
Comparative Analysis of Chloroplast Genome Between Widely Distributed and Locally Distributed Lysionotus (Gesneriaceae) Related Members


Abstract
1. Introduction
2. Results
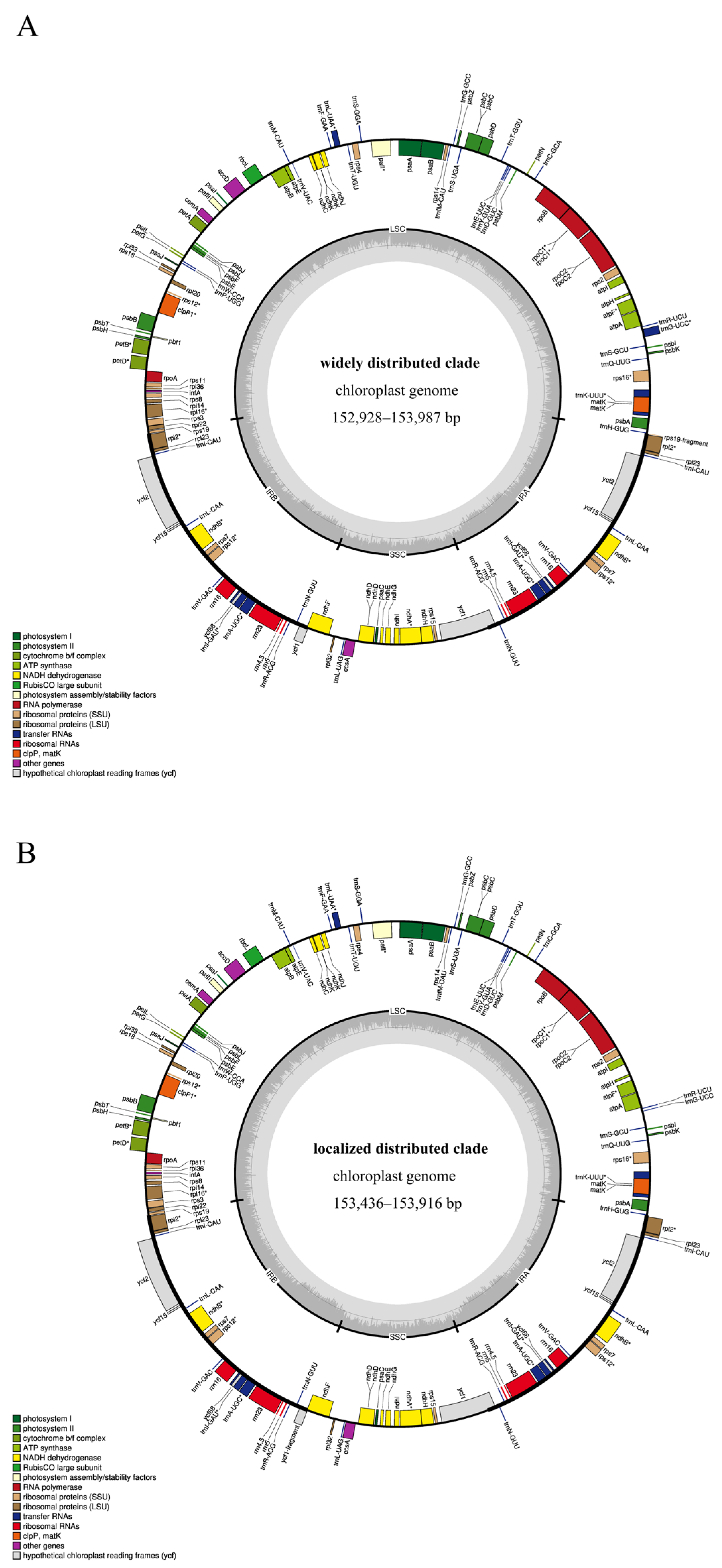
2.1. Features of the Plastome Genome
2.2. Genome Annotation
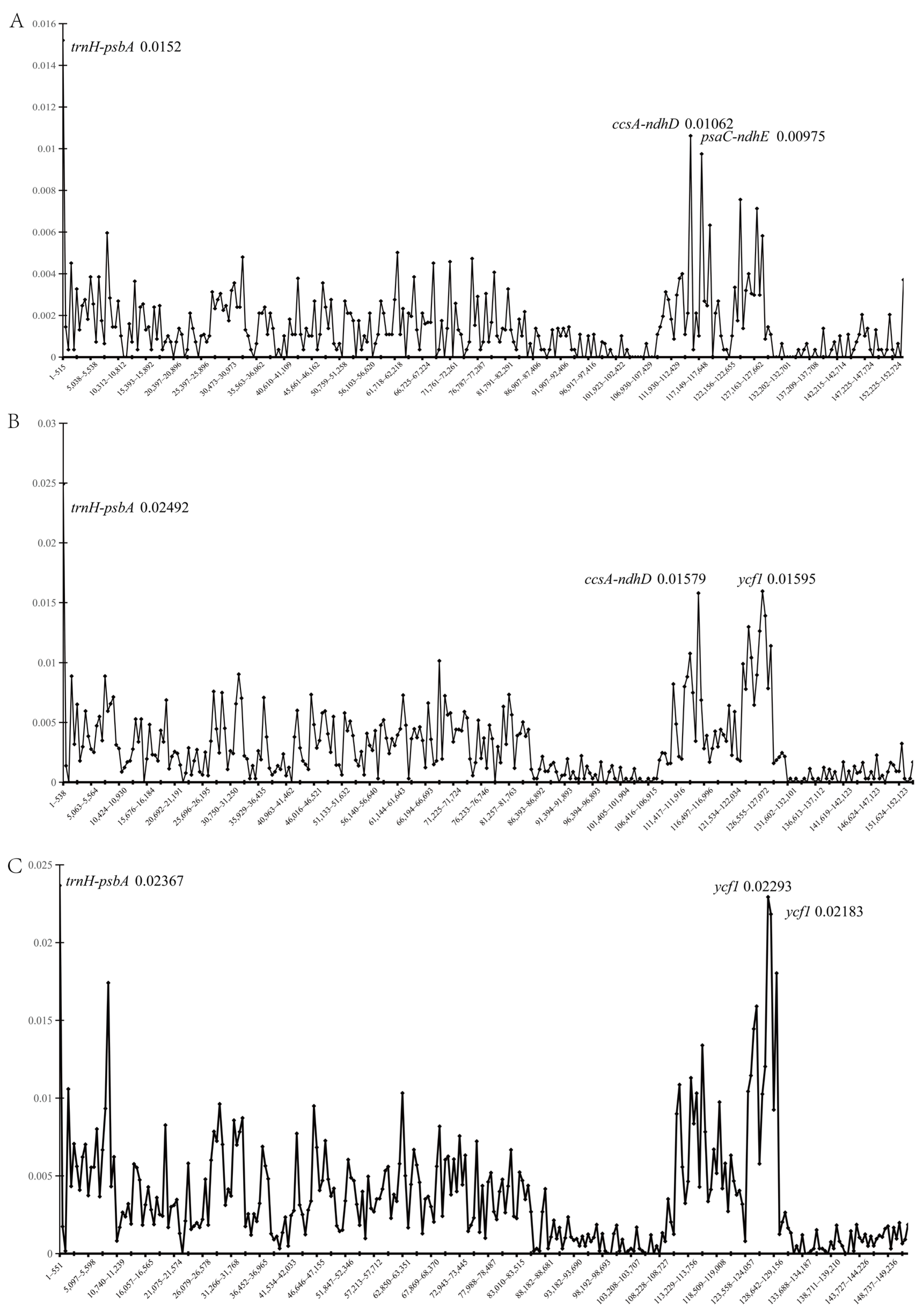
2.3. Genomic Divergence
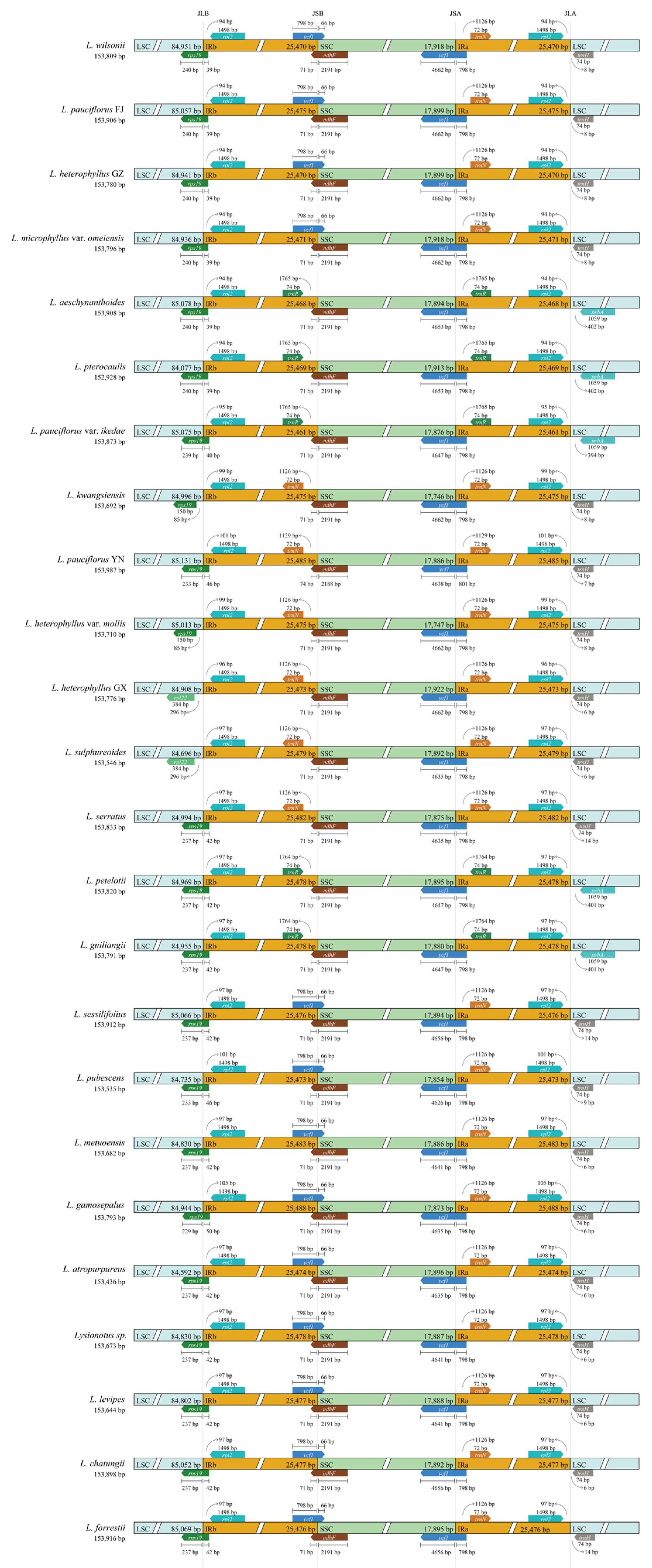
2.4. Contraction and Expansion Analysis of IR Region
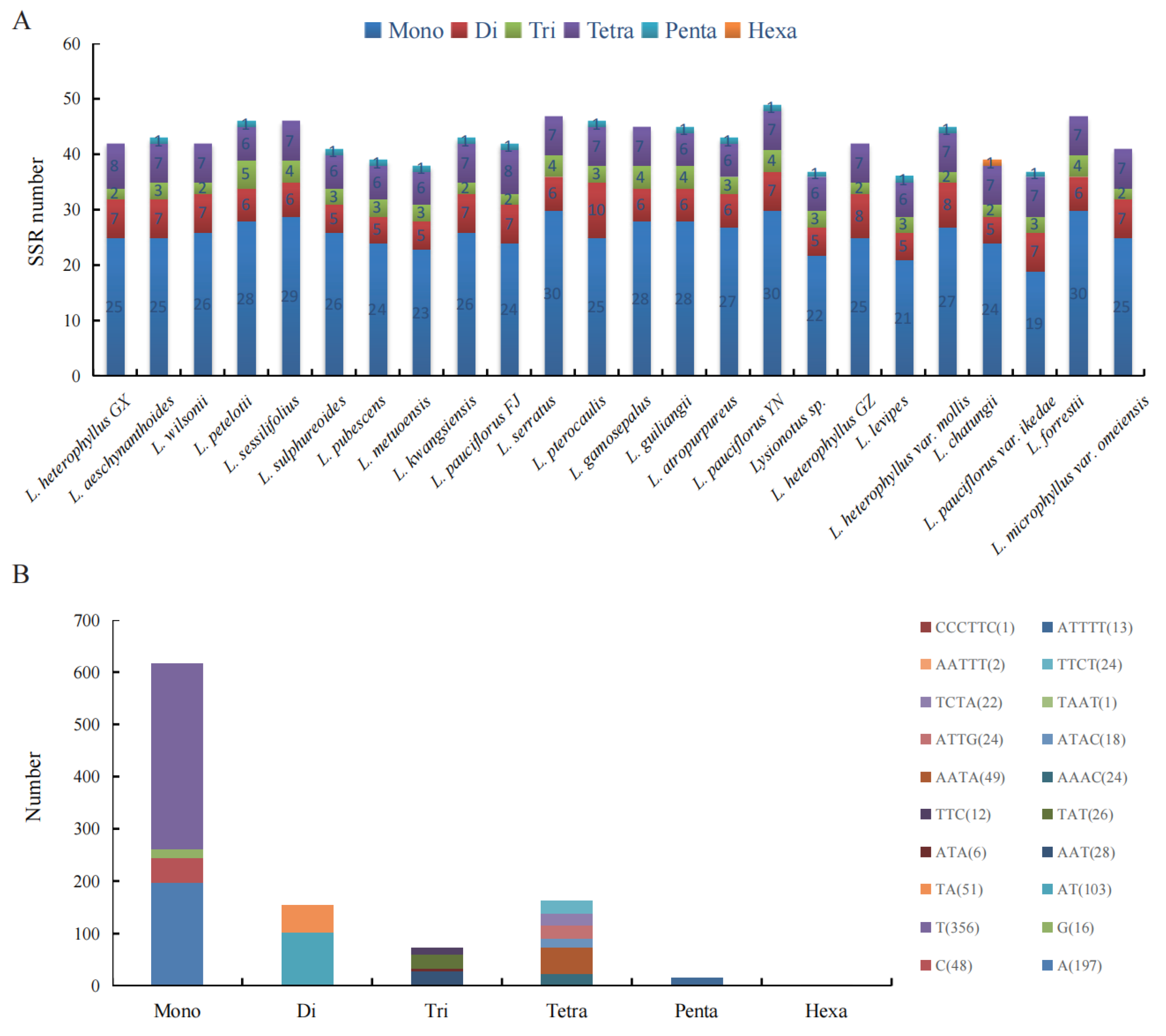
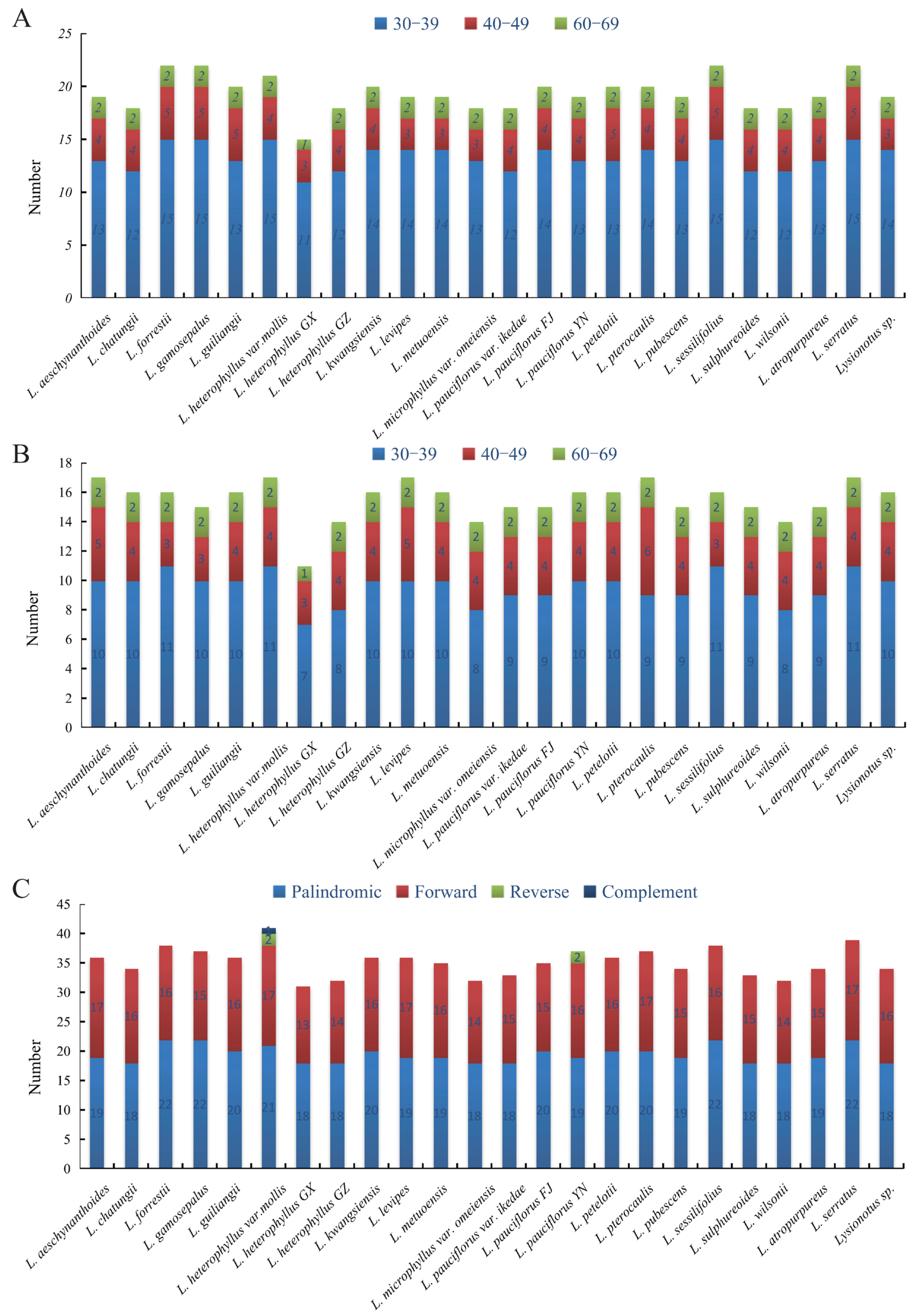
2.5. Repeat Analysis
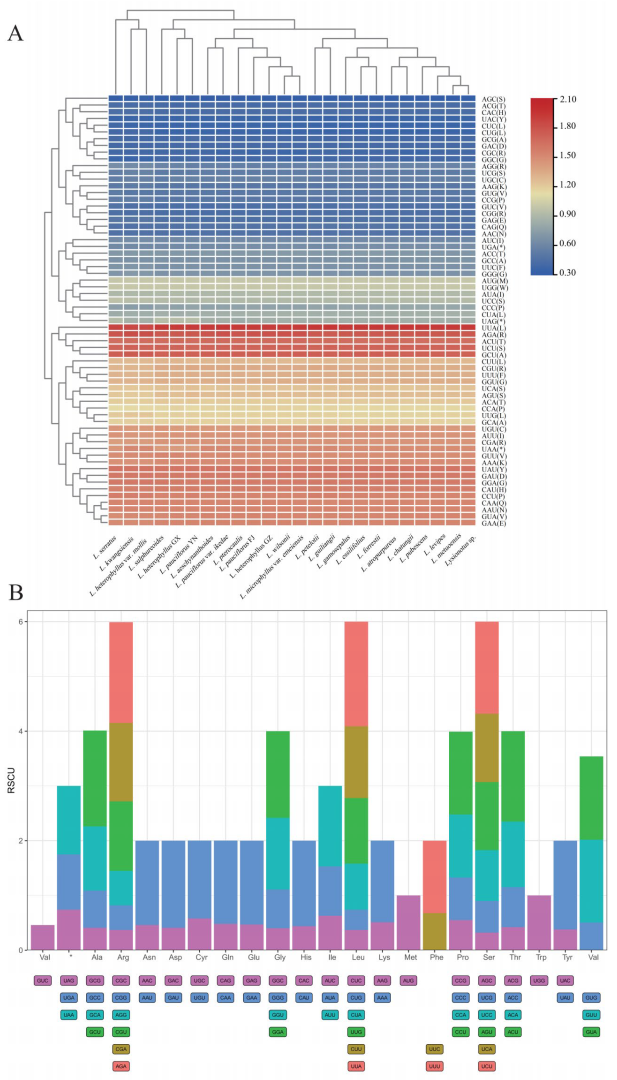
2.6. Codon Usage Bias Pattern
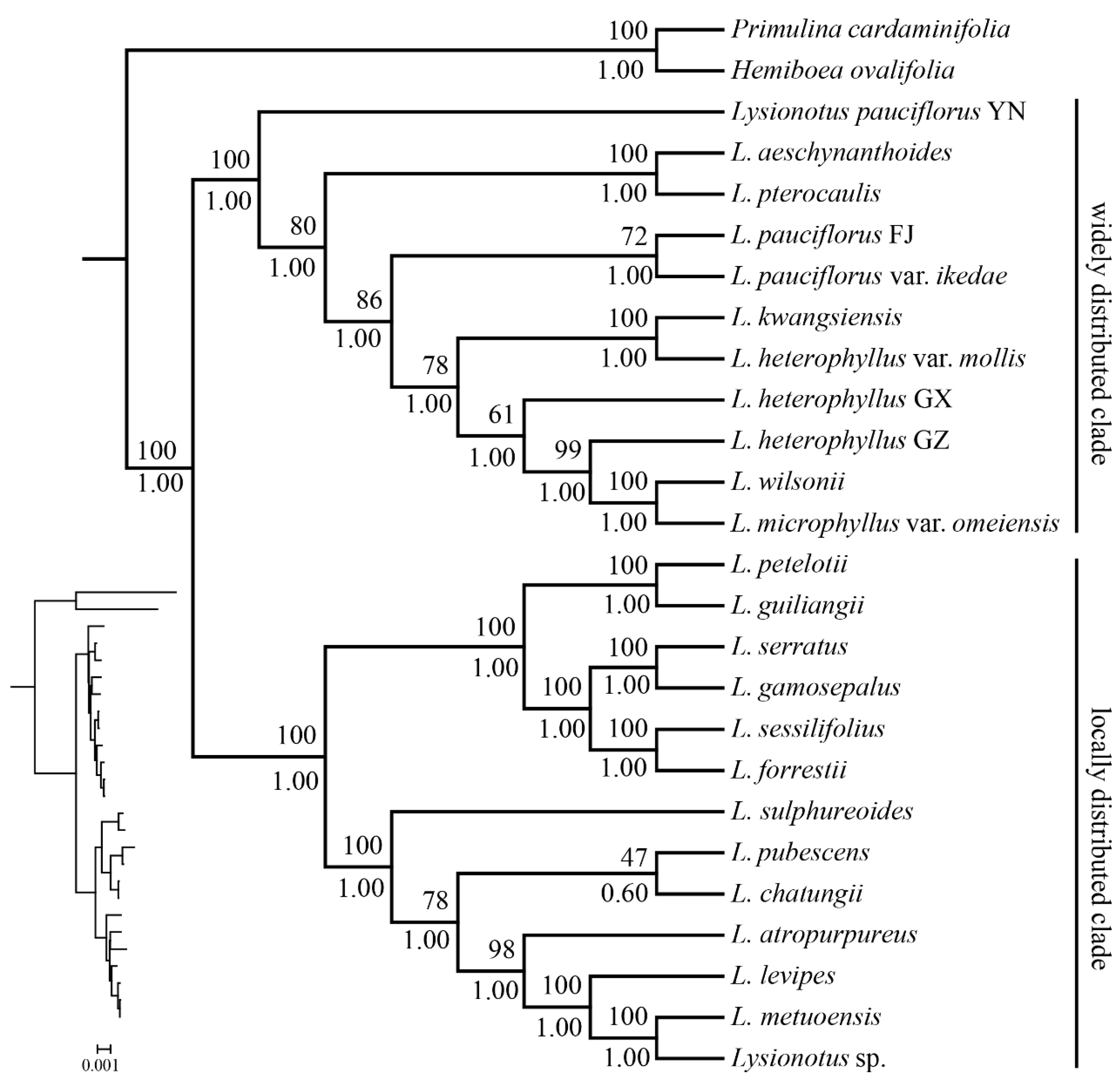
2.7. Phylogenetic Relationships
2.8. Positive Selection Based on Chloroplast
3. Discussion
3.1. Chloroplast Genome Structure and Evolutionary Conservation
3.2. Hypervariable Regions and Ecological Adaptation
3.3. Phylogenetic Insights: Comparison with Previous Studies
3.4. Research Limitations and Future Directions
3.5. Novel Contributions and Conservation Implications
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction and Sequencing
4.3. Plastome Genome Assembly and Annotation
4.4. Genome Comparison
4.5. Repeats Analysis
4.6. Phylogenetic Analysis
4.7. Positive Selected Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Harlan, J.R. Agricultural Origins: Centers and Noncenters. Science 1971, 174, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Tanksley, S.D.; McCouch, S.R. Seed Banks and Molecular Maps: Unlocking Genetic Potential from the Wild. Science 1997, 277, 1063–1066. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A.; Emshwiller, E.; Smith, B.D.; Bradley, D.G. Documenting domestication: The intersection of genetics and archaeology. Trends Genet. 2006, 22, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Meyer, R.S.; Purugganan, M.D. Evolution of crop species: Genetics of domestication and diversification. Nat. Rev. Genet. 2013, 14, 840–852. [Google Scholar] [CrossRef]
- Ford-Lloyd, B.V.; Schmidt, M.; Armstrong, S.J.; Barazani, O.Z.; Engels, J.; Hadas, R.; Hammer, K.; Kell, S.P.; Kang, D.; Khoshbakht, K.; et al. Crop Wild Relatives—Undervalued, Underutilized and under Threat? BioScience 2011, 61, 559–565. [Google Scholar] [CrossRef]
- Ma, J.; Li, Z.-Y.; Wang, Y.-Z.; Wang, W.-T.; Pan, K.-Y. Plants of Gesneriaceae in China. Taxon 2005, 54, 574. (In Chinese) [Google Scholar] [CrossRef]
- Wang, K.; Zhang, C.; Chen, H.; Yue, Y.; Zhang, W.; Zhang, M.; Qi, X.; Fu, Z. Karst landscapes of China: Patterns, ecosystem processes and services. Landsc. Ecol. 2019, 34, 2743–2763. [Google Scholar] [CrossRef]
- Liu, C.; Huang, Y.; Wu, F.; Liu, W.; Ning, Y.; Huang, Z.; Tang, S.; Liang, Y. Plant adaptability in karst regions. J. Plant Res. 2021, 134, 889–906. [Google Scholar] [CrossRef]
- Yan, W.; Zhou, Q.; Peng, D.; Wei, X.; Tang, X.; Yuan, E.; Wang, Y.; Shi, C. Soil moisture responses under different vegetation types to winter rainfall events in a humid karst region. Environ. Sci. Pollut. Res. 2021, 28, 56984–56995. [Google Scholar] [CrossRef]
- Möller, M.; Chen, W.; Shui, Y.; Atkins, H.; Middleton, D.J. A new genus of Gesneriaceae in China and the transfer of Briggsia species to other genera. Gard. Bull. 2014, 66, 195–205. [Google Scholar]
- Hu, R.C.; Hu, Q.M.; Qin, Y.; Xu, W.B.; Huang, Y.F. Oreocharis tianlinensis, a new species of Gesneriaceae from the limestone area in northwestern Guangxi, China. Taiwania 2022, 67, 479–483. [Google Scholar]
- Meyer, R.S.; DuVal, A.E.; Jensen, H.R. Patterns and processes in crop domestication: An historical review and quantitative analysis of 203 global food crops. New Phytol. 2012, 196, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.Y.; Liu, Y.L.; Shen, Y.; Zhang, Q.Y.; Liu, S.; Ren, Q.R.; Qin, L.P.; Sun, Y.Q. Phylogeography of cultivated and wild ophiopogon japonicus based on chloroplast DNA: Exploration of the origin and sustainable cultivation. BMC Plant Biol. 2023, 23, 242. [Google Scholar] [CrossRef]
- Riaz, S.; De Lorenzis, G.; Velasco, D.; Koehmstedt, A.; Maghradze, D.; Bobokashvili, Z.; Musayev, M.; Zdunic, G.; Laucou, V.; Andrew Walker, M.; et al. Genetic diversity analysis of cultivated and wild grapevine (Vitis vinifera L.) accessions around the Mediterranean basin and Central Asia. BMC Plant Biol. 2018, 18, 137. [Google Scholar] [CrossRef]
- Wright, S. Isolation by distance. Genetics 1943, 28, 114. [Google Scholar] [CrossRef]
- Petit, R.; Abdelhamid, E.M.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol. 2008, 12, 844–855. [Google Scholar] [CrossRef]
- Wu, Q.; Dong, S.; Zhao, Y.; Yang, L.; Qi, X.; Ren, Z.; Dong, S.; Cheng, J. Genetic diversity, population genetic structure and gene flow in the rare and endangered wild plant Cypripedium macranthos revealed by genotyping-by-sequencing. BMC Plant Biol. 2023, 23, 254. [Google Scholar] [CrossRef]
- Takeshima, R.; Yabe, S.; Matsui, K. Genetic basis of maturity time is independent from that of flowering time and contributes to ecotype differentiation in common buckwheat (Fagopyrum esculentum Moench). BMC Plant Biol. 2022, 22, 353. [Google Scholar] [CrossRef]
- Gao, L.-Z.; Innan, H. Very low gene duplication rate in the yeast genome. Science 2004, 306, 1367–1370. [Google Scholar] [CrossRef]
- Tilman, D.; Clark, M. Global diets link environmental sustainability and human health. Nature 2014, 515, 518–522. [Google Scholar] [CrossRef]
- Kang, L.; Qian, L.; Zheng, M.; Chen, L.; Chen, H.; Yang, L.; You, L.; Yang, B.; Yan, M.; Gu, Y.; et al. Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 2021, 53, 1392–1402. [Google Scholar] [CrossRef]
- Raven, J.A.; Allen, J.F. Genomics and chloroplast evolution: What did cyanobacteria do for plants? Genome Biol. 2003, 4, 209. [Google Scholar] [CrossRef]
- Nelson, N.; Yocum, C.F. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006, 57, 521–565. [Google Scholar] [CrossRef]
- Darracq, A.; Varré, J.S.; Maréchal-Drouard, L.; Courseaux, A.; Castric, V.; Saumitou-Laprade, P.; Oztas, S.; Lenoble, P.; Vacherie, B.; Barbe, V.; et al. Structural and content diversity of mitochondrial genome in beet: A comparative genomic analysis. Genome Biol. Evol. 2011, 3, 723–736. [Google Scholar] [CrossRef]
- Straub, S.C.; Parks, M.; Weitemier, K.; Fishbein, M.; Cronn, R.C.; Liston, A. Navigating the tip of the genomic iceberg: Next-generation sequencing for plant systematics. Am. J. Bot. 2012, 99, 349–364. [Google Scholar] [CrossRef]
- Varshney, R.K.; Terauchi, R.; McCouch, S.R. Harvesting the promising fruits of genomics: Applying genome sequencing technologies to crop breeding. PLoS Biol. 2014, 12, e1001883. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The third revolution in sequencing technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.; Lickey, E.B.; Beck, J.T.; Farmer, S.B.; Liu, W.; Miller, J.; Siripun, K.C.; Winder, C.T.; Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar] [CrossRef]
- Wicke, S.; Schneeweiss, G.M.; Depamphilis, C.W.; Müller, K.F.; Quandt, D. The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 2011, 76, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Ruhfel, B.R.; Gitzendanner, M.A.; Soltis, P.S.; Soltis, D.E.; Burleigh, J.G. From algae to angiosperms–inferring the phylogeny of green plants (Viridiplantae) from 360 plastid genomes. BMC Evol. Biol. 2014, 14, 23. [Google Scholar] [CrossRef]
- Xu, W.B.; Guo, J.; Pan, B.; Zhang, Q.; Liu, Y. Diversity and distribution of Gesneriaceae in China. Guihaia 2017, 37, 1219–1226. [Google Scholar] [CrossRef]
- Yang, L.-H.; Shi, X.-Z.; Wen, F.; Kang, M. Phylogenomics reveals widespread hybridization and polyploidization in Henckelia (Gesneriaceae). Ann. Bot. 2023, 131, 953–966. [Google Scholar] [CrossRef]
- Monro, A.K.; Bystriakova, N.; Fu, L.; Wen, F.; Wei, Y. Discovery of a diverse cave flora in China. PLoS ONE 2018, 13, e0190801. [Google Scholar] [CrossRef] [PubMed]
- Hilliard, O.; Burtt, B. Old World Gesneriaceae. IV. Notes on Didymocarpus and Lysionotus. Edinb. J. Bot. 1995, 52, 215–224. [Google Scholar] [CrossRef]
- Cai, L.; Huang, Z.-J.; Wen, F.; Dao, Z.-L. Two new species of Oreocharis (Gesneriaceae) from karst regions in Yunnan and notes on O. tetraptera and O. brachypoda from China. PhytoKeys 2020, 162, 1. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Zhang, C.F.; Peng, S.; Wang, J.J.; Wang, Y.; Hu, G.W.; Wang, Q.F. Lysionotus coccinus (Gesneriaceae), a new species from southwestern Yunnan, China. Nord. J. Bot. 2020, 38, e02912. [Google Scholar] [CrossRef]
- Kokubugata, G.; Hirayama, Y.; Peng, C.-I.; Yokota, M.; Möller, M. Phytogeographic aspects of Lysionotus pauciflorus sensu lato (Gesneriaceae) in the China, Japan and Taiwan regions: Phylogenetic and morphological relationships and taxonomic consequences. Plant Syst. Evol. 2011, 292, 177–188. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Lan, Y.; Sun, J.; Tian, R.; Bartlett, D.H.; Li, R.; Wong, Y.H.; Zhang, W.; Qiu, J.W.; Xu, T.; He, L.S.; et al. Molecular adaptation in the world’s deepest-living animal: Insights from transcriptome sequencing of the hadal amphipod Hirondellea gigas. Mol. Ecol. 2017, 26, 3732–3743. [Google Scholar] [CrossRef]
- Ren, T.; Zheng, W.; Han, K.; Zeng, S.; Zhao, J.; Liu, Z.-L. Characterization of the complete chloroplast genome sequence of Lysionotus pauciflorus (Gesneriaceae). Conserv. Genet. Resour. 2017, 9, 185–187. [Google Scholar] [CrossRef]
- Pakrashi, A.; Kumar, V.; Stanford-Beale, D.A.C.; Cameron, S.L.; Tyagi, K. Gene arrangement, phylogeny and divergence time estimation of mitogenomes in Thrips. Mol. Biol. Rep. 2022, 49, 6269–6283. [Google Scholar] [CrossRef]
- Huang, J.; Condamine, F.L.; Han, M.Q.; Cai, L.; Nguyen, K.S.; Zou, C.Y.; Xu, W.B. Miocene climatic fluctuations likely explain the species diversification of Lysionotus (Gesneriaceae) in Pan-Himalaya. J. Syst. Evol. 2025, 63, 134–147. [Google Scholar] [CrossRef]
- Huang, Z.P.; Lu, Z.C.; Zou, C.Y.; Han, M.Q.; Zhang, Q.; Xu, W.B. Confirmation of the taxonomic status of Lysionotus pterocaulis (Gesneriaceae) supported by morphological and molecular evidence. Taiwania 2022, 67, 455–460. [Google Scholar]
- Jin, J.J.; Yu, W.B.; Yang, J.B.; Song, Y.; DePamphilis, C.W.; Yi, T.S.; Li, D.Z. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Krumsiek, J.; Arnold, R.; Rattei, T. Gepard: A rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 2007, 23, 1026–1028. [Google Scholar] [CrossRef]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Li, H.; Guo, Q.; Xu, L.; Gao, H.; Liu, L.; Zhou, X. CPJSdraw: Analysis and visualization of junction sites of chloroplast genomes. PeerJ 2023, 11, e15326. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Su, Y.; Wang, T. Comparative genomic analysis of Polypodiaceae chloroplasts reveals fine structural features and dynamic insertion sequences. BMC Plant Biol. 2021, 21, 31. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.; Wang, W.; Wagutu, G.K.; Li, W.; Li, X.; Chen, Y. Fifteen complete chloroplast genomes of Trapa species (Trapaceae): Insight into genome structure, comparative analysis and phylogenetic relationships. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef] [PubMed]
- Moghaddam, M.; Ohta, A.; Shimizu, M.; Terauchi, R.; Kazempour-Osaloo, S. The complete chloroplast genome of Onobrychis gaubae (Fabaceae-Papilionoideae): Comparative analysis with related IR-lacking clade species. BMC Plant Biol. 2022, 22, 75. [Google Scholar] [CrossRef]
- Jiang, H.; Tian, J.; Yang, J.; Dong, X.; Zhong, Z.; Mwachala, G.; Zhang, C.; Hu, G.; Wang, Q. Comparative and phylogenetic analyses of six Kenya Polystachya (Orchidaceae) species based on the complete chloroplast genome sequences. BMC Plant Biol. 2022, 22, 177. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Li, J.; Tang, J.; Zeng, S.; Han, F.; Yuan, J.; Yu, J. Comparative plastid genomics of four Pilea (Urticaceae) species: Insight into interspecific plastid genome diversity in Pilea. BMC Plant Biol. 2021, 21, 25. [Google Scholar] [CrossRef]
- Wen, F.; Wu, X.; Li, T.; Jia, M.; Liu, X.; Liao, L. The complete chloroplast genome of Stauntonia chinensis and compared analysis revealed adaptive evolution of subfamily Lardizabaloideae species in China. BMC Genom. 2021, 22, 161. [Google Scholar] [CrossRef]
- Duan, N.; Deng, L.; Zhang, Y.; Shi, Y.; Liu, B. Comparative and phylogenetic analysis based on chloroplast genome of Heteroplexis (Compositae), a protected rare genus. BMC Plant Biol. 2022, 22, 605. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma Ki Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.; Von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.; Nielsen, R. Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol. Biol. Evol. 2002, 19, 908–917. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample Name | Voucher Specimen | Sample Location | Genbank Accession |
|---|---|---|---|---|
| Lysionotus pauciflorus | L. pauciflorus FJ | 14217 | Nanping, Fujian | PQ433130 |
| Lysionotus pauciflorus | L. pauciflorus YN | 14073 | Funing, Yunnan | PQ468987 |
| Lysionotus pauciflorus var. ikedae | L. pauciflorus var. ikedae | GCCC | Lanyu, Taiwan | PQ468992 |
| Lysionotus aeschynanthoides | L. aeschynanthoides | 15256 | Napo, Guangxi | PQ468968 |
| Lysionotus pterocaulis | L. pterocaulis | 14228 | Maguan, Yunnan | PQ468977 |
| Lysionotus heterophyllus | L. heterophyllus GZ | HMQ1814 | Panzhou, Guizhou | PQ468988 |
| Lysionotus wilsonii | L. wilsonii | 14206 | Emeishan, Sichuan | PQ468969 |
| Lysionotus microphyllus var. omeiensis | L. microphyllus var. omeiensis | WF180921 | Jinfoshan, Chongqing | PQ468994 |
| Lysionotus heterophyllus | L. heterophyllus GX | s.n. | Leye, Guangxi | PQ468967 |
| Lysionotus kwangsiensis | L. kwangsiensis | JWS | Rongshui, Guangxi | PQ468975 |
| Lysionotus heterophyllus var. mollis | L. heterophyllus var. mollis | GCCC | Rongshui, Guangxi | PQ468990 |
| Lysionotus gamosepalus | L. gamosepalus | HMQ | Motuo, Tibet | PQ468978 |
| Lysionotus pubescens | L. pubescens | 14185 | Tengchong, Yunnan | PQ468973 |
| Lysionotus atropurpureus | atropurpureus | HMQ | Motuo, Tibet | PQ468986 |
| Lysionotus sessilifolius | L. sessilifolius | 14190 | Gongshan, Yunnan | PQ468971 |
| Lysionotus forrestii | L. forrestii | ZX16177 | Gongshan, Yunnan | PQ468993 |
| Lysionotus levipes | L. levipes | GCCC | Bomi, Tibet | PQ468989 |
| Lysionotus chatungii | L. chatungii | GCCC | southeast Tibet | PQ468991 |
| Lysionotus guiliangii ined. | L. guiliangii | 2020136 | Pingbian, Yunnan | PQ468983 |
| Lysionotus sp. | L. sp. | HMQ | Motuo, Tibet | PQ468996 |
| Lysionotus petelotii | L. petelotii | ZCY313 | Malipo, Yunnan | PQ468970 |
| Lysionotus sulphureoides | L. sulphureoides | 14184 | Baoshan, Yunnan | PQ468972 |
| Lysionotus serratus | L.serratus | JZS | Longlin, Guangxi | PQ468976 |
| Lysionotus metuoensis | L. metuoensis | HMQ | Motuo, Tibet | PQ468974 |
| Species | LSC | SSC | IR | Total | CDs | rRNA | tRNA | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Length | GC Content | Length | GC Content | Length | GC Content | Length | GC Content | ||||
| L. pauciflorus FJ | 85,057 | 0.3535 | 17,899 | 0.3124 | 25,475 | 0.4317 | 153,906 | 0.3746 | 80 | 4 | 36 |
| L. pauciflorus YN | 85,131 | 0.3531 | 17,886 | 0.3125 | 25,485 | 0.4315 | 153,987 | 0.3743 | 80 | 4 | 36 |
| L. pauciflorus var. ikedae | 85,075 | 0.3532 | 17,876 | 0.3120 | 25,461 | 0.4314 | 153,873 | 0.3743 | 80 | 4 | 36 |
| L. aeschynanthoides | 85,078 | 0.3533 | 17,894 | 0.3124 | 25,468 | 0.4317 | 153,908 | 0.3745 | 80 | 4 | 36 |
| L. pterocaulis | 84,077 | 0.3539 | 17,913 | 0.3121 | 25,469 | 0.4316 | 152,928 | 0.3749 | 80 | 4 | 36 |
| L. heterophyllus GZ | 84,941 | 0.3537 | 17,899 | 0.3118 | 25,470 | 0.4318 | 153,780 | 0.3747 | 80 | 4 | 36 |
| L. wilsonii | 84,951 | 0.3538 | 17,918 | 0.3124 | 25,470 | 0.4318 | 153,809 | 0.3748 | 80 | 4 | 36 |
| L. microphyllus var. omeiensis | 84,936 | 0.3539 | 17,918 | 0.3124 | 25,471 | 0.4319 | 153,796 | 0.3749 | 80 | 4 | 36 |
| L. heterophyllus GX | 84,908 | 0.3539 | 17,922 | 0.3117 | 25,473 | 0.4318 | 153,776 | 0.3748 | 79 | 4 | 36 |
| L. kwangsiensis | 84,996 | 0.3536 | 17,746 | 0.3125 | 25,475 | 0.4316 | 153,692 | 0.3747 | 80 | 4 | 36 |
| L. heterophyllus var. mollis | 85,013 | 0.3535 | 17,747 | 0.3125 | 25,475 | 0.4316 | 153,710 | 0.3747 | 80 | 4 | 36 |
| L. gamosepalus | 84,944 | 0.3535 | 17,873 | 0.3110 | 25,488 | 0.4317 | 153,793 | 0.3745 | 80 | 4 | 36 |
| L. pubescens | 84,735 | 0.3532 | 17,854 | 0.3123 | 25,473 | 0.4316 | 153,535 | 0.3745 | 80 | 4 | 36 |
| L. atropurpureus | 84,592 | 0.3538 | 17,896 | 0.3123 | 25,474 | 0.4316 | 153,436 | 0.3748 | 80 | 4 | 36 |
| L. sessilifolius | 85,066 | 0.3535 | 17,894 | 0.3117 | 25,476 | 0.4319 | 153,912 | 0.3746 | 80 | 4 | 36 |
| L. forrestii | 85,069 | 0.3535 | 17,895 | 0.3117 | 25,476 | 0.4318 | 153,916 | 0.3746 | 80 | 4 | 36 |
| L. levipes | 84,802 | 0.3537 | 17,888 | 0.3128 | 25,477 | 0.4316 | 153,644 | 0.3748 | 80 | 4 | 36 |
| L. chatungii | 85,052 | 0.3531 | 17,892 | 0.3127 | 25,477 | 0.4316 | 153,898 | 0.3744 | 80 | 4 | 36 |
| L. guiliangii | 84,955 | 0.3533 | 17,780 | 0.3105 | 25,478 | 0.4317 | 153,791 | 0.3743 | 80 | 4 | 36 |
| L. sp. | 84,830 | 0.3537 | 17,887 | 0.3129 | 25,478 | 0.4316 | 153,673 | 0.3748 | 80 | 4 | 36 |
| L. petelotii | 84,969 | 0.3535 | 17,895 | 0.3098 | 25,478 | 0.4317 | 153,820 | 0.3743 | 80 | 4 | 36 |
| L. sulphureoides | 84,696 | 0.3533 | 17,892 | 0.3119 | 25,479 | 0.4317 | 153,546 | 0.3745 | 79 | 4 | 36 |
| L. serratus | 84,994 | 0.3534 | 17,875 | 0.3112 | 25,482 | 0.4318 | 153,833 | 0.3745 | 80 | 4 | 36 |
| L. metuoensis | 84,936 | 0.3539 | 17,918 | 0.3124 | 25,471 | 0.4319 | 153,796 | 0.3749 | 80 | 4 | 36 |
| Gene Name | Null Hypothesis | Alternative Hypothesis | Significance Test | ||||||
|---|---|---|---|---|---|---|---|---|---|
| lnL | df | Omega (ω = 1) | lnL | df | Omega (ω > 1) | BEB | NEB | p-Value | |
| accD | −2248.632758 | 47 | 1 | −2248.632741 | 48 | 1 | NA | NA | 9.95 × 10−1 |
| atpA | −2058.307032 | 47 | 1 | −2058.307032 | 48 | 1 | 191, N, 0.525 | NA | 1.00 × 100 |
| atpB | −2105.775158 | 47 | 1 | −2105.775222 | 48 | 1 | NA | NA | 9.91 × 10−1 |
| atpE | −533.527045 | 47 | 1 | −533.527046 | 48 | 3.11641 | NA | NA | 9.99 × 10−1 |
| atpF | −801.456315 | 47 | 1 | −801.456315 | 48 | 2.89696 | NA | NA | 1.00 × 100 |
| atpH | −332.181884 | 47 | 1 | −332.181918 | 48 | 1 | NA | NA | 9.93 × 10−1 |
| atpI | −1009.601571 | 47 | 1 | −1009.601571 | 48 | 1.43147 | NA | NA | 1.00 × 100 |
| cemA | −987.48884 | 47 | 1 | −987.376114 | 48 | 999 | 73, L, 0.550 | NA | 6.35 × 10−1 |
| clpP | −866.539298 | 47 | 1 | −866.539298 | 48 | 1 | 56, N, 0.762/73, M, 0.763 | NA | 1.00 × 100 |
| infA | −324.952445 | 47 | 1 | −324.952445 | 48 | 3.36607 | NA | NA | 1.00 × 100 |
| matK | −2289.050163 | 47 | 1 | −2289.040516 | 48 | 62.13547 | NA | NA | 8.90 × 10−1 |
| ndhA | −1615.94473 | 47 | 1 | −1615.71938 | 48 | 74.33031 | 188, S, 0.945 | NA | 5.02 × 10−1 |
| ndhB | −2056.657197 | 47 | 1 | −2056.425012 | 48 | 455555 | 300, T, 0.760 | NA | 4.96 × 10−1 |
| ndhC | −470.93447 | 47 | 1 | −470.691545 | 48 | 2.537453 | 30, F, 0.723 | NA | 4.86 × 10−1 |
| ndhD | −1657.695752 | 47 | 1 | −1657.695665 | 48 | 5.37607 | NA | NA | 9.89 × 10−1 |
| ndhE | −797.350973 | 47 | 1 | −796.338806 | 48 | 1 | 6, V, 0.667/92, G, 0.667 | NA | 1.55 × 10−1 |
| ndhF | −3479.498086 | 47 | 1 | −3479.495536 | 48 | 268.69832 | NA | NA | 9.43 × 10−1 |
| ndhG | −745.874227 | 47 | 1 | −745.874237 | 48 | 3.45598 | NA | NA | 9.96 × 10−1 |
| ndhH | −1658.650947 | 47 | 1 | −1658.544271 | 48 | 183.30161 | 321, L, 0.652 | NA | 6.44 × 10−1 |
| ndhI | −686.641873 | 47 | 1 | −686.641873 | 48 | 1 | NA | NA | 1.00 × 100 |
| ndhJ | −648.355572 | 47 | 1 | −648.355572 | 48 | 3.96959 | NA | NA | 1.00 × 100 |
| ndhK | −928.671632 | 47 | 1 | −928.671632 | 48 | 2.73846 | NA | NA | 1.00 × 100 |
| petA | −1410.28301 | 47 | 1 | −1410.282903 | 48 | 12.57637 | NA | NA | 9.88 × 10−1 |
| petB | −648.640314 | 47 | 1 | −648.640433 | 48 | 3.49232 | NA | NA | 9.88 × 10−1 |
| petD | −636.531041 | 47 | 1 | −636.531041 | 48 | 1.73442 | NA | NA | 1.00 × 100 |
| petG | −144.952513 | 47 | 1 | −144.952513 | 48 | 3.96269 | NA | NA | 1.00 × 100 |
| petL | −110.86864 | 47 | 1 | −110.868657 | 48 | 1.0009 | NA | NA | 9.95 × 10−1 |
| petN | −110.133659 | 47 | 1 | −110.133721 | 48 | 4.82041 | NA | NA | 9.91 × 10−1 |
| psaA | −3206.269402 | 47 | 1 | −3206.269403 | 48 | 2.98077 | NA | NA | 9.99 × 10−1 |
| psaB | −3062.739322 | 47 | 1 | −3062.738803 | 48 | 3.12016 | NA | NA | 9.74 × 10−1 |
| psaC | −323.719242 | 47 | 1 | −323.719242 | 48 | 4.44198 | NA | NA | 1.00 × 100 |
| psaI | −131.283698 | 47 | 1 | −131.283628 | 48 | 2.33483 | NA | NA | 9.91 × 10−1 |
| psaJ | −179.165779 | 47 | 1 | −179.165779 | 48 | 3.35833 | NA | NA | 1.00 × 100 |
| psbA | −1466.554562 | 47 | 1 | −1466.554562 | 48 | 3.87052 | NA | NA | 1.00 × 100 |
| psbB | −2109.542106 | 47 | 1 | −2109.536245 | 48 | 2.43776 | NA | NA | 9.14 × 10−1 |
| psbC | −1953.325342 | 47 | 1 | −1953.224272 | 48 | 2.594515 | NA | NA | 6.53 × 10−1 |
| psbE | −326.594616 | 47 | 1 | −326.593561 | 48 | 1 | NA | NA | 9.63 × 10−1 |
| psbF | −160.673626 | 47 | 1 | −160.673626 | 48 | 3.75946 | NA | NA | 1.00 × 100 |
| psbH | −287.330874 | 47 | 1 | −287.330874 | 48 | 1 | NA | NA | 1.00 × 100 |
| psbI | −142.497656 | 47 | 1 | −142.497705 | 48 | 3.67788 | NA | NA | 9.92 × 10−1 |
| psbJ | −142.497656 | 47 | 1 | −154.654 | 48 | 3.6036 | NA | NA | 9.98 × 10−1 |
| psbK | −217.948136 | 47 | 1 | −217.948136 | 48 | 3.03544 | NA | NA | 1.00 × 100 |
| psbL | −154.805662 | 47 | 1 | −154.805678 | 48 | 3.42968 | NA | NA | 9.95 × 10−1 |
| psbM | −129.554891 | 47 | 1 | −129.554864 | 48 | 1.61771 | NA | NA | 9.94 × 10−1 |
| psbN | −171.012236 | 47 | 1 | −171.011114 | 48 | 1 | NA | NA | 9.62 × 10−1 |
| psbT | −134.337509 | 47 | 1 | −134.337528 | 48 | 1 | NA | NA | 9.95 × 10−1 |
| rbcL | −2076.746457 | 47 | 1 | −2071.211324 | 48 | 3.145512 | NA | NA | 8.77 × 10−4 |
| rpl14 | −483.945572 | 47 | 1 | −483.945572 | 48 | 3.13388 | NA | NA | 1.00 × 100 |
| rpl16 | −416.903274 | 47 | 1 | −416.903274 | 48 | 3.56892 | NA | NA | 1.00 × 100 |
| rpl2 | −1103.495182 | 47 | 1 | −1103.495439 | 48 | 3.32559 | NA | NA | 9.82 × 10−1 |
| rpl20 | −568.82109 | 47 | 1 | −568.604892 | 48 | 43.34374 | NA | NA | 5.11 × 10−1 |
| rpl22 | −523.228276 | 47 | 1 | −523.040286 | 48 | 41.52047 | NA | NA | 5.40 × 10−1 |
| rpl23 | −363.661484 | 47 | 1 | −363.660276 | 48 | 5.72649 | NA | NA | 9.61 × 10−1 |
| rpl32 | −249.962411 | 47 | 1 | −249.957138 | 48 | 3.364021 | NA | NA | 9.18 × 10−1 |
| rpl33 | −282.055455 | 47 | 1 | −282.055454 | 48 | 3.49801 | NA | NA | 9.99 × 10−1 |
| rpl36 | −147.104844 | 47 | 1 | −147.104845 | 48 | 3.68001 | NA | NA | 9.99 × 10−1 |
| rpoA | −1644.775529 | 47 | 1 | −1644.372116 | 48 | 73.59795 | NA | NA | 3.69 × 10−1 |
| rpoB | −4556.139932 | 47 | 1 | −4556.139933 | 48 | 3.54957 | NA | NA | 9.99 × 10−1 |
| rps11 | −607.498798 | 47 | 1 | −607.498786 | 48 | 1 | NA | NA | 9.96 × 10−1 |
| rps14 | −462.578581 | 47 | 1 | −462.578586 | 48 | 3.43329 | NA | NA | 9.97 × 10−1 |
| rps15 | −425.650605 | 47 | 1 | −425.650263 | 48 | 2.96167 | NA | NA | 9.79 × 10−1 |
| rps16 | −301.101204 | 47 | 1 | −301.101204 | 48 | 3.32542 | NA | NA | 1.00 × 100 |
| rps18 | −301.10126 | 47 | 1 | −301.101204 | 48 | 3.73522 | NA | NA | 9.92 × 10−1 |
| rps2 | −1060.616195 | 47 | 1 | −1060.616195 | 48 | 1 | NA | NA | 1.00 × 100 |
| rps3 | −972.616253 | 47 | 1 | −972.616106 | 48 | 3.28734 | NA | NA | 9.86 × 10−1 |
| rps4 | −858.00646 | 47 | 1 | −858.027768 | 48 | 3.12 | NA | NA | 8.36 × 10−1 |
| rps7 | −601.835587 | 47 | 1 | −601.836044 | 48 | 3.28237 | NA | NA | 9.76 × 10−1 |
| rps8 | −615.141664 | 47 | 1 | −614.894166 | 48 | 3.489105 | 24, V, 0.840 | NA | 4.82 × 10−1 |
| ycf1 | −1433.829235 | 47 | 1 | −1433.570645 | 48 | 83.60908 | 11, L, 0.510 | NA | 4.72 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.-H.; Xu, W.-B.; Guo, C.-H. Comparative Analysis of Chloroplast Genome Between Widely Distributed and Locally Distributed Lysionotus (Gesneriaceae) Related Members. Int. J. Mol. Sci. 2025, 26, 7031. https://doi.org/10.3390/ijms26157031
Li J-H, Xu W-B, Guo C-H. Comparative Analysis of Chloroplast Genome Between Widely Distributed and Locally Distributed Lysionotus (Gesneriaceae) Related Members. International Journal of Molecular Sciences. 2025; 26(15):7031. https://doi.org/10.3390/ijms26157031
Chicago/Turabian StyleLi, Jia-Hui, Wei-Bin Xu, and Chang-Hong Guo. 2025. "Comparative Analysis of Chloroplast Genome Between Widely Distributed and Locally Distributed Lysionotus (Gesneriaceae) Related Members" International Journal of Molecular Sciences 26, no. 15: 7031. https://doi.org/10.3390/ijms26157031
APA StyleLi, J.-H., Xu, W.-B., & Guo, C.-H. (2025). Comparative Analysis of Chloroplast Genome Between Widely Distributed and Locally Distributed Lysionotus (Gesneriaceae) Related Members. International Journal of Molecular Sciences, 26(15), 7031. https://doi.org/10.3390/ijms26157031