
Gene Therapy Approaches for Atherosclerosis Focusing on Targeting Lipid Metabolism and Inflammation

 ,
,  , , , ,
, , , ,
Abstract
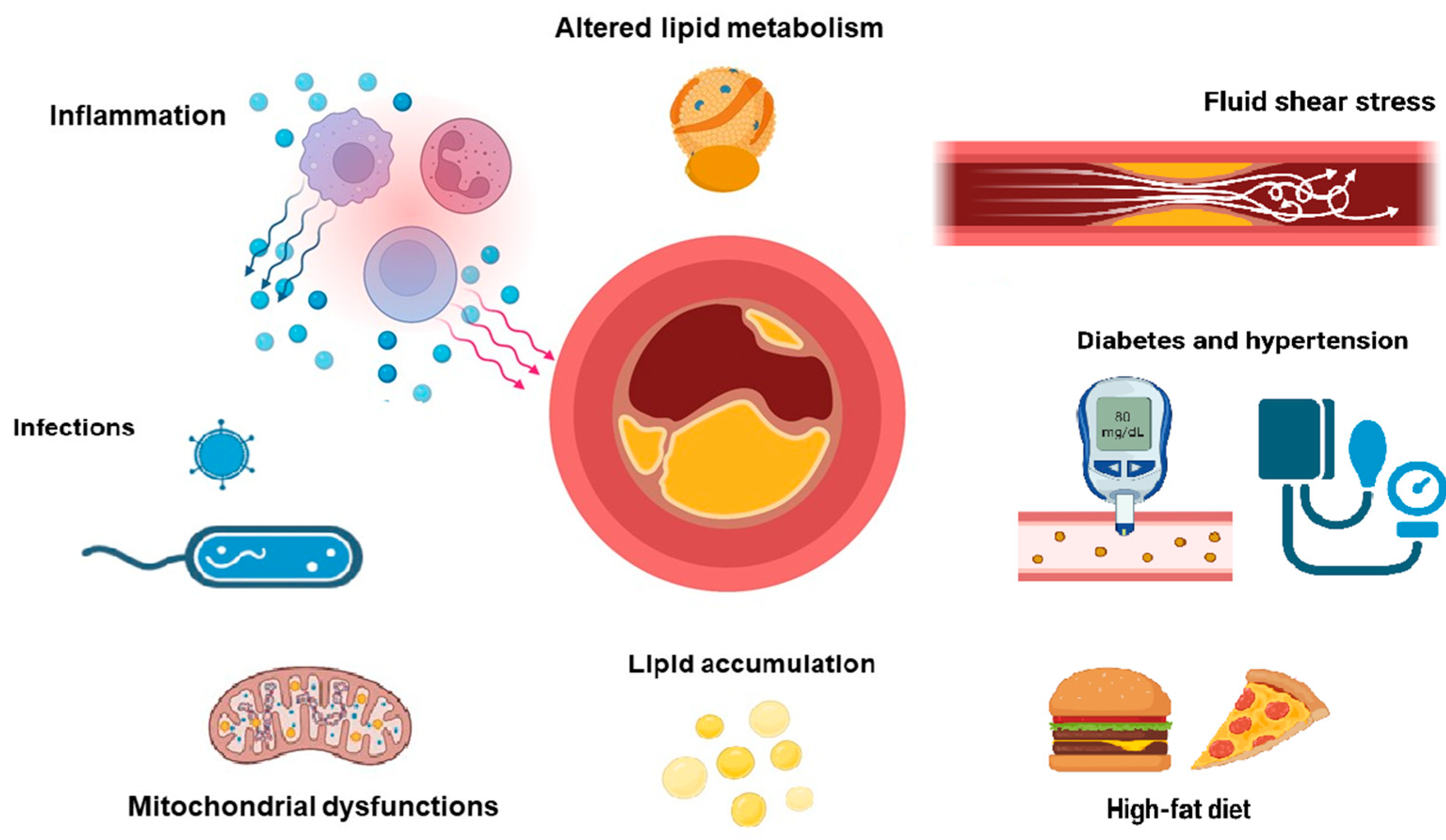
1. Introduction
1.1. LDL Role in Atherosclerosis Pathogenicity
1.2. LDL Metabolism and Functions
- Synthesis in the liver (using excess triglycerides derived from free carbohydrates, plasma fatty acids, and chylomicron remnants). VLDL synthesis increases with an increase in intrahepatic free fatty acids, such as with high-fat diets and when excess adipose tissue releases free fatty acids directly into the bloodstream (e.g., obesity, diabetes) [21].
- As a part of the lipid core of VLDL with the integral protein B-100, they enter the liver capillaries. There, apolipoproteins C II and E are transferred from high-density lipoproteins to VLDL. Apolipoprotein C II, located on the surface of VLDL, activates endothelial lipoprotein lipase, which breaks down triglycerides into free fatty acids and glycerol, which are absorbed by cells.
- The interaction of VLDL with lipoprotein lipase in tissue capillaries leads to the formation of residual cholesterol-rich VLDL. In this case, their size decreases several-fold and their density grows.
- Getting into the liver through apolipoprotein E and B-100 receptors, VLDL are either destroyed (approximately half of the particles) or, under the action of liver lipase, are converted into LDL, while apolipoproteins C II and E return to HDL, and only apolipoprotein B 100 remains on LDL. LDL contains ¾ of all plasma cholesterol, and the diameter of the particles decreases again. Their main function is to deliver cholesterol to the cells of the adrenal glands, skeletal muscles, lymphocytes, gonads and kidneys (Figure 2).
- (1)
- Are removed from the bloodstream by liver cells via hepatic LDL receptors with the participation of apoprotein B 100 within 2–6 h (40–60% of particles).
- (2)
- The remaining LDL is taken up either by the liver or extrahepatic cells via scavenger receptors (SR). Conversely, with a decrease in fat and cholesterol in the diet, the number of these receptors increases (“up-regulation”). With the entry of chylomicron cholesterol into the liver and an increased content of saturated fats in the diet, the number and binding capacity of hepatic LDL receptors decrease (“down-regulation”). The excess LDL that is not taken up by the liver LDL receptors (LDL-R) is removed from the bloodstream via extrahepatic SR “scavenger receptors” (mainly in macrophages).
2. Atherosclerosis Gene Therapies Targeting Lipid and Lipoprotein Metabolism
2.1. LDL Receptor Targets
2.2. Targeting Apolipoproteins
2.3. Proprotein Convertase Subtilisin/Kexin Type 9 Gene Therapy Target
- -
- Reduction in systemic inflammation—improvement of the lipid profile leads to a decrease in atherosclerotic vascular lesions and the accompanying inflammatory response.
- -
- Improvement of endothelial function—normalization of vascular function can reduce atrial ischemia and electrical instability.
- -
- Direct effect on cardiomyocytes—some studies indicate a possible direct antiarrhythmic effect of Pcsk9 inhibitors.
2.4. Angiopoietin-like 3
2.5. Apolipoprotein C III (APOC3)
2.6. Microsomal Triacylglycerol Transfer Protein
2.7. Sterol Transporters
2.8. Conclusion
3. Alternative Gene Targets for Atherosclerosis Treatment
3.1. Approaches of Gene Therapy Related to the Reduction of Inflammation Connected with Atherosclerosis
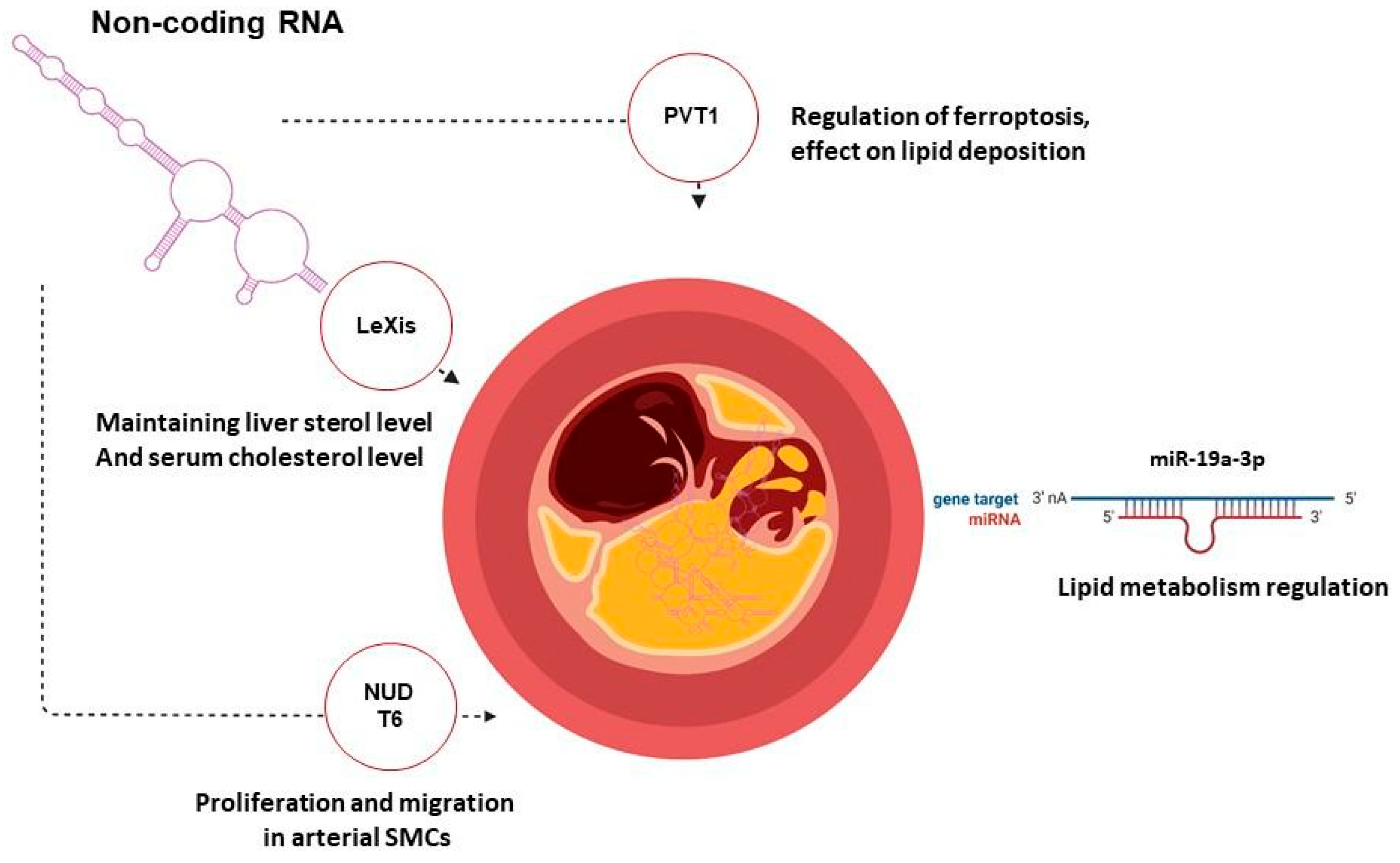
3.2. Gene Therapy of Atherosclerosis Targeting Non-Coding RNA
4. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Luca, A.C.; David, S.G.; David, A.G.; Țarcă, V.; Pădureț, I.-A.; Mîndru, D.E.; Roșu, S.T.; Roșu, E.V.; Adumitrăchioaiei, H.; Bernic, J.; et al. Atherosclerosis from Newborn to Adult—Epidemiology, Pathological Aspects, and Risk Factors. Life 2023, 13, 2056. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez Melo, M.; Von Eckardstein, A.; Robert, J. Modeling human atherosclerotic lesions in the test tube: Are we there yet? Atherosclerosis 2024, 398, 118560. [Google Scholar] [CrossRef] [PubMed]
- Gaggini, M.; Gorini, F.; Vassalle, C. Lipids in Atherosclerosis: Pathophysiology and the Role of Calculated Lipid Indices in Assessing Cardiovascular Risk in Patients with Hyperlipidemia. Int. J. Mol. Sci. 2022, 24, 75. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.-Y.; Huang, X.-F.; Zhang, D.-D.; Guo, R.-J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Giacca, M. SARS-CoV-2 infection boosts inflammation in atherosclerotic plaques. Nat. Cardiovasc. Res. 2023, 2, 966–967. [Google Scholar] [CrossRef] [PubMed]
- Poznyak, A.V.; Bezsonov, E.E.; Borisov, E.E.; Grechko, A.V.; Kartuesov, A.G.; Orekhov, A.N. Atherosclerosis in HIV Patients: What Do We Know so Far? Int. J. Mol. Sci. 2022, 23, 2504. [Google Scholar] [CrossRef] [PubMed]
- Vendrov, A.E.; Lozhkin, A.; Hayami, T.; Levin, J.; Silveira Fernandes Chamon, J.; Abdel-Latif, A.; Runge, M.S.; Madamanchi, N.R. Mitochondrial dysfunction and metabolic reprogramming induce macrophage pro-inflammatory phenotype switch and atherosclerosis progression in aging. Front. Immunol. 2024, 15, 1410832. [Google Scholar] [CrossRef] [PubMed]
- Natarajan, N.; Florentin, J.; Johny, E.; Xiao, H.; O’Neil, S.P.; Lei, L.; Shen, J.; Ohayon, L.; Johnson, A.R.; Rao, K.; et al. Aberrant mitochondrial DNA synthesis in macrophages exacerbates inflammation and atherosclerosis. Nat. Commun. 2024, 15, 7337. [Google Scholar] [CrossRef] [PubMed]
- Dabravolski, S.A.; Bezsonov, E.E.; Baig, M.S.; Popkova, T.V.; Orekhov, A.N. Mitochondrial Lipid Homeostasis at the Crossroads of Liver and Heart Diseases. Int. J. Mol. Sci. 2021, 22, 6949. [Google Scholar] [CrossRef] [PubMed]
- Solanki, K.; Rajpoot, S.; Bezsonov, E.E.; Orekhov, A.N.; Saluja, R.; Wary, A.; Axen, C.; Wary, K.; Baig, M.S. The expanding roles of neuronal nitric oxide synthase (NOS1). PeerJ 2022, 10, e13651. [Google Scholar] [CrossRef] [PubMed]
- Hutter, N.; Baena, M.; Sangüesa, G.; Dávalos, A.; Latasa, M.J.; Escolà-Gil, J.C.; Sánchez, R.M.; Roglans, N.; Alegret, M.; Laguna, J.C. Liquid fructose supplementation in LDL-R−/−mice fed a western-type diet enhances lipid burden and atherosclerosis despite identical calorie consumption. IJC Metab. Endocr. 2015, 9, 12–21. [Google Scholar] [CrossRef]
- Kolderup, A.; Svihus, B. Fructose Metabolism and Relation to Atherosclerosis, Type 2 Diabetes, and Obesity. J. Nutr. Metab. 2015, 2015, 823081. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, Z.; Chang, R.; Zeng, C.; Zhao, Y. High-Fructose Diet Induces Cardiac Dysfunction via Macrophage Recruitment in Adult Mice. J. Cardiovasc. Pharmacol. Ther. 2023, 28, 10742484231162249. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Sun, H.; Gandhi, A.; Du, Y.; Ebrahimi, S.; Jiang, Y.; Xu, S.; Uwase, H.; Seidel, A.; Bingaman, S.S.; et al. Role of shear stress-induced red blood cell released ATP in atherosclerosis. Am. J. Physiol. Heart Circ. Physiol. 2025, 328, H774–H791. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Junho, C.V.C.; Schieren, L.; Wollenhaupt, J.; Sluimer, J.C.; Van der Vorst, E.P.C.; Noels, H. Cellular metabolism changes in atherosclerosis and the impact of comorbidities. Front. Cell Dev. Biol. 2024, 12, 1446964. [Google Scholar] [CrossRef] [PubMed]
- Bezsonov, E.E.; Groenning, M.; Galzitskaya, O.V.; Gorkovskii, A.A.; Semisotnov, G.V.; Selyakh, I.O.; Ziganshin, R.H.; Rekstina, V.V.; Kudryashova, I.B.; Kuznetsov, S.A.; et al. Amyloidogenic peptides of yeast cell wall glucantransferase Bgl2p as a model for the investigation of its pH-dependent fibril formation. Prion 2013, 7, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.K. Anti-Prion Systems in Yeast and Inositol Polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Rubel, M.S.; Fedotov, S.A.; Grizel, A.V.; Sopova, J.V.; Malikova, O.A.; Chernoff, Y.O.; Rubel, A.A. Functional Mammalian Amyloids and Amyloid-Like Proteins. Life 2020, 10, 156. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Feng, X.; Shen, B.; Ma, J.; Zhao, W. Is Vascular Amyloidosis Intertwined with Arterial Aging, Hypertension and Atherosclerosis? Front. Genet. 2017, 8, 126. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Watanabe, T. Atherosclerosis: Known and unknown. Pathol. Int. 2022, 72, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Lent-Schochet, D.; Jialal, I. Biochemistry, Lipoprotein Metabolism. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK553193/ (accessed on 17 October 2024).
- Chan, Y.-H.; Ramji, D.P. Key Roles of Inflammation in Atherosclerosis: Mediators Involved in Orchestrating the Inflammatory Response and Its Resolution in the Disease Along with Therapeutic Avenues Targeting Inflammation. Methods Mol. Biol. 2022, 2419, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Shao, B.; Heinecke, J.W. HDL, lipid peroxidation, and atherosclerosis. J. Lipid Res. 2009, 50, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Egorova, I.E.; Gutsol, L.O.; Kuzmenko, V.V. Metabolizm Lipoproteidov v Norme i Patologii. Metody Issledovaniy Pokazateley Lipidnogo Obmena: Uchebnoe Posobie [Metabolism of Lipoproteins in Norm and Pathology. Methods of Research Indicators of Lipid Metabolism: A Tutorial]; IGMU Publishing: Irkutsk, Russia, 2024; 59p. (In Russian) [Google Scholar]
- Fenyo, I.M.; Gafencu, A.V. The involvement of the monocytes/macrophages in chronic inflammation associated with atherosclerosis. Immunobiology 2013, 218, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, P.; Zhang, Y.; Wang, J.; Wang, C.; Liu, Y.; Yang, G.; Yuan, L. Exosome-based Ldlr gene therapy for familial hypercholesterolemia in a mouse model. Theranostics 2021, 11, 2953–2965. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, S.A.; Buening, H.; Dishart, K.L.; De Alwis, M.; Girod, A.; Hacker, U.; Thrasher, A.J.; Ali, R.R.; Hallek, M.; Baker, A.H. Efficient and Selective AAV2-Mediated Gene Transfer Directed to Human Vascular Endothelial Cells. Mol. Ther. 2001, 4, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Shichinohe, T.; Bochner, B.H.; Mizutani, K.; Nishida, M.; Hegerich-Gilliam, S.; Naldini, L.; Kasahara, N. Development of lentiviral vectors for antiangiogenic gene delivery. Cancer Gene Ther. 2001, 8, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.B.; Xu, B.C.; Chen, Y.T.; Yuan, X.; Liu, J.Y.; Liu, T.; Du, G.Z.; Jiang, W.; Yang, Y.; Zhu, Y.; et al. Directed evolution of AAV accounting for long-term and enhanced transduction of cardiovascular endothelial cells in vivo. Mol. Ther. Methods Clin. Dev. 2021, 22, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Snyder, R.O.; Miao, C.H.; Patijn, G.A.; Spratt, S.K.; Danos, O.; Nagy, D.; Gown, A.M.; Winther, B.; Meuse, L.; Cohen, L.K.; et al. Persistent and therapeutic concentrations of human factor IX in mice after hepatic gene transfer of recombinant AAV vectors. Nat. Genet. 1997, 16, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Follenzi, A.; Sabatino, G.; Lombardo, A.; Boccaccio, C.; Naldini, L. Efficient Gene Delivery and Targeted Expression to Hepatocytes In Vivo by Improved Lentiviral Vectors. Hum. Gene Ther. 2002, 13, 243–260. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.; Schepelmann, S.; Athanasopoulos, T.; Graham, I.; Stannard, A.; Mohri, Z.; Hill, V.; Hassall, D.; Owen, J.; Dickson, G. Inhibition of atherosclerosis in apolipoprotein-E-deficient mice following muscle transduction with adeno-associated virus vectors encoding human apolipoprotein-E. Gene Ther. 2002, 9, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, J.L.; Brown, M.S. Familial hypercholesterolemia. Am. J. Med. 1975, 58, 147–150. [Google Scholar] [CrossRef] [PubMed]
- Soria, L.F.; Ludwig, E.H.; Clarke, H.R.; Vega, G.L.; Grundy, S.M.; McCarthy, B.J. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc. Natl. Acad. Sci. USA 1989, 86, 587–591. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.-P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef] [PubMed]
- Varret, M.; Abifadel, M.; Rabès, J.; Boileau, C. Genetic heterogeneity of autosomal dominant hypercholesterolemia. Clin. Genet. 2008, 73, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Iacocca, M.A.; Chora, J.R.; Carrié, A.; Freiberger, T.; Leigh, S.E.; Defesche, J.C.; Kurtz, C.L.; DiStefano, M.T.; Santos, R.D.; Humphries, S.E.; et al. on behalf of the ClinGen FH Variant Curation Expert Panel. ClinVar database of global familial hypercholesterolemia-associated DNA variants. Hum. Mutat. 2018, 39, 1631–1640. [Google Scholar] [CrossRef] [PubMed]
- Berberich, A.J.; Hegele, R.A. The complex molecular genetics of familial hypercholesterolaemia. Nat. Rev. Cardiol. 2019, 16, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Balling, M.; Afzal, S.; Davey Smith, G.; Varbo, A.; Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. Elevated LDL Triglycerides and Atherosclerotic Risk. J. Am. Coll. Cardiol. 2023, 81, 136–152. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Cohen, J.; Hobbs, H.H. Monogenic hypercholesterolemia: New insights in pathogenesis and treatment. J. Clin. Invest. 2003, 111, 1795–1803. [Google Scholar] [CrossRef] [PubMed]
- Grossman, M.; Rader, D.J.; Muller, D.W.M.; Kolansky, D.M.; Kozarsky, K.; Clark, B.J.; Stein, E.A.; Lupien, P.J.; Brewer, H.B.; Raper, S.E.; et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat. Med. 1995, 1, 1148–1154. [Google Scholar] [CrossRef] [PubMed]
- Graves, L.E.; Horton, A.; Alexander, I.E.; Srinivasan, S. Gene Therapy for Paediatric Homozygous Familial Hypercholesterolaemia. Heart Lung Circ. 2023, 32, 769–779. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Duan, L.; Lu, J.; Xia, J. Engineering exosomes for targeted drug delivery. Theranostics 2021, 11, 3183–3195. [Google Scholar] [CrossRef] [PubMed]
- Sawhney, J.P.S.; Madan, K. Familial hypercholesterolemia. Indian Heart J. 2024, 76, S108–S112. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, Y.; He, L.; Pu, W.; Yu, W.; Li, Y.; Wu, Y.-T.; Xu, C.; Wei, Y.; Ding, Q.; et al. In Vivo AAV-CRISPR/Cas9–Mediated Gene Editing Ameliorates Atherosclerosis in Familial Hypercholesterolemia. Circulation 2020, 141, 67–79. [Google Scholar] [CrossRef] [PubMed]
- Owczarek, J.; Rychlik-Sych, M.; Barańska, M.; Dudarewicz, M. The importance of the APOB gene expression as a marker in assessing the severity of atherosclerosis in coronary vessels. Pol. Arch. Intern. Med. 2023, 133, 16540. [Google Scholar] [CrossRef] [PubMed]
- Koornneef, A.; Maczuga, P.; Van Logtenstein, R.; Borel, F.; Blits, B.; Ritsema, T.; Van Deventer, S.; Petry, H.; Konstantinova, P. Apolipoprotein B Knockdown by AAV-delivered shRNA Lowers Plasma Cholesterol in Mice. Mol. Ther. 2011, 19, 731–740. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Oxidized phospholipids as a unifying theory for lipoprotein(a) and cardiovascular disease. Nat. Rev. Cardiol. 2019, 16, 305–318. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk: Insights From the FOURIER Trial. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Zanoni, P.; Khetarpal, S.A.; Larach, D.B.; Hancock-Cerutti, W.F.; Millar, J.S.; Cuchel, M.; DerOhannessian, S.; Kontush, A.; Surendran, P.; Saleheen, D.; et al. CHD Exome+ Consortium, CARDIoGRAM Exome Consortium, Global Lipids Genetics Consortium. Rare variant in scavenger receptor BI raises HDL cholesterol and increases risk of coronary heart disease. Science 2016, 351, 1166–1171. [Google Scholar] [CrossRef] [PubMed]
- Helgadottir, A.; Sulem, P.; Thorgeirsson, G.; Gretarsdottir, S.; Thorleifsson, G.; Jensson, B.Ö.; Arnadottir, G.A.; Olafsson, I.; Eyjolfsson, G.I.; Sigurdardottir, O.; et al. Rare SCARB1 mutations associate with high-density lipoprotein cholesterol but not with coronary artery disease. Eur. Heart J. 2018, 39, 2172–2178. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.-C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Gaudreault, N.; Kumar, N.; Posada, J.M.; Stephens, K.B.; Reyes De Mochel, N.S.; Eberlé, D.; Olivas, V.R.; Kim, R.Y.; Harms, M.J.; Johnson, S.; et al. ApoE Suppresses Atherosclerosis by Reducing Lipid Accumulation in Circulating Monocytes and the Expression of Inflammatory Molecules on Monocytes and Vascular Endothelium. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 264–272. [Google Scholar] [CrossRef] [PubMed]
- Hasty, A.H.; Linton, M.F.; Brandt, S.J.; Babaev, V.R.; Gleaves, L.A.; Fazio, S. Retroviral Gene Therapy in ApoE-Deficient Mice: ApoE Expression in the Artery Wall Reduces Early Foam Cell Lesion Formation. Circulation 1999, 99, 2571–2576. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liu, H.; Liu, M.; Li, F.; Liu, L.; Du, F.; Fan, D.; Yu, H. A human apolipoprotein E mimetic peptide reduces atherosclerosis in aged apolipoprotein E null mice. Am. J. Transl. Res. 2016, 8, 3482–3492. [Google Scholar] [PubMed]
- Hegele, R.A.; Little, J.A.; Vezina, C.; Maguire, G.F.; Tu, L.; Wolever, T.S.; Jenkins, D.J.; Connelly, P.W. Hepatic lipase deficiency. Clinical, biochemical, and molecular genetic characteristics. Arterioscler. Thromb. 1993, 13, 720–728. [Google Scholar] [CrossRef] [PubMed]
- Connelly, P.W.; Maguire, G.F.; Lee, M.; Little, J.A. Plasma lipoproteins in familial hepatic lipase deficiency. Arteriosclerosis 1990, 10, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, C.; Xu, Y.; Lu, G.; Hu, Y.; Mao, W.; Ke, L.; Tong, Z.; Xia, Y.; Ma, S.; Dong, X.; et al. AAV-mediated hepatic LPL expression ameliorates severe hypertriglyceridemia and acute pancreatitis in Gpihbp1 deficient mice and rats. Mol. Ther. 2024, 32, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Kuhbandner, K.; Herz, J.; Pohlkamp, T. Lipoprotein receptors. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2021; pp. 583–622. [Google Scholar] [CrossRef]
- Burke, A.C.; Dron, J.S.; Hegele, R.A.; Huff, M.W. PCSK9: Regulation and Target for Drug Development for Dyslipidemia. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 223–244. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef] [PubMed]
- Macchi, C.; Sirtori, C.R.; Corsini, A.; Santos, R.D.; Watts, G.F.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef] [PubMed]
- Katzmann, J.L.; Packard, C.J.; Chapman, M.J.; Katzmann, I.; Laufs, U. Targeting RNA With Antisense Oligonucleotides and Small Interfering RNA in Dyslipidemias. J. Am. Coll. Cardiol. 2020, 76, 563–579. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Correction to: Inclisiran: First Approval. Drugs 2021, 81, 1129. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, Y.; Pantea Stoian, A.; Cicero, A.F.G.; Fogacci, F.; Nikolic, D.; Sachinidis, A.; Rizvi, A.A.; Janez, A.; Rizzo, M. Inclisiran: A small interfering RNA strategy targeting PCSK9 to treat hypercholesterolemia. Expert Opin. Drug Saf. 2022, 21, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. Familial Hypercholesterolemia: Genes Beyond. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Boyce, A., Chrousos, G., Corpas, E., de Herder, W.W., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK343488/ (accessed on 18 October 2024).
- Ding, Q.; Strong, A.; Patel, K.M.; Ng, S.-L.; Gosis, B.S.; Regan, S.N.; Cowan, C.A.; Rader, D.J.; Musunuru, K. Permanent Alteration of PCSK9 With In Vivo CRISPR-Cas9 Genome Editing. Circ. Res. 2014, 115, 488–492. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, A.C.; Musunuru, K. Treatment of Dyslipidemia Using CRISPR/Cas9 Genome Editing. Curr. Atheroscler. Rep. 2017, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Smith, J.; Breton, C.; Clark, P.; Zhang, J.; Ying, L.; Che, Y.; Lape, J.; Bell, P.; Calcedo, R.; et al. Meganuclease targeting of PCSK9 in macaque liver leads to stable reduction in serum cholesterol. Nat. Biotechnol. 2018, 36, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Su, J.; Liu, Y.; Jin, X.; Zhong, X.; Mo, L.; Wang, Q.; Deng, H.; Yang, Y. In vivo PCSK9 gene editing using an all-in-one self-cleavage AAV-CRISPR system. Mol. Ther. Methods Clin. Dev. 2021, 20, 652–659. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Raghavan, A.; Chen, T.; Qiao, L.; Zhang, Y.; Ding, Q.; Musunuru, K. CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo—Brief Report. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 783–786. [Google Scholar] [CrossRef] [PubMed]
- Ilahibaks, N.F.; Kluiver, T.A.; De Jong, O.G.; De Jager, S.C.A.; Schiffelers, R.M.; Vader, P.; Peng, W.C.; Lei, Z.; Sluijter, J.P.G. Extracellular vesicle-mediated delivery of CRISPR/Cas9 ribonucleoprotein complex targeting proprotein convertase subtilisin-kexin type 9 (Pcsk9) in primary mouse hepatocytes. J. Extracell. Vesicle 2024, 13, 12389. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Zhang, C.; Zhou, Y.; Li, H.; Xiao, W.; Herzog, R.W.; Xu, J.; Zhang, J.; Chen, Y.E.; Han, R. Liver-specific in vivo base editing of Angptl3 via AAV delivery efficiently lowers blood lipid levels in mice. Cell Biosci. 2023, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Corbett, A.L.; Taatizadeh, E.; Tasnim, N.; Little, J.P.; Garnis, C.; Daugaard, M.; Guns, E.; Hoorfar, M.; Li, I.T.S. Challenges and opportunities in exosome research-Perspectives from biology, engineering, and cancer therapy. APL Bioeng. 2019, 3, 011503. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kennedy, T.L.; Russell, A.J.; Riley, P. Experimental limitations of extracellular vesicle-based therapies for the treatment of myocardial infarction. Trends Cardiovasc. Med. 2021, 31, 405–415. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Carreras, A.; Pane, L.S.; Nitsch, R.; Madeyski-Bengtson, K.; Porritt, M.; Akcakaya, P.; Taheri-Ghahfarokhi, A.; Ericson, E.; Bjursell, M.; Perez-Alcazar, M.; et al. In vivo genome and base editing of a human PCSK9 knock-in hypercholesterolemic mouse model. BMC Biol. 2019, 17, 4. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Rothgangl, T.; Dennis, M.K.; Lin, P.J.C.; Oka, R.; Witzigmann, D.; Villiger, L.; Qi, W.; Hruzova, M.; Kissling, L.; Lenggenhager, D.; et al. In vivo adenine base editing of PCSK9 in macaques reduces LDL cholesterol levels. Nat. Biotechnol. 2021, 39, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Cullis, P.R.; Hope, M.J. Lipid Nanoparticle Systems for Enabling Gene Therapies. Mol. Ther. 2017, 25, 1467–1475. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting PCSK9 in Nonhuman Primate and Mouse Models. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Hooper, A.J.; Tang, X.L.; Burnett, J.R. VERVE-101, a CRISPR base-editing therapy designed to permanently inactivate hepatic PCSK9 and reduce LDL-cholesterol. Expert Opin. Investig. Drugs 2024, 33, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Castiello, D.S.; Buongiorno, F.; Manzi, L.; Narciso, V.; Forzano, I.; Florimonte, D.; Sperandeo, L.; Canonico, M.E.; Avvedimento, M.; Paolillo, R.; et al. Procedural and Antithrombotic Therapy Optimization in Patients with Atrial Fibrillation Undergoing Percutaneous Coronary Intervention: A Narrative Review. J. Cardiovasc. Dev. Dis. 2025, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Lang, W.; Frishman, W.H. Angiopoietin-Like 3 Protein Inhibition: A New Frontier in Lipid-Lowering Treatment. Cardiol. Rev. 2019, 27, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Huff, M.W.; Rayner, K.J. Atherosclerosis. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2021; pp. 623–665. ISBN 9780128240489. [Google Scholar]
- Jørgensen, A.B.; Frikke-Schmidt, R.; Nordestgaard, B.G.; Tybjærg-Hansen, A. Loss-of-Function Mutations in APOC3 and Risk of Ischemic Vascular Disease. N. Engl. J. Med. 2014, 371, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Zha, Y.; Lu, Y.; Zhang, T.; Yan, K.; Zhuang, W.; Liang, J.; Cheng, Y.; Wang, Y. CRISPR/Cas9-mediated knockout of APOC3 stabilizes plasma lipids and inhibits atherosclerosis in rabbits. Lipids Health Dis. 2021, 20, 180. [Google Scholar] [CrossRef] [PubMed]
- Stitziel, N.O.; Khera, A.V.; Wang, X.; Bierhals, A.J.; Vourakis, A.C.; Sperry, A.E.; Natarajan, P.; Klarin, D.; Emdin, C.A.; Zekavat, S.M.; et al. ANGPTL3 Deficiency and Protection Against Coronary Artery Disease. J. Am. Coll. Cardiol. 2017, 69, 2054–2063. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.-X.; Redon, V.; Yu, H.; Querbes, W.; Pirruccello, J.; Liebow, A.; Deik, A.; Trindade, K.; Wang, X.; Musunuru, K.; et al. Role of angiopoietin-like 3 (ANGPTL3) in regulating plasma level of low-density lipoprotein cholesterol. Atherosclerosis 2018, 268, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-H.; So, J.-H.; Kim, H.-T.; Choi, J.-H.; Lee, M.-S.; Choi, S.-Y.; Kim, C.-H.; Kim, M.J. Angiopoietin-like 3 regulates hepatocyte proliferation and lipid metabolism in zebrafish. Biochem. Biophys. Res. Commun. 2014, 446, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118, e2020401118. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Ma, X.; Gao, F.; Guo, Y. Off-target effects in CRISPR/Cas9 gene editing. Front. Bioeng. Biotechnol. 2023, 11, 1143157. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Watts, G.F.; Chan, D.C. RNA interference therapy for targeting ANGPTL3 and atherogenic lipoproteins: Findings and implications of a recent phase I study. Clin. Transl. Med. 2023, 13, e1484. [Google Scholar] [CrossRef] [PubMed]
- Palaschak, B.; Herzog, R.W.; Markusic, D.M. AAV-Mediated Gene Delivery to the Liver: Overview of Current Technologies and Methods. Methods Mol. Biol. 2019, 1950, 333–360. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Musunuru, K. Angiopoietin-Like 3: From Discovery to Therapeutic Gene Editing. JACC Basic Transl. Sci. 2019, 4, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Ramms, B.; Patel, S.; Sun, X.; Pessentheiner, A.R.; Ducasa, G.M.; Mullick, A.E.; Lee, R.G.; Crooke, R.M.; Tsimikas, S.; Witztum, J.L.; et al. Interventional hepatic apoC-III knockdown improves atherosclerotic plaque stability and remodeling by triglyceride lowering. JCI Insight 2022, 7, e158414. [Google Scholar] [CrossRef] [PubMed]
- Kohan, A.B. Apolipoprotein C-III: A potent modulator of hypertriglyceridemia and cardiovascular disease. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Xu, Y.; Dong, Z.; Zhou, Z.; Cong, N.; Gao, M.; Huang, W.; Wang, Y.; Liu, G.; Xian, X. Inactivation of ApoC3 by CRISPR/Cas9 Protects Against Atherosclerosis in Hamsters. Circ. Res. 2020, 127, 1456–1458. [Google Scholar] [CrossRef] [PubMed]
- Uddin, F.; Rudin, C.M.; Sen, T. CRISPR Gene Therapy: Applications, Limitations, and Implications for the Future. Front. Oncol. 2020, 10, 1387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, G.; Sharma, A.R.; Bhattacharya, M.; Lee, S.S.; Chakraborty, C. CRISPR-Cas9: A Preclinical and Clinical Perspective for the Treatment of Human Diseases. Mol Ther. 2021, 29, 571–586. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gouni-Berthold, I.; Alexander, V.J.; Yang, Q.; Hurh, E.; Steinhagen-Thiessen, E.; Moriarty, P.M.; Hughes, S.G.; Gaudet, D.; Hegele, R.A.; O’Dea, L.S.L.; et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): A multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021, 9, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Gaudet, D.; Pall, D.; Watts, G.F.; Nicholls, S.J.; Rosenson, R.S.; Modesto, K.; San Martin, J.; Hellawell, J.; Ballantyne, C.M. Plozasiran (ARO-APOC3) for Severe Hypertriglyceridemia: The SHASTA-2 Randomized Clinical Trial. JAMA Cardiol. 2024, 9, 620. [Google Scholar] [CrossRef] [PubMed]
- Vance, J.E.; Adeli, K. Assembly and secretion of triacylglycerol-rich lipoproteins. In Biochemistry of Lipids, Lipoproteins and Membranes; Elsevier: Amsterdam, The Netherlands, 2008; pp. 507–531. [Google Scholar] [CrossRef]
- Khatun, I.; Zeissig, S.; Iqbal, J.; Wang, M.; Curiel, D.; Shelness, G.S.; Blumberg, R.S.; Hussain, M.M. Phospholipid transfer activity of microsomal triglyceride transfer protein produces apolipoprotein B and reduces hepatosteatosis while maintaining low plasma lipids in mice. Hepatology 2012, 55, 1356–1368. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.A.; Wetterau, J.R.; Gregg, R.E. Microsomal triglyceride transfer protein: A protein complex required for the assembly of lipoprotein particles. Trends Cell Biol. 1995, 5, 317–321. [Google Scholar] [CrossRef] [PubMed]
- Soh, J.; Iqbal, J.; Queiroz, J.; Fernandez-Hernando, C.; Hussain, M.M. MicroRNA-30c reduces hyperlipidemia and atherosclerosis in mice by decreasing lipid synthesis and lipoprotein secretion. Nat. Med. 2013, 19, 892–900. [Google Scholar] [CrossRef] [PubMed]
- Hewing, B.; Parathath, S.; Mai, C.K.; Fiel, M.I.; Guo, L.; Fisher, E.A. Rapid regression of atherosclerosis with MTP inhibitor treatment. Atherosclerosis 2013, 227, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Horie, T.; Nishino, T.; Baba, O.; Kuwabara, Y.; Nakao, T.; Nishiga, M.; Usami, S.; Izuhara, M.; Sowa, N.; Yahagi, N.; et al. MicroRNA-33 regulates sterol regulatory element-binding protein 1 expression in mice. Nat. Commun. 2013, 4, 2883. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Barrett, T.J.; Fisher, E.A. HDL and Reverse Cholesterol Transport: Basic Mechanisms and Their Roles in Vascular Health and Disease. Circ. Res. 2019, 124, 1505–1518. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; Parhofer, K.G.; Ginsberg, H.N.; Hegele, R.A. Clinical review on triglycerides. Eur. Heart J. 2020, 41, 99–109c. [Google Scholar] [CrossRef] [PubMed]
- Arndt, L.; Hernandez-Resendiz, I.; Moos, D.; Dokas, J.; Müller, S.; Jeromin, F.; Wagner, R.; Ceglarek, U.; Heid, I.M.; Höring, M.; et al. Trib1 Deficiency Promotes Hyperlipidemia, Inflammation, and Atherosclerosis in LDL Receptor Knockout Mice. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 979–994. [Google Scholar] [CrossRef] [PubMed]
- Mehrabian, M.; Schulthess, F.T.; Nebohacova, M.; Castellani, L.W.; Zhou, Z.; Hartiala, J.; Oberholzer, J.; Lusis, A.J.; Maedler, K.; Allayee, H. Identification of ALOX5 as a gene regulating adiposity and pancreatic function. Diabetologia 2008, 51, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Manev, H.; Manev, R. 5-Lipoxygenase as a Possible Biological Link Between Depressive Symptoms and Atherosclerosis. Arch. Gen. Psychiatry 2007, 64, 1333. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Li, J.; Hu, W. The complex interplay between ferroptosis and atherosclerosis. Biomed. Pharmacother. 2024, 178, 117183. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights Into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef] [PubMed]
- Drożdż, D.; Drożdż, M.; Wójcik, M. Endothelial dysfunction as a factor leading to arterial hypertension. Pediatr. Nephrol. 2023, 38, 2973–2985. [Google Scholar] [CrossRef] [PubMed]
- Borén, J.; Chapman, M.J.; Krauss, R.M.; Packard, C.J.; Bentzon, J.F.; Binder, C.J.; Daemen, M.J.; Demer, L.L.; Hegele, R.A.; Nicholls, S.J.; et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: Pathophysiological, genetic, and therapeutic insights: A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur. Heart J. 2020, 41, 2313–2330. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Akyol, O.; Yang, C.Y.; Woodside, D.G.; Chiang, H.H.; Chen, C.H.; Gotto, A.M. Comparative Analysis of Atherogenic Lipoproteins L5 and Lp(a) in Atherosclerotic Cardiovascular Disease. Curr. Atheroscler. Rep. 2024, 26, 317–329. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Itabe, H.; Obama, T. The Oxidized Lipoproteins In Vivo: Its Diversity and Behavior in the Human Circulation. Int. J. Mol. Sci. 2023, 24, 5747. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lara-Guzmán, O.J.; Gil-Izquierdo, Á.; Medina, S.; Osorio, E.; Álvarez-Quintero, R.; Zuluaga, N.; Oger, C.; Galano, J.-M.; Durand, T.; Muñoz-Durango, K. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol. 2018, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Bendszus, M. Inflammation and Atherosclerosis: Novel Insights Into Plaque Formation and Destabilization. Stroke 2006, 37, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P.; Tabas, I. Inflammation and plaque vulnerability. J. Intern. Med. 2015, 278, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Inflammation in Atherosclerosis—No Longer a Theory. Clin. Chem. 2021, 67, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Lerman, A.; Zeiher, A.M. Endothelial function: Cardiac events. Circulation 2005, 111, 363–368. [Google Scholar] [CrossRef] [PubMed]
- Trepels, T.; Zeiher, A.M.; Fichtlscherer, S. The Endothelium and Inflammation. Endothelium 2006, 13, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Luscher, T.F. Anti-inflammatory therapies for cardiovascular disease. Eur. Heart J. 2014, 35, 1782–1791. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The Inflammasome. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Hara, H.; Núñez, G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem. Sci. 2016, 41, 1012–1021. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.; Ann Campbell, L. Pathogens and atherosclerosis: Update on the potential contribution of multiple infectious organisms to the pathogenesis of atherosclerosis. Thromb. Haemost. 2011, 106, 858–867. [Google Scholar] [CrossRef] [PubMed]
- Hoseini, Z.; Sepahvand, F.; Rashidi, B.; Sahebkar, A.; Masoudifar, A.; Mirzaei, H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J. Cell. Physiol. 2018, 233, 2116–2132. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Guan, Y.; Liang, B.; Ding, P.; Hou, X.; Wei, W.; Ma, Y. Therapeutic potential of MCC950, a specific inhibitor of NLRP3 inflammasome. Eur. J. Pharmacol. 2022, 928, 175091. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xing, S.; Gong, Z.; Mu, W.; Xing, Q. Silence of NLRP3 Suppresses Atherosclerosis and Stabilizes Plaques in Apolipoprotein E-Deficient Mice. Mediat. Inflamm. 2014, 2014, 507208. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Zhong, X.; Li, J.; Liu, H.; Ma, X.; He, R.; Zhao, Y. MicroRNA-30c-5p inhibits NLRP3 inflammasome-mediated endothelial cell pyroptosis through FOXO3 down-regulation in atherosclerosis. Biochem. Biophys. Res. Commun. 2018, 503, 2833–2840. [Google Scholar] [CrossRef] [PubMed]
- Emanuel, R.; Sergin, I.; Bhattacharya, S.; Turner, J.N.; Epelman, S.; Settembre, C.; Diwan, A.; Ballabio, A.; Razani, B. Induction of Lysosomal Biogenesis in Atherosclerotic Macrophages Can Rescue Lipid-Induced Lysosomal Dysfunction and Downstream Sequelae. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1942–1952. [Google Scholar] [CrossRef] [PubMed]
- Stamatikos, A.; Dronadula, N.; Ng, P.; Palmer, D.; Knight, E.; Wacker, B.K.; Tang, C.; Kim, F.; Dichek, D.A. ABCA1 Overexpression in Endothelial Cells In Vitro Enhances ApoAI-Mediated Cholesterol Efflux and Decreases Inflammation. Hum. Gene Ther. 2019, 30, 236–248. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.; Gochman, A.; He, M.; Certain, N.; Wollmuth, L.P. NMDA receptors require multiple pre-opening gating steps for efficient synaptic activity. Neuron 2020, 109, 488–501.e4. [Google Scholar] [CrossRef] [PubMed]
- Hogan-Cann, A.D.; Anderson, C.M. Physiological Roles of Non-Neuronal NMDA Receptors. Trends Pharmacol. Sci. 2016, 37, 750–767. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Lee, S.J.; Seo, K.W.; Bae, J.U.; Park, S.Y.; Kim, C.D. Homocysteine induces COX-2 expression in macrophages through ROS generated by NMDA receptor-calcium signaling pathways. Free Radic. Res. 2013, 47, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Cheng, Q.; Bao, X.; Luo, Y.; Zhou, Y.; Li, Y.; Hua, Q.; Liu, W.; Tang, S.; Feng, D.; et al. Over-activation of NMDA receptors promotes ABCA1 degradation and foam cell formation. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2020, 1865, 158778. [Google Scholar] [CrossRef] [PubMed]
- Bae, D.-H.; Jansson, P.J.; Huang, M.L.; Kovacevic, Z.; Kalinowski, D.; Lee, C.S.; Sahni, S.; Richardson, D.R. The role of NDRG1 in the pathology and potential treatment of human cancers. J. Clin. Pathol. 2013, 66, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Chekmarev, J.; Azad, M.G.; Richardson, D.R. The Oncogenic Signaling Disruptor, NDRG1: Molecular and Cellular Mechanisms of Activity. Cells 2021, 10, 2382. [Google Scholar] [CrossRef] [PubMed]
- Ellen, T.P.; Ke, Q.; Zhang, P.; Costa, M. NDRG1, a growth and cancer related gene: Regulation of gene expression and function in normal and disease states. Carcinogenesis 2007, 29, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Qin, Q.; Zhang, C.; Sun, X.; Kazama, K.; Yi, B.; Cheng, F.; Guo, Z.-F.; Sun, J. NDRG1 Signaling Is Essential for Endothelial Inflammation and Vascular Remodeling. Circ. Res. 2023, 132, 306–319. [Google Scholar] [CrossRef] [PubMed]
- Bursill, C.A. NDRG1: A New Regulator of Vascular Inflammation and Atherothrombosis. Circ. Res. 2023, 132, 320–322. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, H.; Yue, Y.; Li, S.; Zhang, D.; He, R. TUG1 knockdown ameliorates atherosclerosis via up-regulating the expression of miR-133a target gene FGF1. Cardiovasc. Pathol. 2018, 33, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Wassler, M.; Zhang, L.; Li, Y.; Wang, J.; Zhang, Y.; Shelat, H.; Williams, J.; Geng, Y.-J. MicroRNA-133a regulates insulin-like growth factor-1 receptor expression and vascular smooth muscle cell proliferation in murine atherosclerosis. Atherosclerosis 2014, 232, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, T. LncRNA TUG1 regulates ApoM to promote atherosclerosis progression through miR-92a/FXR1 axis. J. Cell. Mol. Med. 2020, 24, 8836–8848. [Google Scholar] [CrossRef] [PubMed]
- Borup, A.; Christensen, P.M.; Nielsen, L.B.; Christoffersen, C. Apolipoprotein M in lipid metabolism and cardiometabolic diseases. Curr. Opin. Lipidol. 2015, 26, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [PubMed]
- Mills, C. M1 and M2 Macrophages: Oracles of Health and Disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur. J. Pharmacol. 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Tarique, A.A.; Logan, J.; Thomas, E.; Holt, P.G.; Sly, P.D.; Fantino, E. Phenotypic, Functional, and Plasticity Features of Classical and Alternatively Activated Human Macrophages. Am. J. Respir. Cell Mol. Biol. 2015, 53, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Hörhold, F.; Eisel, D.; Oswald, M.; Kolte, A.; Röll, D.; Osen, W.; Eichmüller, S.B.; König, R. Reprogramming of macrophages employing gene regulatory and metabolic network models. PLoS Comput. Biol. 2020, 16, e1007657. [Google Scholar] [CrossRef] [PubMed]
- Spiller, K.L.; Nassiri, S.; Witherel, C.E.; Anfang, R.R.; Ng, J.; Nakazawa, K.R.; Yu, T.; Vunjak-Novakovic, G. Sequential delivery of immunomodulatory cytokines to facilitate the M1-to-M2 transition of macrophages and enhance vascularization of bone scaffolds. Biomaterials 2015, 37, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Wang, S.Y.; Kwak, G.; Yang, Y.; Kwon, I.C.; Kim, S.H. Exosome-Guided Phenotypic Switch of M1 to M2 Macrophages for Cutaneous Wound Healing. Adv. Sci. 2019, 6, 1900513. [Google Scholar] [CrossRef] [PubMed]
- Puylaert, P.; Zurek, M.; Rayner, K.J.; De Meyer, G.R.Y.; Martinet, W. Regulated Necrosis in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1283–1306. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Nguyen, M.-A.; Geoffrion, M.; Vreeken, D.; Lister, Z.; Cheng, H.S.; Otte, N.; Essebier, P.; Wyatt, H.; Kandiah, J.W.; et al. RIPK1 Expression Associates With Inflammation in Early Atherosclerosis in Humans and Can Be Therapeutically Silenced to Reduce NF-κB Activation and Atherogenesis in Mice. Circulation 2021, 143, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Tian, S.; Liu, Z.; Dong, W. Knockdown of RIPK2 Inhibits Proliferation and Migration, and Induces Apoptosis via the NF-κB Signaling Pathway in Gastric Cancer. Front. Genet. 2021, 12, 627464. [Google Scholar] [CrossRef] [PubMed]
- Olivares, A.L.; González Ballester, M.A.; Noailly, J. Virtual exploration of early stage atherosclerosis. Bioinformatics 2016, 32, 3798–3806. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Qin, X.; Zhou, X.; Zhou, J.; Wen, P.; Chen, S.; Wu, M.; Wu, Y.; Zhuang, J. Analysis of genes and underlying mechanisms involved in foam cells formation and atherosclerosis development. PeerJ 2020, 8, e10336. [Google Scholar] [CrossRef] [PubMed]
- St Laurent, G.; Wahlestedt, C.; Kapranov, P. The Landscape of long noncoding RNA classification. Trends Genet. 2015, 31, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef] [PubMed]
- Pelechano, V.; Steinmetz, L.M. Gene regulation by antisense transcription. Nat. Rev. Genet. 2013, 14, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Winter, H.; Winski, G.; Busch, A.; Chernogubova, E.; Fasolo, F.; Wu, Z.; Bäcklund, A.; Khomtchouk, B.B.; Van Booven, D.J.; Sachs, N.; et al. Targeting long non-coding RNA NUDT6 enhances smooth muscle cell survival and limits vascular disease progression. Mol. Ther. 2023, 31, 1775–1790. [Google Scholar] [CrossRef] [PubMed]
- Nugent, M.A.; Iozzo, R.V. Fibroblast growth factor-2. Int. J. Biochem. Cell Biol. 2000, 32, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Nguyen, L.T.; Zhang, Z.W.; Moodie, K.L.; Carmeliet, P.; Stan, R.V.; Simons, M. The FGF system has a key role in regulating vascular integrity. J. Clin. Investig. 2008, 118, 3355–3366. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.-H.; Tang, Z.-H.; Li, G.-H.; Qu, S.-L.; Zhang, Y.; Ren, Z.; Liu, L.-S.; Jiang, Z.-S. Janus-like role of fibroblast growth factor 2 in arteriosclerotic coronary artery disease: Atherogenesis and angiogenesis. Atherosclerosis 2013, 229, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Barillari, G.; Iovane, A.; Bonuglia, M.; Albonici, L.; Garofano, P.; Di Campli, E.; Falchi, M.; Condò, I.; Manzari, V.; Ensoli, B. Fibroblast growth factor-2 transiently activates the p53 oncosuppressor protein in human primary vascular smooth muscle cells: Implications for atherogenesis. Atherosclerosis 2010, 210, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Wang, Q.; Ma, H.; Yan, W.; Yang, J. Knockout of Low Molecular Weight FGF2 Attenuates Atherosclerosis by Reducing Macrophage Infiltration and Oxidative Stress in Mice. Cell Physiol. Biochem. 2018, 45, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Liu, X.Q.; Song, Y.; Zhai, C.G.; Xu, X.L.; Zhang, L.; Zhang, Y. Fibroblast growth factor-2/platelet-derived growth factor enhances atherosclerotic plaque stability. J. Cell. Mol. Med. 2020, 24, 1128–1140. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Yang, S.; Xia, Y.; Hu, R.; Chen, S.; Li, B.; Chen, S.; Luo, X.; Mao, L.; Li, Y.; et al. LncRNA MIAT sponges miR-149-5p to inhibit efferocytosis in advanced atherosclerosis through CD47 upregulation. Cell Death Dis. 2019, 10, 138. [Google Scholar] [CrossRef] [PubMed]
- Quan, W.; Hu, P.-F.; Zhao, X.; Lianhua, C.-G.; Batu, B.-R. Expression level of lncRNA PVT1 in serum of patients with coronary atherosclerosis disease and its clinical significance. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 6333–6337. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Zhang, J.; Xu, N.; Han, G.; Geng, Q.; Song, J.; Li, S.; Zhao, J.; Chen, H. Signature of Circulating MicroRNAs as Potential Biomarkers in Vulnerable Coronary Artery Disease. PLoS ONE 2013, 8, e80738. [Google Scholar] [CrossRef] [PubMed]
- Latunde-Dada, G.O. Ferroptosis: Role of lipid peroxidation, iron and ferritinophagy. Biochim. Biophys. Acta (BBA)-Gen. Subj. 2017, 1861, 1893–1900. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhao, Y.; Ye, T.; Yang, L.; Shen, Y.; Li, H. Ferroptosis Signaling and Regulators in Atherosclerosis. Front. Cell Dev. Biol. 2021, 9, 809457. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Li, M.; Liu, Y.; Qiao, Z.; Wang, Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free. Radic. Biol. Med. 2020, 160, 92–102. [Google Scholar] [CrossRef] [PubMed]
- He, G.-N.; Bao, N.-R.; Wang, S.; Xi, M.; Zhang, T.-H.; Chen, F.-S. Ketamine Induces Ferroptosis of Liver Cancer Cells by Targeting lncRNA PVT1/miR-214-3p/GPX4. Drug Des. Dev. Ther. 2021, 15, 3965–3978. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Yu, Z.; Zhao, L.; Luo, H. Long non-coding RNA PVT1 regulates atherosclerosis progression via the microRNA-106b-5p/ACSL4 axis. Biochem. Biophys. Res. Commun. 2023, 667, 170–179. [Google Scholar] [CrossRef] [PubMed]
- Dexheimer, P.J.; Cochella, L. MicroRNAs: From Mechanism to Organism. Front. Cell Dev. Biol. 2020, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Li, X.; Liu, H.; Zhong, D.; Yin, K.; Li, Y.; Zhu, L.; Xu, C.; Li, M.; Wang, C. Bone marrow stromal cell-derived exosomal circular RNA improves diabetic foot ulcer wound healing by activating the nuclear factor erythroid 2-related factor 2 pathway and inhibiting ferroptosis. Diabet. Med. 2023, 40, e15031. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, N.; Sun, L.; Zheng, D.; Shao, G. Non-coding RNAs: The key detectors and regulators in cardiovascular disease. Genomics 2021, 113, 1233–1246. [Google Scholar] [CrossRef] [PubMed]
- Bao, M.-H.; Li, J.-M.; Zhou, Q.-L.; Li, G.-Y.; Zeng, J.; Zhao, J.; Zhang, Y.-W. Effects of miR-590 on oxLDL-induced endothelial cell apoptosis: Roles of p53 and NF-κB. Mol. Med. Rep. 2016, 13, 867–873. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Chen, J.; Zheng, H.; Lin, A.; Li, J.; Wu, W.; Jie, Q. MiR-199a-5p-containing macrophage-derived extracellular vesicles inhibit SMARCA4 and alleviate atherosclerosis by reducing endothelial cell pyroptosis. Cell Biol. Toxicol. 2023, 39, 591–605. [Google Scholar] [CrossRef] [PubMed]
- Jiang, F.; Chen, Q.; Wang, W.; Ling, Y.; Yan, Y.; Xia, P. Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J. Hepatol. 2020, 72, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Shang, F.; Wang, S.-C.; Gongol, B.; Han, S.Y.; Cho, Y.; Schiavon, C.R.; Chen, L.; Xing, Y.; Zhao, Y.; Ning, M.; et al. Obstructive Sleep Apnea–induced Endothelial Dysfunction Is Mediated by miR-210. Am. J. Respir. Crit. Care Med. 2023, 207, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Liu, L.; Jiang, Z.; Hao, X. Inhibition of microRNA-17-5p reduces the inflammation and lipid accumulation, and up-regulates ATP-binding cassette transporterA1 in atherosclerosis. J. Pharmacol. Sci. 2019, 139, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Li, X.; Liu, S.; Gu, L.; Zhou, X. MircroRNA-19a promotes vascular inflammation and foam cell formation by targeting HBP-1 in atherogenesis. Sci. Rep. 2017, 7, 12089. [Google Scholar] [CrossRef] [PubMed]
- Mayer, O.; Seidlerová, J.; Černá, V.; Kučerová, A.; Vaněk, J.; Karnosová, P.; Bruthans, J.; Wohlfahrt, P.; Cífková, R.; Pešta, M.; et al. The low expression of circulating microRNA-19a represents an additional mortality risk in stable patients with vascular disease. Int. J. Cardiol. 2019, 289, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Kim, C.W.; Simmons, R.D.; Jo, H. Role of Flow-Sensitive microRNAs in Endothelial Dysfunction and Atherosclerosis: Mechanosensitive Athero-miRs. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, S.; Hartmann, P.; Karshovska, E.; Rinderknecht, F.-A.; Subramanian, P.; Gremse, F.; Grommes, J.; Jacobs, M.; Kiessling, F.; Weber, C.; et al. Endothelial Hypoxia-Inducible Factor-1α Promotes Atherosclerosis and Monocyte Recruitment by Upregulating MicroRNA-19a. Hypertension 2015, 66, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Wang, L.; Cui, C.; Chen, H.; Zeng, W.; Li, X. MicroRNA-19a-3p inhibits endothelial dysfunction in atherosclerosis by targeting JCAD. BMC Cardiovasc. Disord. 2024, 24, 394. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Wu, X.; Jones, M.; Zhang, Z.; Salisbury, D.; Sallam, T. Long Noncoding RNA Facilitated Gene Therapy Reduces Atherosclerosis in a Murine Model of Familial Hypercholesterolemia. Circulation 2017, 136, 776–778. [Google Scholar] [CrossRef] [PubMed]
- Nazari-Jahantigh, M.; Egea, V.; Schober, A.; Weber, C. MicroRNA-specific regulatory mechanisms in atherosclerosis. J. Mol. Cell. Cardiol. 2015, 89, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Ewe, A.; Aigner, A. Cationic Lipid-Coated Polyplexes (Lipopolyplexes) for DNA and Small RNA Delivery. Methods Mol. Biol. 2016, 1445, 187–200. [Google Scholar] [CrossRef] [PubMed]
- Del Pozo-Acebo, L.; López De Las Hazas, M.-C.; Tomé-Carneiro, J.; Gil-Cabrerizo, P.; San-Cristobal, R.; Busto, R.; García-Ruiz, A.; Dávalos, A. Bovine Milk-Derived Exosomes as a Drug Delivery Vehicle for miRNA-Based Therapy. Int. J. Mol. Sci. 2021, 22, 1105. [Google Scholar] [CrossRef] [PubMed]
- Sobenin, I.A.; Chistiakov, D.A.; Bobryshev, Y.V.; Postnov, A.Y.; Orekhov, A.N. Mitochondrial mutations in atherosclerosis: New solutions in research and possible clinical applications. Curr. Pharm. Des. 2013, 19, 5942–5953. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Li, J.; Liu, X.; Tong, T.; Zhang, Z. Age-dependent accumulation of mitochondrial DNA deletions in the aortic root of atherosclerosis-prone apolipoprotein E-knockout mice. Arch. Gerontol. Geriatr. 2016, 63, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Berardo, A.; Musumeci, O.; Toscano, A. Cardiological manifestations of mitochondrial respiratory chain disorders. Acta Myol. 2011, 30, 9–15. [Google Scholar] [PubMed]
- Sobenin, I.A.; Sazonova, M.A.; Postnov, A.Y.; Salonen, J.T.; Bobryshev, Y.V.; Orekhov, A.N. Association of Mitochondrial Genetic Variation with Carotid Atherosclerosis. PLoS ONE 2013, 8, e68070. [Google Scholar] [CrossRef] [PubMed]
- Halioua-Haubold, C.L.; Peyer, J.G.; Smith, J.A.; Arshad, Z.; Scholz, M.; Brindley, D.A.; MacLaren, R.E. Regulatory considerations for gene therapy products in the US, EU, and Japan. Yale J. Biol. Med. 2017, 90, 683–693. [Google Scholar] [PubMed]
- Wang, J.H.; Gessler, D.J.; Zhan, W.; Gallagher, T.L.; Gao, G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Signal Transduct. Target. Ther. 2024, 9, 78. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Irfan, H.; Fatima, E.; Nazir, Z.; Verma, A.; Akilimali, A. Revolutionary breakthrough: FDA approves CASGEVY, the first CRISPR/Cas9 gene therapy for sickle cell disease. Ann. Med. Surg. 2024, 86, 4555–4559. [Google Scholar] [CrossRef] [PubMed]
- Coller, B.S. Ethics of human genome editing. Annu. Rev. Med. 2019, 70, 289–305. [Google Scholar] [CrossRef] [PubMed]
- Ansah, E.O. Ethical challenges and controversies in the practice and advancement of gene therapy. Adv. Cell Gene Ther. 2022, 2022, 1015996. [Google Scholar] [CrossRef]
- Kimberly, L.; Hunt, C.; Beaverson, K.; James, E.; Bateman-House, A.; McGowan, R.; DeSante-Bertkau, J. The lived experience of pediatric gene therapy: A scoping review. Hum. Gene Ther. 2023, 34, 1180–1189. [Google Scholar] [CrossRef] [PubMed]
- Beswick, L. The health economics of cell and gene therapies. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2024; Volume 205, pp. 155–167. [Google Scholar] [CrossRef]
- Van der Schans, S.; Velikanova, R.; Weidlich, D.; Howells, R.; Patel, A.; Bischof, M.; Postma, M.J.; Boersma, C. Cost comparison analysis of onasemnogene abeparvovec and nusinersen for treatment of patients with spinal muscular atrophy type 1 in the Netherlands. Eur. J. Health Econ. 2025. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Zhao, J.; Duan, D.; Lai, Y. Design of AAV Vectors for Delivery of Large or Multiple Transgenes. Methods Mol. Biol. 2019, 1950, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Canepari, C.; Assanelli, S.; Merlin, S.; Borroni, E.; Starinieri, F.; Biffi, M.; Russo, F.; Fabiano, A.; Zambroni, D.; et al. GP64-pseudotyped lentiviral vectors target liver endothelial cells and correct hemophilia A mice. EMBO Mol. Med. 2024, 16, 1427–1450. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Molecule Size, nm | Function and Role in the Development of Atherosclerosis |
|---|---|---|
| Low-density lipoproteins (LDL) | 18–26 | Transport of cholesterol, triacylglycerides and phospholipids from the liver to peripheral tissues. After penetrating the subendothelial space, it can be subjected to oxidative modification. Oxidized LDL and LDL with other modifications can be captured by: 1. smooth muscle cells of the vascular wall via scavenger receptors, 2. macrophages, which are transformed into foam cells overloaded with cholesterol esters. |
| Intermediate-density lipoproteins (IDL) | 25–35 | Transport of cholesterol, triacylglycerides and phospholipids from the liver to peripheral tissues. Being intermediate particles in LDL metabolism, also contribute to the accumulation of cholesterol in the vascular wall. They participate in the formation of the lipid core of the future atherosclerotic plaque, enhancing the processes of cellular infiltration and inflammation. |
| Very low-density lipoproteins (VLDL) | 30–80 | Contains apolipoprotein B 100 (Apo B-100), which transports cholesterol, triacylglycerides and phospholipids from the liver to peripheral tissues. During lipolysis, under the action of lipoprotein lipase, VLDL is transformed into residual particles that are easily retained in the subendothelium and undergo modifications. The accumulation of foam cells, cellular decay products and extracellular lipid material leads to the formation of a necrotic core of an atherosclerotic plaque—a key element of unstable lesions. |
| Target Gene | Approach | Model/Study | Key Findings | Reference |
|---|---|---|---|---|
| LDLR | Exosome-delivered LDLR mRNA | LDLR−/− mice (FH model) | - Reduced serum cholesterol by 50% and atherosclerotic plaques by 3-fold. | [26] |
| LDLR | CRISPR/Cas9 editing via AAV8 | LDLR E208X mice | - Partial Ldlr restoration (11% of WT); 2-fold reduction in plaque area. | [46] |
| ApoB | shRNA-mediated silencing (AAV8) | C57BL/6 mice | - 95% reduction in ApoB mRNA; 79% lower cholesterol. | [48] |
| ApoE | Lentiviral ApoE mimetic peptide | ApoE−/− mice | - 24% reduction in aortic lesions without cholesterol changes. | [56] |
| Lipoprotein (a) (Lp(a)) | Antisense oligonucleotides | Phase 2b clinical trials completed | Reduction of Lp(a) by 35–58% in clinical trials | [53] |
| PCSK9 | CRISPR/Cas9 (AAV8-SaCas9) | C57BL/6 mice | - 80% reduction in Pcsk9 protein; 35% lower cholesterol. | [71] |
| PCSK9 | Lipid nanoparticles (base editing) | Non-human primates | - 90% PCSK9 knockdown; 60% LDL-C reduction. | [82] |
| PCSK9 | Inclisiran (siRNA) | Approved for clinical use in the EU (2020), UK and US (2021), China (2023). ORION-4 clinical trials are expected to be finished by 2049 | - 51% LDL-C reduction in ASCVD patients with biannual dosing. | [68] |
| ANGPTL3 | CRISPR/Cas9 (AAV9) | C57BL/6 mice | - 65% reduction in Angptl3; 57% lower LDL-C. | [78] |
| ANGPTL3 | siRNA (ARO-ANG3) | Clinical trials (phase 1 basket trial) | - 80% Angptl3 reduction; significant LDL-C and TAG lowering. | [97] |
| APOC3 | CRISPR knockout | Syrian hamsters | - Reduced atherosclerotic lesions and plasma TAG. | [102] |
| APOC3 | siRNA (ARO-APOC3) | Clinical trials (2b placebo-controlled) | - 77% Apoc3 reduction; 57% TAG decrease. | [106] |
| APOC3 | Volanesorsen (antisense oligonucleotide) | Approved for clinical use in the EU (2019) | Blood TAG levels decreased by 77% | [105] |
| MTP | miRNA-30c (inhibits MTP/APOB) | ApoE−/− mice | - Reduced plasma cholesterol (~30%) and ApoB secretion. | [110] |
| MTP | Small-molecule inhibitor (BMS 212122) | LDLR−/− mice | - 94% cholesterol reduction; reduced plaque lipid content. | [111] |
| Target | Method | Model | Results | Reference |
|---|---|---|---|---|
| IL-1β | Antibody therapy | Clinical trials (10,061 patients) | Affecting IL-1β-regulated IL-6 expression and decreasing the level of C-reactive protein expression. In groups receiving Canakinumab, the median of C-reactive protein decline was 26–41% higher than in the control group. | [131] |
| NLRP3 | RNA-interference using lentiviral vector delivery | Murine model | Silencing of the NLRP3 gene halted plaque progression and suppressed the expression of pro-inflammatory cytokines. RNA interference decreased macrophage and lipid content within the plaques, while increasing the presence of smooth muscle cells and collagen, thereby contributing to the stabilization of atherosclerotic plaques. | [137] |
| NLRP3 | Increasing miRNA expression level using synthesized oligonucleotides | Cell lines | It was shown that the addition of miR-30c-5p reversed LDL-induced pyroptosis in the HAEC cell line. The abundance of NLRP3 was decreased at the protein and mRNA levels. | [138] |
| NDRG1 | RNA-interference using lentiviral vector delivery | Murine models and human cell lines | Knockdown of NDRG1 using a lentivirus encoding NDRG1 shRNA reduces IL-1β- and TNF-α-induced expression of cytokines, chemokines, and adhesion molecules. NDRG1 inhibition also significantly decreases the expression of procoagulant factors such as plasminogen activator inhibitor-1 (PAI-1) and tissue factor (TF), while enhancing the expression of antithrombotic molecules like thrombomodulin (TM) and tissue-type plasminogen activator (t-PA), thereby promoting strong antithrombotic effects in endothelial cells. | [148] |
| lncRNA TUG1 | RNA-interference using siRNA against TUG1 | Murine models | Silencing of TUG1 reduced hyperlipidemia, suppressed inflammatory responses, and alleviated atherosclerotic lesions in HFD-treated ApoE−/− mice. Over-expression of TUG1 promoted cell proliferation, enhanced inflammatory cytokine production, and inhibited apoptosis in ox-LDL-exposed cells | [150] |
| Target Gene | Approach | Model/Study | Key Findings | Reference |
|---|---|---|---|---|
| Nucleoside diphosphate-linked moiety X motif 6 (NUDT6) | Antisense oligonucleotides | Atherosclerotic ApoE-deficient mouse model | Site-specific antisense silencing of NUDT6 expression reduced plaque rupture rates and experimental abdominal aortic aneurysm growth | [169] |
| PVT1 | Antisense oligonucleotides | Atherosclerotic ApoE-deficient mouse model | The expression of LncRNA PVT1 in the si-LncRNA PVT1 group was reduced. The aortic arch of this group showed a decreased number and size of atherosclerotic plaque compared with the control group, by 70%. | [185] |
| MicroRNA-19a-3p | Antisense oligonucleotides | HUVEC cell line; atherosclerotic ApoE-deficient mouse model | MiR-19a-3p defines an important regulatory mechanism of endothelial function and the pathogenesis of atherosclerosis. | [198] |
| Liver-expressed LXR-induced sequence (LeXis) | AAV8 expressing LeXis under the control of the human liver-specific thyroxine-binding globulin promoter | Ldlr-deficient mouse model | LDLR−/− animal treatment with AAV8.hTBG.LeXis demonstrated a reduction of almost 2 times in AS load compared to mice in the control group. | [199] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bezsonov, E.; Chernyi, N.; Saruhanyan, M.; Shimchenko, D.; Bondar, N.; Gavrilova, D.; Baig, M.S.; Malogolovkin, A. Gene Therapy Approaches for Atherosclerosis Focusing on Targeting Lipid Metabolism and Inflammation. Int. J. Mol. Sci. 2025, 26, 6950. https://doi.org/10.3390/ijms26146950
Bezsonov E, Chernyi N, Saruhanyan M, Shimchenko D, Bondar N, Gavrilova D, Baig MS, Malogolovkin A. Gene Therapy Approaches for Atherosclerosis Focusing on Targeting Lipid Metabolism and Inflammation. International Journal of Molecular Sciences. 2025; 26(14):6950. https://doi.org/10.3390/ijms26146950
Chicago/Turabian StyleBezsonov, Evgeny, Nikita Chernyi, Mane Saruhanyan, Dariia Shimchenko, Nikolai Bondar, Darina Gavrilova, Mirza S. Baig, and Alexander Malogolovkin. 2025. "Gene Therapy Approaches for Atherosclerosis Focusing on Targeting Lipid Metabolism and Inflammation" International Journal of Molecular Sciences 26, no. 14: 6950. https://doi.org/10.3390/ijms26146950
APA StyleBezsonov, E., Chernyi, N., Saruhanyan, M., Shimchenko, D., Bondar, N., Gavrilova, D., Baig, M. S., & Malogolovkin, A. (2025). Gene Therapy Approaches for Atherosclerosis Focusing on Targeting Lipid Metabolism and Inflammation. International Journal of Molecular Sciences, 26(14), 6950. https://doi.org/10.3390/ijms26146950