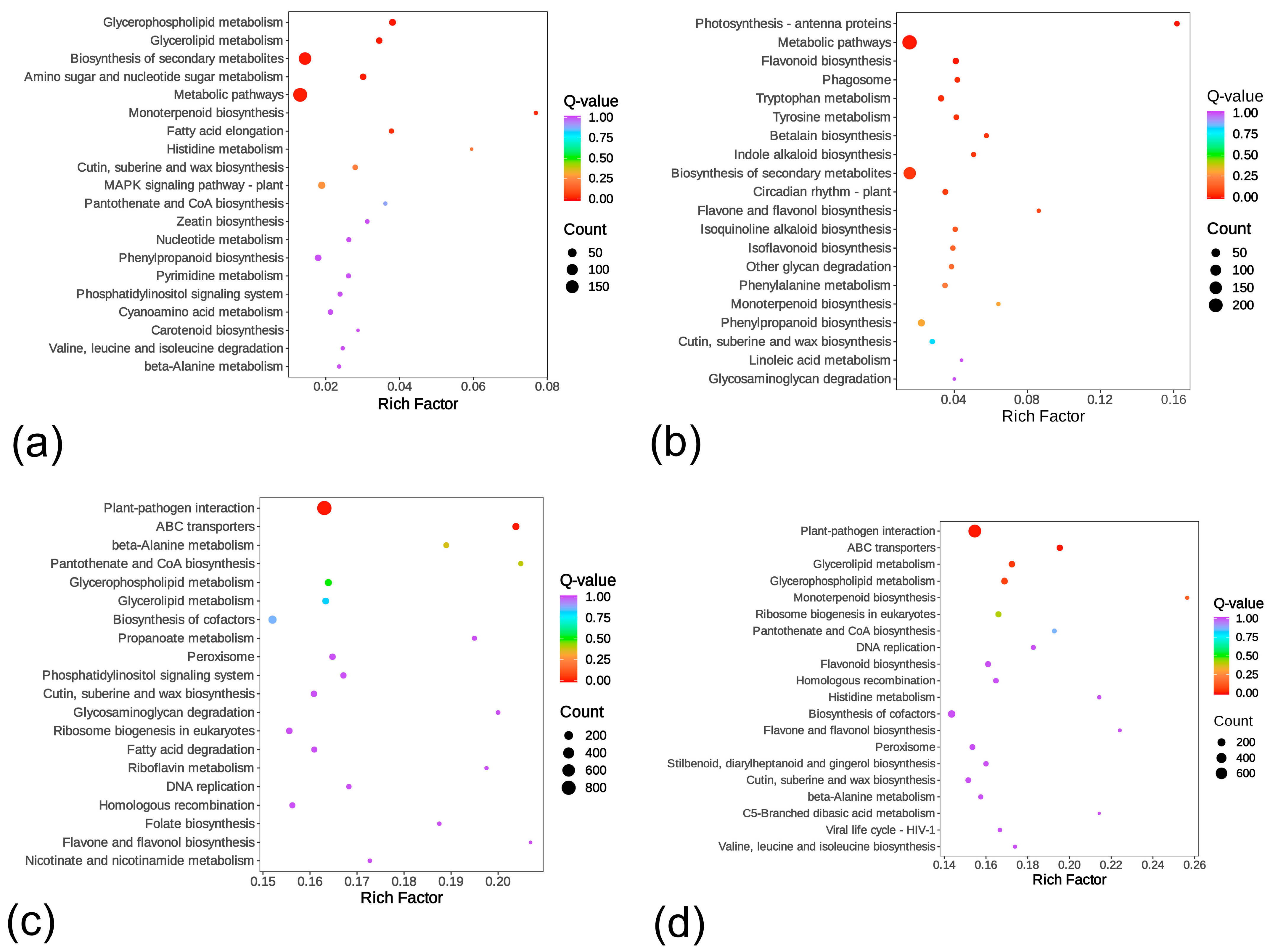
2.2.4. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis of DEGs
2.3. Metabolomic Reprogramming Signatures in Rust-Infected L. chinensis Germplasms
2.3.1. Metabolomic Data and Quality Control (QC) of Samples
2.3.2. Principal Component Analysis (PCA) of Metabolites and Validation of Orthogonal Partial Least Squares-Discriminant Analysis (OPLS-DA) Model
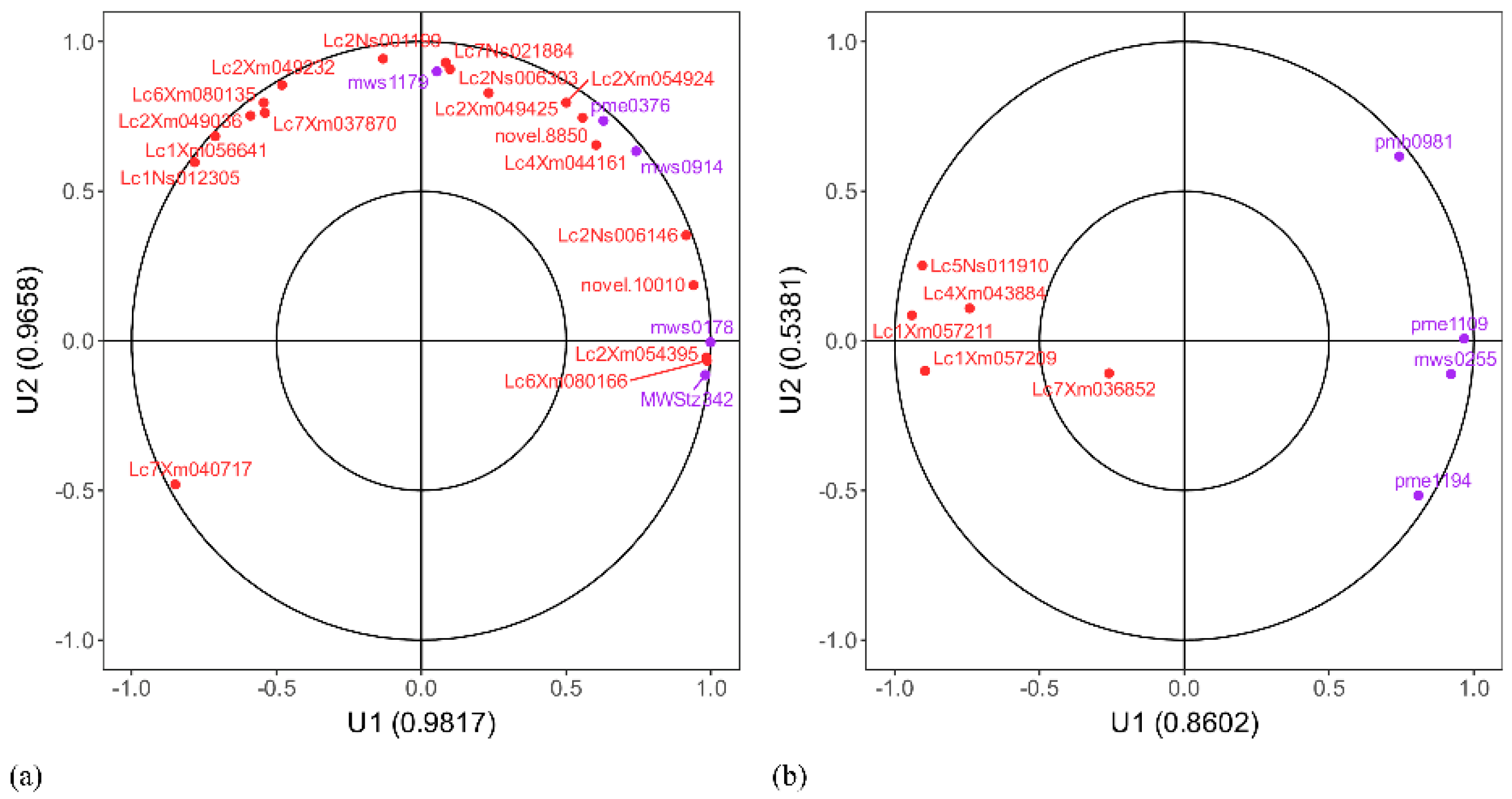
2.3.3. Screening of Differentially Accumulated Metabolites (DAMs)
2.3.4. Kyoto Encyclopedia of Genes and Genomes (KEGG) Functional Annotation and Enrichment Analysis of Differentially Accumulated Metabolites (DAMs)
2.4. Coordinated Transcriptomic Metabolomic Dynamics During Rust Infection
2.4.1. KEGG Pathway Co-Enrichment Analysis
2.4.2. Expression Correlation Analysis
3. Discussion
3.1. Evaluation of Rust Resistance in Leymus chinensis
3.2. Transcriptomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
3.3. Metabolomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
3.4. Integrated Transcriptomic and Metabolomic Analysis of Resistant and Susceptible L. chinensis in Response to Rust Infection
4. Materials and Methods
4.1. Materials
4.2. Evaluation of Rust Resistance in 24 Leymus chinensis Germplasms Under Laboratory Conditions
4.3. Inoculation of Rust Fungi and Sampling in Resistant and Susceptible L. chinensis
4.4. Transcriptome Sequencing and Data Analysis
4.5. Metabolome Sequencing and Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.X.; Ma, H.Y.; Ni, H.W.; Li, S.Y.; Xu, L.; Sun, M.D.; Qi, W.W.; Zhao, D.D. Adaptation Responses of Different Ecotypes of Leymus chinensis to Saline–Alkaline Stress. Front. Ecol. Evol. 2024, 12, 1361124. [Google Scholar] [CrossRef]
- Wu, Z.N.; Hou, X.Y.; Ren, W.B.; Wang, Z.L.; Chang, C.; Yang, Y.P.; Yang, Y.T. Prediction of the Potential Geographic Distribution of Leymus chinensis Based on MaxEnt and Collection and Protection of Germplasm. Acta Pratacult. Sin. 2018, 27, 125–135. [Google Scholar]
- Guo, H.H.; Hao, M.D.; Meng, J.; Xiao, Q.H.; Wu, D.B.; Liu, G.S. Fertilization Effects on Grass Yield and Nutrient Uptake of Leymus chinensis in Artificial Grasslands. J. Ningxia Univ. Nat. Sci. Ed. 2016, 37, 118–124. [Google Scholar]
- Niu, Y.; Yang, S.; Wang, G.; Liu, L.; Hua, L. Effects of Grazing Disturbance on Plant Diversity, Community Structure and Direction of Succession in an Alpine Meadow on Tibet Plateau, China. Acta Ecol. Sin. 2018, 38, 179–185. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate Transcript Quantification from RNA-Seq Data with or without a Reference Genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; You, Y.; Wang, D.; Zhu, L. Quality Evaluation of the Soil-Root Composites Layer of Leymus chinensis Grassland Based on Different Degradation Degrees. Catena 2022, 215, 106330. [Google Scholar] [CrossRef]
- Hovmøller, M.S.; Sørensen, C.K.; Walter, S.; Justesen, A.F. Diversity of Puccinia striiformis on Cereals and Grasses. Annu. Rev. Phytopathol. 2011, 49, 197–217. [Google Scholar] [CrossRef] [PubMed]
- Gultyaeva, E.I.; Bespalova, L.A.; Ablova, I.B.; Shaydayuk, E.L.; Khudokormova, Z.N.; Yakovleva, D.R.; Titova, Y.A. Wild Grasses as the Reservoirs of Infection of Rust Species for Winter Soft Wheat in the Northern Caucasus. Vavilovskii Zhurnal Genet. Selektsii 2021, 25, 638–646. [Google Scholar] [CrossRef] [PubMed]
- Ren, S.; Sun, M.; Yan, H.; Wu, B.; Jing, T.; Huang, L.; Zeng, B. Identification and Distribution of NBS-Encoding Resistance Genes of Dactylis glomerata L. and Its Expression under Abiotic and Biotic Stress. Biochem. Genet. 2020, 58, 824–847. [Google Scholar] [CrossRef] [PubMed]
- Park, R.F.; Boshoff, W.H.P.; Cabral, A.L.; Chong, J.; Martinelli, J.A.; McMullen, M.S.; Fetch, J.W.M.; Paczos-Grzęda, E.; Prats, E.; Roake, J.; et al. Breeding Oat for Resistance to the Crown Rust Pathogen Puccinia coronata f. Sp. Avenae: Achievements and Prospects. TAG Theor. Appl. Genet. Theor. Angew. Genet. 2022, 135, 3709–3734. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Wang, Z.; Shao, X.; Wang, Y. Advances in Genetics and Breeding for Rust Diseases Resistance in Turfgrasses. Pratacult. Sci. 2011, 28, 436–443. [Google Scholar]
- Daudi, H.; Shimelis, H.; Mathew, I.; Rathore, A.; Ojiewo, C.O. Combining Ability and Gene Action Controlling Rust Resistance in Groundnut (Arachis hypogaea L.). Sci. Rep. 2021, 11, 16513. [Google Scholar] [CrossRef] [PubMed]
- An, J.X.; Ma, Y.; Zhao, W.B.; Hu, Y.M.; Wang, Y.R.; Zhang, Z.J.; Luo, X.F.; Zhang, B.Q.; Ding, Y.Y.; Liu, Y.Q. Drug Repurposing Strategy II: From Approved Drugs to Agri-Fungicide Leads. J. Antibiot. 2023, 76, 131–182. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, Y.W.; Nan, Z.B.; Duan, T.Y. Progress of Research into the Effects of Native Grassland Management Practices on Plant Disease. Acta Ecol. Sin. 2016, 36, 4211–4220. [Google Scholar] [CrossRef]
- Islam, T.; Danishuddin; Tamanna, N.T.; Matin, M.N.; Barai, H.R.; Haque, M.A. Resistance Mechanisms of Plant Pathogenic Fungi to Fungicide, Environmental Impacts of Fungicides, and Sustainable Solutions. Plants 2024, 13, 2737. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Wang, B.; Zhang, Q.; Fu, Y.; Huang, L.; Wang, X.; Kang, Z. Exploration of microRNAs and Their Targets Engaging in the Resistance Interaction between Wheat and Stripe Rust. Front. Plant Sci. 2015, 6, 469. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Zhou, X.; Feng, J. Editorial: Wheat Disease Resistance: Diagnosis, Germplasm Mining, and Molecular Breeding. Front. Plant Sci. 2024, 15, 1500414. [Google Scholar] [CrossRef] [PubMed]
- Boller, T.; Felix, G. A Renaissance of Elicitors: Perception of Microbe-Associated Molecular Patterns and Danger Signals by Pattern-Recognition Receptors. Annu. Rev. Plant Biol. 2009, 60, 379–406. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, A.; Hayashi, M. Building the Interaction Interfaces: Host Responses upon Infection with Microorganisms. Curr. Opin. Plant Biol. 2015, 23, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.Q.; Niu, H.Q.; Liu, C.; Wang, H.L.; Yin, W.; Xia, X. PTI-ETI Synergistic Signal Mechanisms in Plant Immunity. Plant Biotechnol. J. 2024, 22, 2113–2128. [Google Scholar] [CrossRef] [PubMed]
- Pfeilmeier, S.; Saur, I.M.-L.; Rathjen, J.P.; Zipfel, C.; Malone, J.G. High Levels of Cyclic-Di-GMP in Plant-Associated Pseudomonas Correlate with Evasion of Plant Immunity. Mol. Plant Pathol. 2016, 17, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xing, Z.; Li, W.; Yang, X. Study on Diversity of Undergrowth Plant Community in Cibagou Nature Reserve. Am. J. Plant Sci. 2017, 8, 2149–2158. [Google Scholar] [CrossRef]
- Shi, H.; Shen, Q.; Qi, Y.; Yan, H.; Nie, H.; Chen, Y.; Zhao, T.; Katagiri, F.; Tang, D. BR-SIGNALING KINASE1 Physically Associates with FLAGELLIN SENSING2 and Regulates Plant Innate Immunity in Arabidopsis. Plant Cell 2013, 25, 1143–1157. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, W.; Xiang, T.; Liu, Z.; Laluk, K.; Ding, X.; Zou, Y.; Gao, M.; Zhang, X.; Chen, S.; et al. Receptor-like Cytoplasmic Kinases Integrate Signaling from Multiple Plant Immune Receptors and Are Targeted by a Pseudomonas syringae Effector. Cell Host Microbe 2010, 7, 290–301. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Cai, S.; Wu, P.; Dai, L.; Li, X.; Ai, N.; Feng, G.; Wang, N.; Zhou, B. General Regulatory Factor7 Regulates Innate Immune Signalling to Enhance Verticillium Wilt Resistance in Cotton. J. Exp. Bot. 2024, 75, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Rüter, C.; Schmidt, M.A. Cell-Penetrating Bacterial Effector Proteins: Better Tools than Targets. Trends Biotechnol. 2017, 35, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Macho, A.P.; Zipfel, C. Targeting of Plant Pattern Recognition Receptor-Triggered Immunity by Bacterial Type-III Secretion System Effectors. Curr. Opin. Microbiol. 2015, 23, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Mu, Q.; Wang, X.; Zhang, J.; Yu, H.; Huang, T.; He, Y.; Dai, S.; Meng, X. Multilayered Synergistic Regulation of Phytoalexin Biosynthesis by Ethylene, Jasmonate, and MAPK Signaling Pathways in Arabidopsis. Plant Cell 2022, 34, 3066–3087. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Tang, D. Synergistic Cooperation Between Cell Surface and Intracellular Immune Receptors Potentiates to Activate Robust Plant Defense. Chin. Bull. Bot. 2021, 56, 142–146. [Google Scholar]
- Ngou, B.P.M.; Ahn, H.-K.; Ding, P.; Jones, J.D.G. Mutual Potentiation of Plant Immunity by Cell-Surface and Intracellular Receptors. Nature 2021, 592, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, F.; Asp, T.; Widmer, F.; Kölliker, R. Transcriptional Responses of Italian Ryegrass during Interaction with Xanthomonas translucens Pv. Graminis Reveal Novel Candidate Genes for Bacterial Wilt Resistance. Theor. Appl. Genet. 2011, 122, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zhong, Q.; Yin, F.; Li, J.; Liu, L.; Zhang, Y.; Wang, B.; Jiang, C.; Cheng, Z.; Zhang, H.; et al. Research Progress on Germplasm Resources and Multi-Omics of Oryza officinalis. Crops 2025, 41, 1–8. [Google Scholar] [CrossRef]
- Marshall, D.D.; Powers, R. Beyond the Paradigm: Combining Mass Spectrometry and Nuclear Magnetic Resonance for Metabolomics. Prog. Nucl. Magn. Reson. Spectrosc. 2017, 100, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.; Durling, M.B.; de Jager, V.; Menezes, R.C.; Nordkvist, E.; Svatoš, A.; Dubey, M.; Lauterbach, L.; Dickschat, J.S.; Karlsson, M.; et al. Deciphering the Genome and Secondary Metabolome of the Plant Pathogen Fusarium culmorum. FEMS Microbiol. Ecol. 2018, 94, fiy078. [Google Scholar] [CrossRef] [PubMed]
- Robison, F.M.; Turner, M.F.; Jahn, C.E.; Schwartz, H.F.; Prenni, J.E.; Brick, M.A.; Heuberger, A.L. Common Bean Varieties Demonstrate Differential Physiological and Metabolic Responses to the Pathogenic Fungus Sclerotinia sclerotiorum. Plant Cell Environ. 2018, 41, 2141–2154. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Ruan, Z.; Fei, Z.; Yan, J.; Tang, G. Integrated Transcriptome and Metabolome Analysis Revealed That Flavonoid Biosynthesis May Dominate the Resistance of Zanthoxylum bungeanum against Stem Canker. J. Agric. Food Chem. 2021, 69, 6360–6378. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Li, H.; Yang, Y. Phenotypic Plasticity in Sexual Reproduction Based on Nutrients Supplied from Vegetative Ramets in a Leymus chinensis Population. Front. Plant Sci. 2019, 10, 1681. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Hou, X.; Wu, Z.; Kong, L.; Guo, H.; Hu, N.; Wan, D.; Zhang, J. De Novo Transcriptomic Profiling of the Clonal Leymus chinensis Response to Long-Term Overgrazing-Induced Memory. Sci. Rep. 2018, 8, 17912. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Ma, Z.H. A Catalogue of Forage Grass Diseases in Ningxia, China. J. Ningxia Agric. Coll. 1994, 57–64. [Google Scholar]
- Zhang, Y.; Nan, Z.; Christensen, M.J.; Xin, X.; Zhang, N. Effects of Rust on Plant Growth and Stoichiometry of Leymus chinensis under Different Grazing Intensities in Hulunber Grassland. Agriculture 2022, 12, 961. [Google Scholar] [CrossRef]
- Han, D.J.; Kang, Z.S. Current Status and Future Strategy in Breeding Wheat for Resistance to Stripe Rust in China. Plant Prot. 2018, 44, 1–12. [Google Scholar] [CrossRef]
- Tao, Y.; Xu, L.J.; Li, F.; Li, W.L.; Sun, Q.Z.; Xu, C.; Lin, K.J. The Leymus chinensis industry in China needs to be urgently revitalized. Acta Pratacult. Sin. 2023, 32, 188–198. [Google Scholar]
- Zhang, Y.; Xin, X.; Matthew, C.; Christensen, M.J.; Nan, Z. Pathogen Identification and Factors Influencing Infection Frequency and Severity of Fungal Rust in Four Native Grasses in Hulunber Grassland, China. Plant Dis. 2022, 106, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Lazaridi, E.; Kapazoglou, A.; Gerakari, M.; Kleftogianni, K.; Passa, K.; Sarri, E.; Papasotiropoulos, V.; Tani, E.; Bebeli, P.J. Crop Landraces and Indigenous Varieties: A Valuable Source of Genes for Plant Breeding. Plants 2024, 13, 758. [Google Scholar] [CrossRef] [PubMed]
- Miedaner, T.; Flath, K.; Starck, N.; Weißmann, S.; Maurer, H.P. Quantitative-Genetic Evaluation of Resistances to Five Fungal Diseases in a Large Triticale Diversity Panel (Triticosecale). Crops 2022, 2, 218–232. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, C.; Yuan, C.; Zhang, L.; Huang, L.; Wu, J.; Wang, J.; Zheng, Y.; Zhang, H.; Liu, D.; et al. Stripe Rust Resistance in Aegilops tauschii Germplasm. Crop Sci. 2013, 53, 2014–2020. [Google Scholar] [CrossRef]
- Xu, C.; Zhan, C.; Huang, S.; Xu, Q.; Tang, T.; Wang, Y.; Luo, J.; Zeng, X. Resistance to Powdery Mildew in Qingke Involves the Accumulation of Aromatic Phenolamides through Jasmonate-Mediated Activation of Defense-Related Genes. Front. Plant Sci. 2022, 13, 900345. [Google Scholar] [CrossRef] [PubMed]
- Zeng, X.Q.; Luo, X.M.; Wang, Y.L.; Xu, Q.J.; Bai, L.J.; Yuan, H.J.; Tashi, N. Transcriptome Sequencing in a Tibetan Barley Landrace with High Resistance to Powdery Mildew. Sci. World J. 2014, 2014, 594579. [Google Scholar] [CrossRef] [PubMed]
- Saha, G.C.; Sarker, A.; Chen, W.; Vandemark, G.J.; Muehlbauer, F.J. Identification of Markers Associated with Genes for Rust Resistance in Lens culinaris Medik. Euphytica 2010, 175, 261–265. [Google Scholar] [CrossRef]
- Yan, X.-C.; Liu, Q.; Yang, Q.; Wang, K.-L.; Zhai, X.-Z.; Kou, M.-Y.; Liu, J.-L.; Li, S.-T.; Deng, S.-H.; Li, M.-M.; et al. Single-Cell Transcriptomic Profiling of Maize Cell Heterogeneity and Systemic Immune Responses Against Puccinia polysora Underw. Plant Biotechnol. J. 2025, 23, 549–563. [Google Scholar] [CrossRef] [PubMed]
- Meng, Q.J.; Wen, P.; Shen, H.; Yu, L.J.; Yu, Y.S.; Wu, J.J.; Xu, Y.J.; Hu, T.G. Research Progress on Active Substances and Their Processing and Application of Bamboo Shoot Shells. J. Food Saf. Qual. 2025, 16, 110–118. [Google Scholar] [CrossRef]
- Liu, X.; Cui, X.; Ji, D.; Zhang, Z.; Li, B.; Xu, Y.; Chen, T.; Tian, S. Luteolin-Induced Activation of the Phenylpropanoid Metabolic Pathway Contributes to Quality Maintenance and Disease Resistance of Sweet Cherry. Food Chem. 2021, 342, 128309. [Google Scholar] [CrossRef] [PubMed]
- Carletti, G.; Nervo, G.; Cattivelli, L. Flavonoids and Melanins: A Common Strategy across Two Kingdoms. Int. J. Biol. Sci. 2014, 10, 1159–1170. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Hao, L.; Wang, Z.J.; Jia, J.; Shao, Y.T. Advances in Understanding How Branches of the Phenylpropanoid Pathway Respond to Biotic Stress. China Plant Prot. Guide 2023, 43, 23–30. [Google Scholar]
- Huang, Q.X.; Qu, Y.Y.; Yao, Z.P.; Li, M.Y.; Chen, Q.J. Correlation between Resistance to Fusarium Wilt and Expression of Flavonoid Metabolism Related Genes in Gossypium barbadense L. Acta Agron. Sin. 2017, 43, 1791–1801. [Google Scholar] [CrossRef]
- Zuo, T.; Zhao, S.T.; Lu, M.Z.; Sun, A.D.; Wang, Y.W.; He, W. Cloning Dihydroflavonol-4-Reductase Gene (DFR) of Poplar and Its Antisense Expression Effects on Catechin Synthesis. J. Northeast. For. Univ. 2016, 44, 49–55. [Google Scholar]
- Lee, S.; Seo, Y.E.; Choi, J.; Yan, X.; Kim, T.; Choi, D.; Lee, J.H. Nucleolar Actions in Plant Development and Stress Responses. Plant Cell Environ. 2024, 47, 5189–5204. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Zhang, Y.; Liu, J.; Zhang, H.; Wang, T. The Regulatory Roles of Plant miRNAs in Biotic Stress Responses. Biochem. Biophys. Res. Commun. 2025, 755, 151568. [Google Scholar] [CrossRef] [PubMed]
- Witte, C.P.; Herde, M. Nucleotide Metabolism in Plants. Plant Physiol. 2020, 182, 63–78. [Google Scholar] [CrossRef] [PubMed]
- Mashabela, M.D.; Piater, L.A.; Steenkamp, P.A.; Dubery, I.A.; Tugizimana, F.; Mhlongo, M.I. Comparative Metabolite Profiling of Wheat Cultivars (Triticum aestivum) Reveals Signatory Markers for Resistance and Susceptibility to Stripe Rust and Aluminium (Al3+) Toxicity. Metabolites 2022, 12, 98. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.Y.; Liu, J.; Li, Z.Y.; Ma, J.F.; Wang, Y.F.; Quan, J.Z.; Liu, L.; Bai, H.; Dong, Z.P. Cloning and Expression Analysis of SibHLH19 Gene in Foxtail millet. J. North China Univ. Agric. 2022, 37, 16–24. [Google Scholar]
- NY/T 1443.1-2007; Technical Specification for Evaluation of Wheat Resistance to Diseases and Pests Part 1: Technical Specification for Evaluation of Wheat Resistance to Stripe Rust. Ministry of Agriculture of the People’s Republic of China: Beijing, China, 2007.
- Yan, H.D.; Zhang, X.Q.; Zeng, B.; Cheng, L.; Nie, G.; Ji, Y.; Huang, L.K. Evaluating Rust Resistance and Field Investigating Over-summer Conditions of Orchardgrass (Dactylis glomerata L.) Germplasms. Acta Agrestia Sin. 2013, 21, 720–728. [Google Scholar]
- Wu, Q.F.; Ma, X.Y.; Xia, K.; Lu, Q.; Liu, Q.; Zhao, Z.G.; Zheng, W.J.; Qiu, S. Identification of the Pathogen of Bletilla striata Rust and Screening of Rust-Resistant Resources. Guihaia 2024, 44, 1129–1137. [Google Scholar]
- Dong, W.K.; Ma, X.; Mao, C.H.; Deng, J.H.; Jia, X.X.; Zhang, S.P.; Guo, S.S.; Ma, H.L. Resistance Evaluation and Physiological Characteristic Analysis of Ten Poa pratensis Varieties to Powdery mildew. Grassl. Turf 2020, 40, 47–56. [Google Scholar] [CrossRef]
- Jones, J.D.G.; Dangl, J.L. The Plant Immune System. Nature 2006, 444, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Dorostkar, S.; Dadkhodaie, A.; Ebrahimie, E.; Heidari, B.; Ahmadi-Kordshooli, M. Comparative Transcriptome Analysis of Two Contrasting Resistant and Susceptible Aegilops tauschii Accessions to Wheat Leaf Rust (Puccinia triticina) Using RNA-Sequencing. Sci. Rep. 2022, 12, 821. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Tang, S.; Li, W.; Zhang, S.; Wang, J.; Pan, D.; Lin, Z.; Ma, X.; Chang, Y.; Liu, B.; et al. Genome Evolution and Initial Breeding of the Triticeae Grass Leymus chinensis Dominating the Eurasian Steppe. Proc. Natl. Acad. Sci. USA 2023, 120, e2308984120. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Material | Incidence Rate | Disease Index | Resistance Level |
|---|---|---|---|---|
| 1 | Lc3 | 54.44 ± 5.09% bcdefjh | 36.89 ± 2.52 cdef | HS |
| 2 | Lc5 | 66.21 ± 3.35% abcde | 56.36 ± 6.30 ab | ES |
| 3 | Lc9 | 42.65 ± 27.52% defjhi | 28.31 ± 16.60 defgh | MS |
| 4 | Lc16 | 33.79 ± 21,53% ghij | 10.46 ± 6.99 hij | MR |
| 5 | Lc19 | 88.33 ± 13.64% a | 46.61 ± 15.92 abcd | ES |
| 6 | Lc23 | 67.95 ± 6.16% abcd | 26.22 ± 13.67 efgh | MS |
| 7 | Lc24 | 51.06 ± 15.44% cdefgh | 18.58 ± 6.86 fghij | MR |
| 8 | Lc27 | 75.44 ± 14.06% abc | 34.16 ± 12.49 cdefg | HS |
| 9 | Lc28 | 71.22 ± 13.16% abcd | 35.25 ± 4.55 cdefg | HS |
| 10 | Lc29 | 68.57 ± 13.25% abcd | 39.20 ± 11.05 bcde | HS |
| 11 | Lc30 | 12.45 ± 2.93% j | 3.26 ± 1.36 j | HR |
| 12 | Lc39 | 56.60 ± 21.77% bcdefg | 25.62 ± 15.52 efghi | MS |
| 13 | Lc40 | 36.66 ± 27.28% fghij | 16.22 ± 13.67 ghij | MR |
| 14 | Lc45 | 50.75 ± 20.76% cdefgh | 16.31 ± 7.87 ghij | MR |
| 15 | Lc49 | 87.43 ± 10.96% a | 62.96 ± 16.63 a | ES |
| 16 | Lc53 | 34.68 ± 24.96% ghij | 8.15 ± 6.62 ij | HR |
| 17 | Lc55 | 71.19 ± 10.84% abcd | 49.95 ± 9.49 abc | ES |
| 18 | Lc57 | 82.10 ± 10.00% ab | 60.78 ± 16.90 a | ES |
| 19 | Lc62 | 21.48 ± 11.18% ij | 4.30 ± 2.24 j | HR |
| 20 | Lc66 | 64.16 ± 2.57% abcdef | 36.16 ± 6.20 cdef | HS |
| 21 | Lc67 | 37.96 ± 15.80% | 11.30 ± 5.56 hij | MR |
| 22 | Lc68 | 17.05 ± 7.95% ij | 3.41 ± 1.59 j | HR |
| 23 | Lc70 | 12.26 ± 2.15% j | 2.45 ± 0.43 j | HR |
| 24 | Lc71 | 26.39 ± 10.49% hij | 7.22 ± 2.55 ij | HR |
| Group | R2X (cum) | R2Y (cum) | Q2 (cum) |
|---|---|---|---|
| 5ST vs. 5SN | 0.674 | 1.000 | 0.937 |
| 71RT vs. 71RN | 0.602 | 1.000 | 0.904 |
| 71RT vs. 5ST | 0.723 | 1.000 | 0.977 |
| 71RN vs. 5SN | 0.729 | 1.000 | 0.985 |
| Material | Longitude (°E) | Latitude (°N) | Elevation (m) |
|---|---|---|---|
| Lc3 | 117°36′ | 48°44′ | 81 |
| Lc5 | 118°41′ | 49°47′ | 152 |
| Lc9 | 113°29′ | 48°35′ | 613 |
| Lc16 | 117°49′ | 43°17′ | 830 |
| Lc19 | 124°14′ | 47°33′ | 934 |
| Lc23 | 118°37′ | 44°51′ | 1016 |
| Lc24 | 117°41′ | 44°41′ | 1019 |
| Lc27 | 106°45′ | 47°40′ | 1043 |
| Lc28 | 106°35′ | 46°54′ | 1045 |
| Lc29 | 113°17′ | 40°49′ | 1047 |
| Lc30 | 107°48′ | 47°38′ | 1050 |
| Lc39 | 115°37′ | 43°38′ | 1170 |
| Lc40 | 113°35′ | 41°39′ | 1195 |
| Lc45 | 110°19′ | 40°53′ | 1262 |
| Lc49 | 113°15′ | 44°34′ | 1349 |
| Lc53 | 115°42′ | 42°33′ | 1445 |
| Lc55 | 103°50′ | 46°39′ | 1504 |
| Lc57 | 106°21′ | 48°29′ | 1541 |
| Lc62 | 102°47′ | 47°50′ | 1668 |
| Lc66 | 113°11′ | 35°31′ | 1701 |
| Lc67 | 113°22′ | 37°13′ | 1706 |
| Lc70 | 118°57′ | 36°52′ | 2000 |
| Lc71 | 116°24′ | 39°54′ | 1100 |
| Level | Disease Index | Resistance Level |
|---|---|---|
| 1 | DI = 0 | Immunity (I) |
| 2 | 0 < DI ≤ 10 | Highly Resistant (HR) |
| 3 | 10 < DI ≤ 20 | Moderately Resistant (MR) |
| 4 | 20 < DI ≤ 30 | Moderately Susceptible (MS) |
| 5 | 30 < DI ≤ 45 | Highly Susceptible (HS) |
| 6 | DI > 45 | Extremely Susceptible (ES) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, W.; Gao, P.; Guo, F.; Hou, X. Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms. Int. J. Mol. Sci. 2025, 26, 7042. https://doi.org/10.3390/ijms26157042
Gao W, Gao P, Guo F, Hou X. Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms. International Journal of Molecular Sciences. 2025; 26(15):7042. https://doi.org/10.3390/ijms26157042
Chicago/Turabian StyleGao, Wenxin, Peng Gao, Fenghui Guo, and Xiangyang Hou. 2025. "Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms" International Journal of Molecular Sciences 26, no. 15: 7042. https://doi.org/10.3390/ijms26157042
APA StyleGao, W., Gao, P., Guo, F., & Hou, X. (2025). Comparative Transcriptomics and Metabolomics Uncover the Molecular Basis of Leaf Rust Resistance in Contrasting Leymus chinensis Germplasms. International Journal of Molecular Sciences, 26(15), 7042. https://doi.org/10.3390/ijms26157042