
A Quest for Effective 19F NMR Spectra Modeling: What Brings a Good Balance Between Accuracy and Computational Cost in Fluorine Chemical Shift Calculations?



Abstract
1. Introduction
2. Results and Discussion
2.1. Studying the Performance of Different Exchange-Correlation Functionals and One-Electron Basis Sets in the Calculation of Equilibrium Geometry Parameters for Fluorine Compounds Within the DFT Method
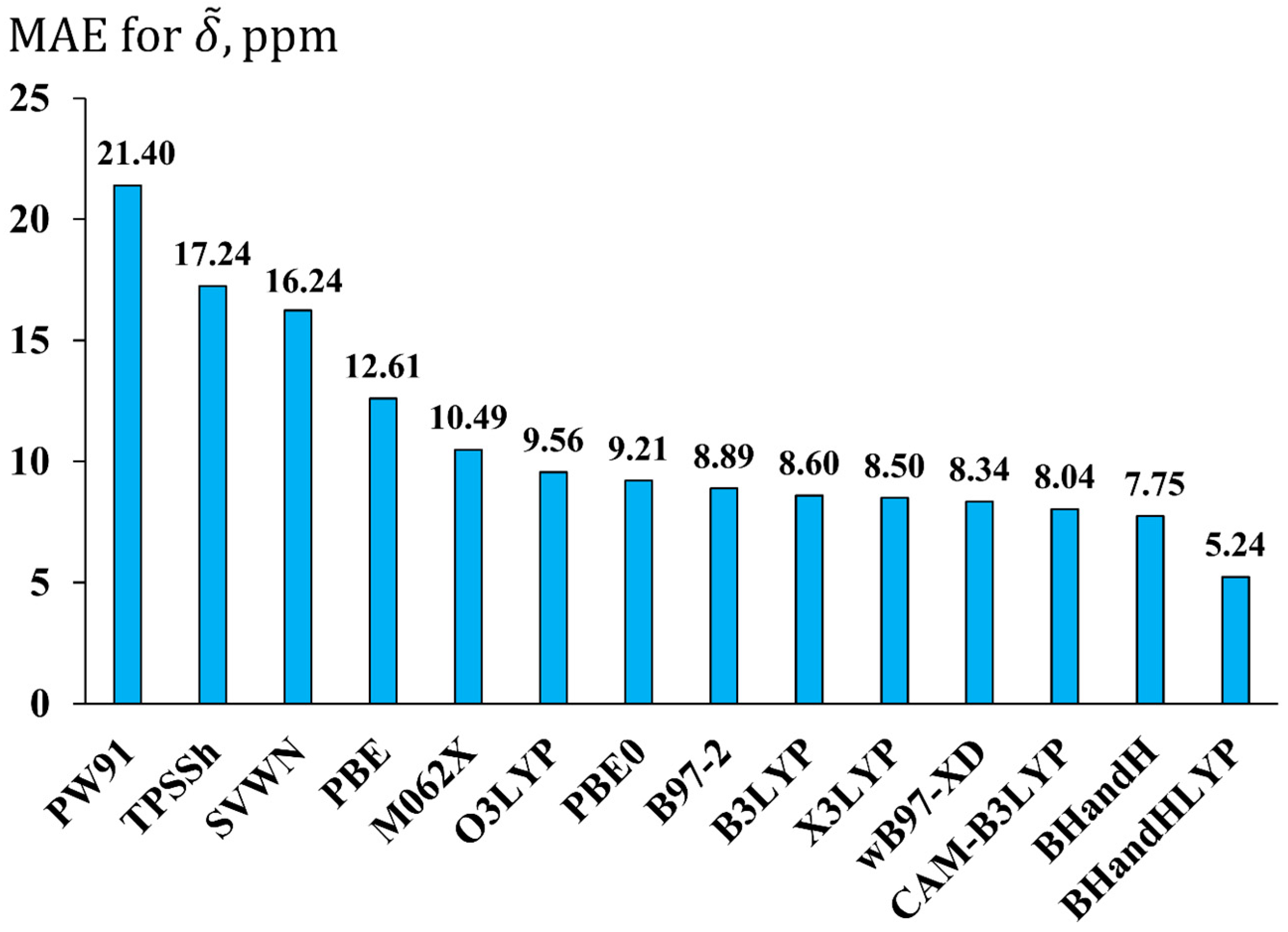
2.2. Studying the Performance of Different Exchange-Correlation DFT Functionals and One-Electron Basis Sets in the Calculation of 19F NMR Chemical Shifts
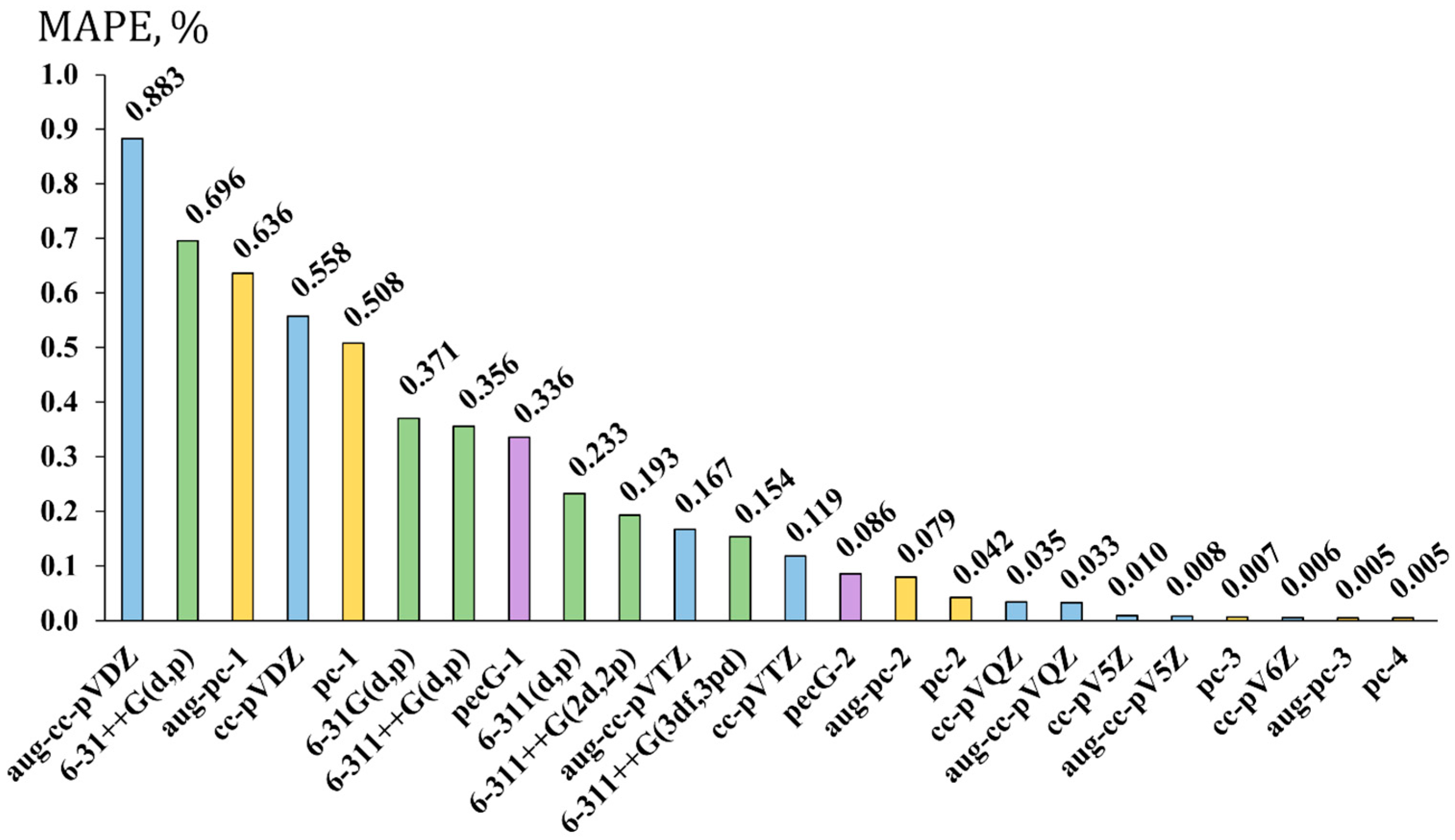
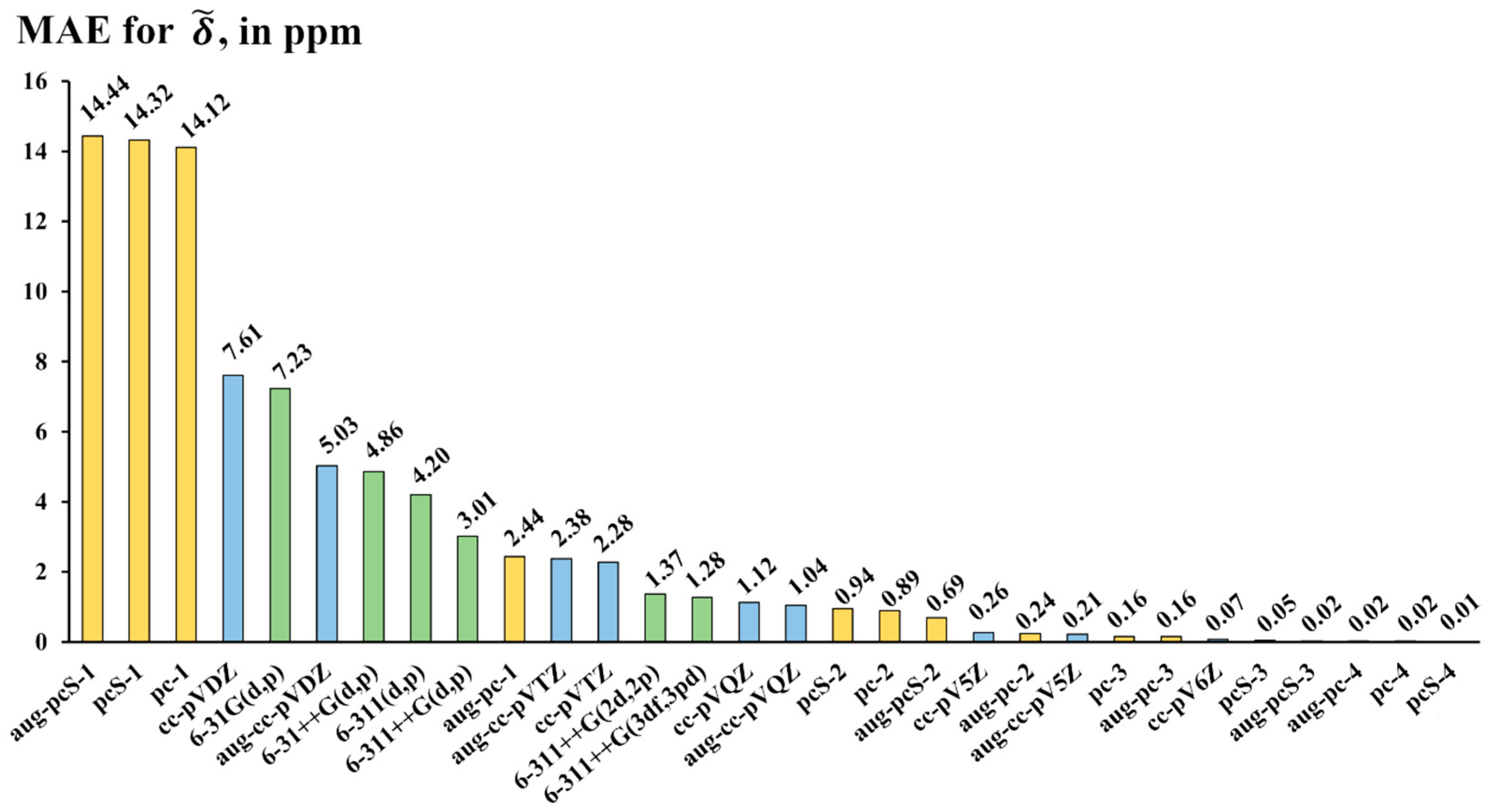
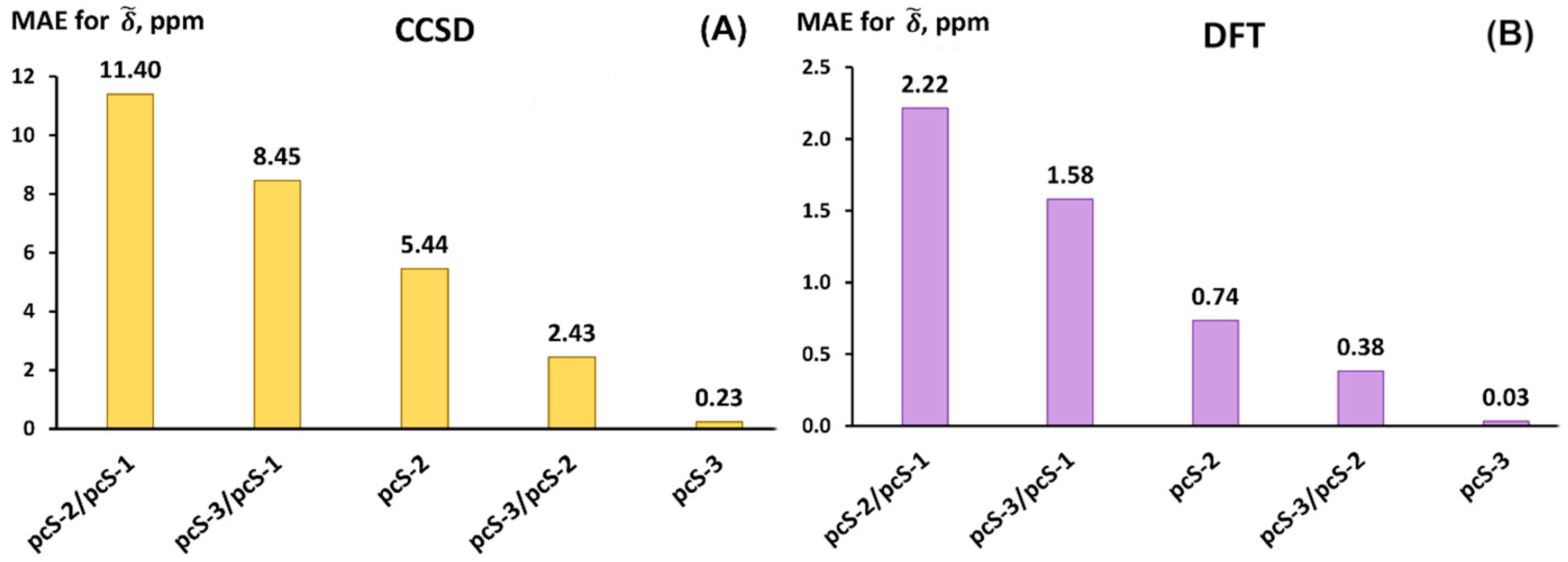
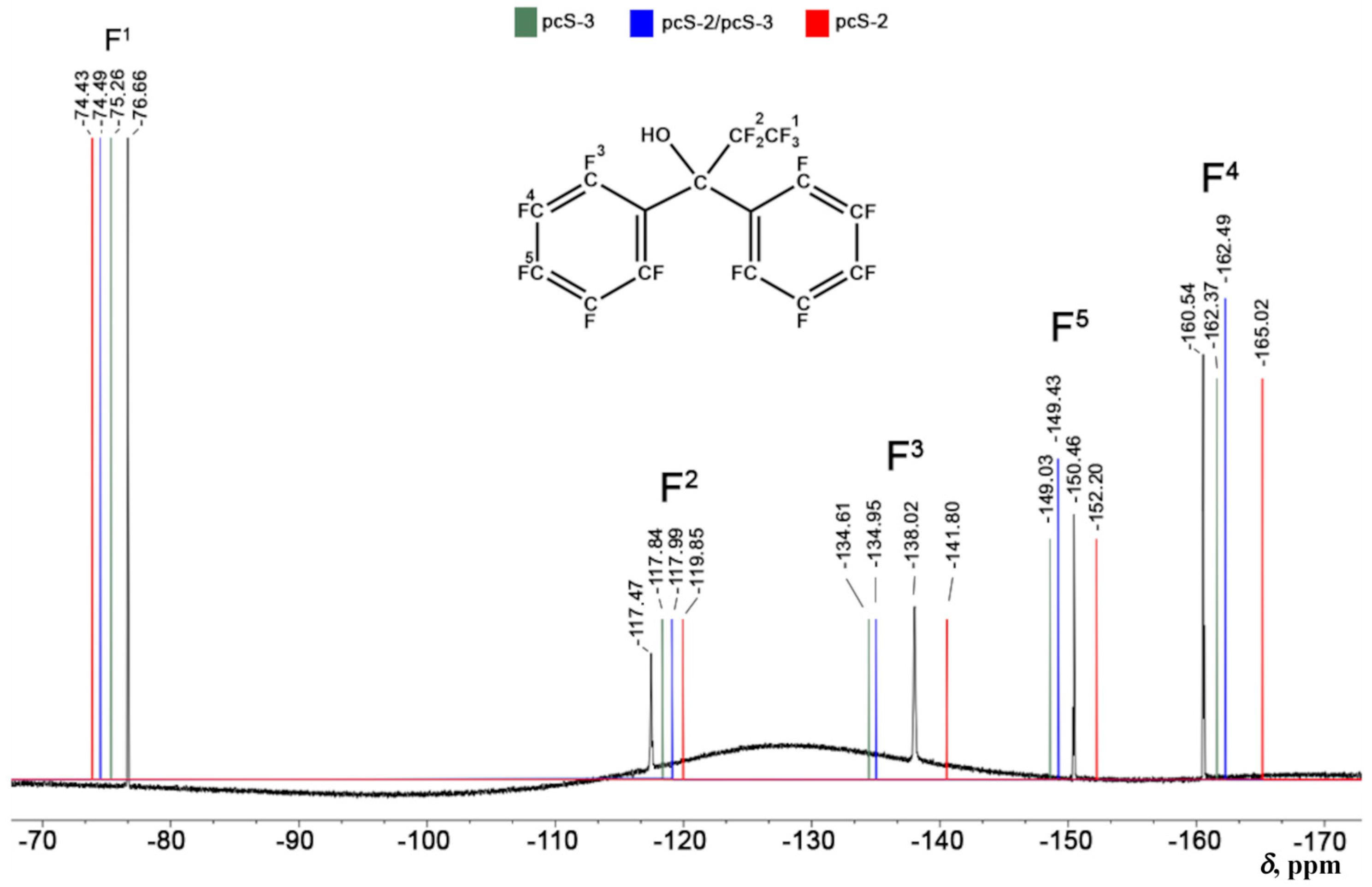
2.3. Testing Different Basis Set Schemes in the Calculations of 19NMR Chemical Shifts Against the Experimental Values
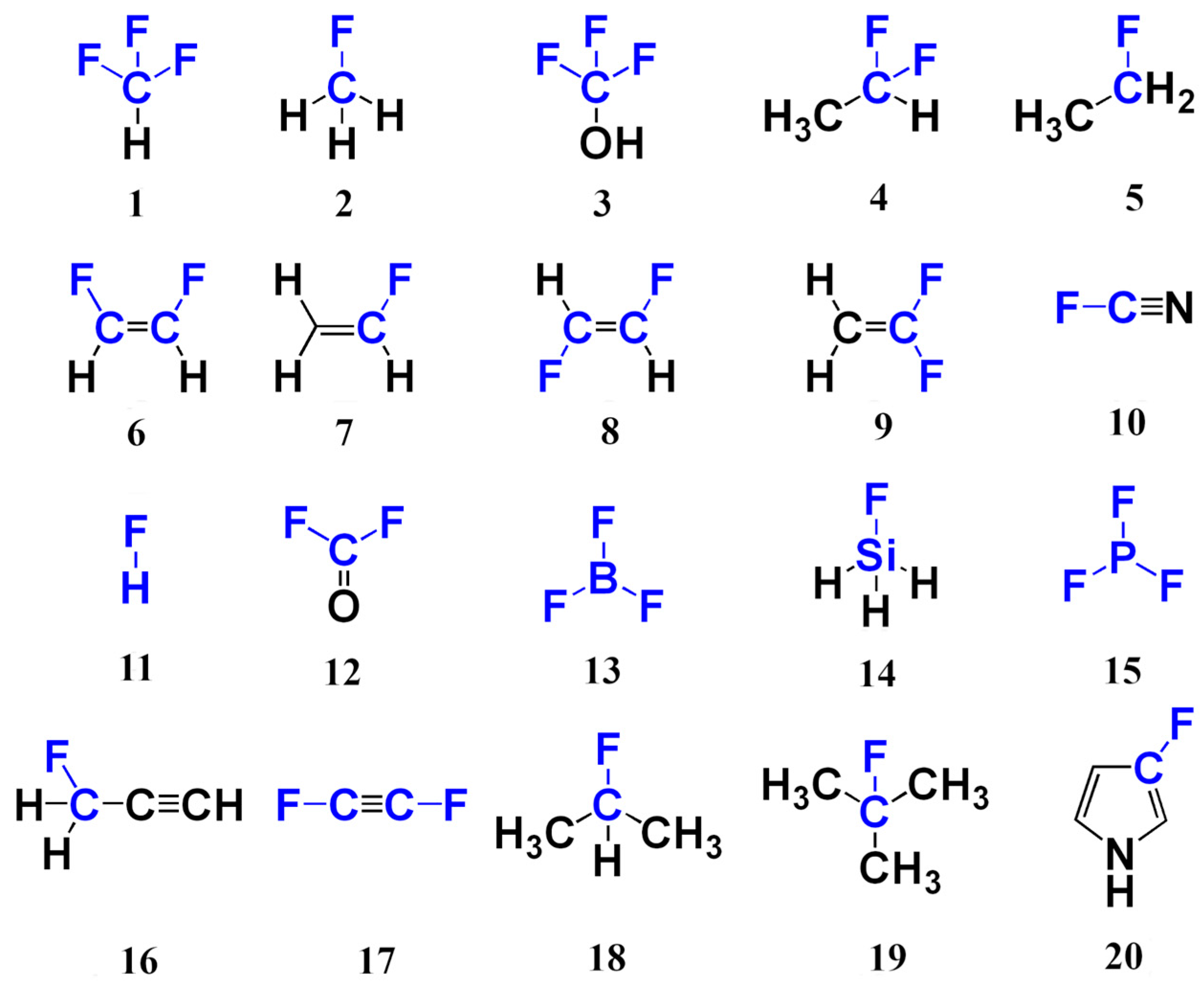
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, C.; Yan, K.; Fu, C.; Peng, H.; Hawker, C.J.; Whittaker, A.K. Biological Utility of Fluorinated Compounds: From Materials Design to Molecular Imaging, Therapeutics and Environmental Remediation. Chem. Rev. 2022, 122, 167–208. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Remete, A.M.; Dobson, L.S.; Kiss, L.; Izawa, K.; Moriwaki, H.; Soloshonok, V.A.; O’Hagan, D.J. Next generation organofluorine containing blockbuster drugs. J. Fluor. Chem. 2020, 239, 109639. [Google Scholar] [CrossRef]
- Abbas, A.A.; Farghaly, T.A.; Dawood, K.M. Recent progress in therapeutic applications of fluorinated five-membered heterocycles and their benzo-fused systems. RSC Adv. 2024, 14, 33864–33905. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Lu, Y. Fluorine in anti-HIV drugs approved by FDA from 1981 to 2023. Eur. J. Med. Chem 2023, 258, 115586. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, A.J.; El-Achkar, G.A.; Ghayad, S.E.; Hariss, L.; Haidar, R.H.; Antar, L.M.; Mallah, Z.I.; Badran, B.; Grée, R.; Hachem, A.; et al. Fluorinated Benzofuran and Dihydrobenzofuran as Anti-Inflammatory and Potential Anticancer Agents. Int. J. Mol. Sci. 2023, 24, 10399. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, C.; Chaudhary, M.; De Oliveira, R.N.; Borbas, A.; Kempaiah, P.; Rathi, P.S.; Rathi, P.S.; Rathi, B. Fluorinated scaffolds for antimalarial drug discovery. Expert Opin. Drug Discov. 2020, 15, 705–718. [Google Scholar] [CrossRef] [PubMed]
- Yoon, M.-H.; Dibenedetto, S.A.; Facchetti, A.; Marks, T.J. Organic thin-film transistors based on carbonyl-functionalized quaterthiophenes: High mobility N-channel semiconductors and ambipolar transport. J. Am. Chem. Soc. 2005, 127, 1348–1349. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, S.; Park, S.K.; Parkin, S.R.; Podzorov, V.; Jackson, T.N.; Anthony, J.E. Chromophore Fluorination Enhances Crystallization and Stability of Soluble Anthradithiophene Semiconductors. J. Am. Chem. Soc. 2008, 130, 2706–2707. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.L.; Reichardt, A.D.; Wei, P.; Bao, Z. Correlating Carrier Type with Frontier Molecular Orbital Energy Levels in Organic Thin Film Transistors of Functionalized Acene Derivatives. J. Am. Chem. Soc. 2009, 131, 5264–5273. [Google Scholar] [CrossRef] [PubMed]
- Ie, Y.; Umemoto, Y.; Kaneda, T.; Aso, Y. Electronegative Oligothiophenes Based on a Hexafluorocyclopentene-Annelated Thiophene Unit. Org. Lett. 2006, 8, 5381–5384. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Collard, D.M. Tuning the Electronic Structure of Conjugated Polymers with Fluoroalkyl Substitution: Alternating Alkyl/Perfluoroalkyl-Substituted Polythiophene. Macromolecules 2005, 38, 372–378. [Google Scholar] [CrossRef]
- Facchetti, A.; Yoon, M.-H.; Stern, C.L.; Hutchison, G.R.; Ratner, M.A.; Marks, T.J. Building Blocks for N-Type Molecular and Polymeric Electronics. Perfluoroalkyl- versus Alkyl-Functionalized Oligothiophenes (nTs; n = 2–6). Systematic Synthesis, Spectroscopy, Electrochemistry, and Solid-State Organization. J. Am. Chem. Soc. 2004, 126, 13480–13501. [Google Scholar] [CrossRef] [PubMed]
- Facchetti, A.; Mushrush, M.; Yoon, M.-H.; Hutchison, G.R.; Ratner, M.A.; Marks, T.J. Building Blocks for n-Type Molecular and Polymeric Electronics. Perfluoroalkyl- versus Alkyl-Functionalized Oligothiophenes (nT; n = 2–6). Systematics of Thin Film Microstructure, Semiconductor Performance, and Modeling of Majority Charge Injection in Field-Effect Transistors. J. Am. Chem. Soc. 2004, 126, 13859–13874. [Google Scholar] [CrossRef] [PubMed]
- Geramita, K.; McBee, J.; Shen, Y.; Radu, N.; Don Tilley, T. Synthesis and Characterization of Perfluoroaryl-Substituted Siloles and Thiophenes: A Series of Electron-Deficient Blue Light Emitting Materials. Chem. Mat. 2006, 18, 3261–3269. [Google Scholar] [CrossRef]
- Matharu, A.; Cowling, S.J.; Wright, G. Laterally fluorinated liquid crystals containing the 2,2′-bithiophene moiety. Liq. Cryst. 2007, 34, 489–506. [Google Scholar] [CrossRef]
- Popkov, S.V.; Kuzenkov, A.V. Synthesis of trifluoromethyl-substituted heteroaromatic aldehydes. Russ. Chem. Bull. Int. Ed. 2005, 54, 1672–1674. [Google Scholar] [CrossRef]
- Ning, X.; Seo, W.; Lee, S.; Takemiya, K.; Rafi, M.; Feng, X.; Weiss, D.; Wang, X.; Williams, L.; Camp, V.M.; et al. PET imaging of bacterial infections with fluorine-18-labeled maltohexaose. Angew. Chem. Int. Ed. 2014, 53, 14096–14101. [Google Scholar] [CrossRef] [PubMed]
- Dolbier, W.R., Jr. Guide to Fluorine NMR for Organic Chemists, 2nd ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; ISBN 978-1-118-83109-0. [Google Scholar]
- Zhao, Y.; Swager, T.M. Detection and differentiation of neutral organic compounds by 19F NMR with a tungsten calix [4]arene imido complex. J. Am. Chem. Soc. 2013, 135, 18770–18773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Markopoulos, G.; Swager, T.M. 19F NMR fingerprints: Identification of neutral organic compounds in a molecular container. J. Am. Chem. Soc. 2014, 136, 10683–10690. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-X.; Hallac, R.R.; Chiguru, S.; Mason, R.P. New frontiers and developing applications in 19F NMR. Prog. Nucl. Magn. Reson. Spectrosc. 2013, 70, 25–49. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.L.; Murphy, C.D. 19F NMR applications in chemical biology. J. Fluor. Chem. 2009, 130, 132–143. [Google Scholar] [CrossRef]
- Chen, H.; Viel, S.; Ziarelli, F.; Peng, L. 19F NMR: A valuable tool for studying biological events. Chem. Soc. Rev. 2013, 42, 7971–7982. [Google Scholar] [CrossRef] [PubMed]
- Gimenez, D.; Phelan, A.; Murphy, C.D.; Cobb, S.L. 19F NMR as a tool in chemical biology. Beilstein J. Org. Chem. 2021, 17, 293–318. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Liu, C.; Zhao, S.; Chen, S.; Zhao, Y. Molecular Sensors for NMR-Based Detection. Chem. Rev. 2019, 119, 195–230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Swager, T.M. Simultaneous chirality sensing of multiple amines by (19)F NMR. J. Am. Chem. Soc. 2015, 137, 3221–3224. [Google Scholar] [CrossRef] [PubMed]
- Kuroki, S.A. 19F NMR Signal Assignment and a Detailed Structural Study of Alternating Tetrafluoroethylene-Propylene Copolymer by High Resolution 19F NMR Spectroscopy and Computational Chemistry. Polym. J. 2009, 41, 449–454. [Google Scholar] [CrossRef]
- Martino, R.; Gilard, V.; Desmoulin, F.; Malet-Martino, M. Fluorine-19 or phosphorus-31 NMR spectroscopy: A suitable analytical technique for quantitative in vitro metabolic studies of fluorinated or phosphorylated drugs. J. Pharm. Biomed. Anal. 2005, 38, 871–891. [Google Scholar] [CrossRef] [PubMed]
- Krivdin, L.B. Computational aspects of 19F NMR. Russ. Chem. Rev. 2020, 89, 104. [Google Scholar] [CrossRef]
- Dreizler, R.M.; Gross, E.K.U. Density Functional Theory. In An Approach to the Quantum Many-Body Problem; Dreizler, R.M., E. Gross, K.U., Eds.; Springer: Berlin/Heidelberg, Germany, 1990. [Google Scholar] [CrossRef]
- Kasireddy, C.; Bann, J.G.; Mitchell-Koch, K.R. Demystifying fluorine chemical shifts: Electronic structure calculations address origins of seemingly anomalous 19F-NMR spectra of fluorohistidine isomers and analogues. Phys. Chem. Chem. Phys. 2015, 17, 30606–30612. [Google Scholar] [CrossRef]
- Dahanayake, J.N.; Kasireddy, C.; Ellis, J.M.; Hildebrandt, D.; Hull, O.A.; Karnes, J.P.; Morlan, D.; Mitchell-Koch, K.R. Evaluating electronic structure methods for accurate calculation of 19F chemical shifts in fluorinated amino acids. J. Comput. Chem. 2017, 38, 2605–2617. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, S.V.; Krivdin, L.B. Computational Protocols for the 19F NMR Chemical Shifts. Part 1: Methodological Aspects. J. Fluor. Chem. 2020, 238, 109625. [Google Scholar] [CrossRef]
- Mifkovic, M.; Pauling, J.; Vyas, S. Computational protocol for predicting 19F NMR chemical shifts for PFAS and connection to PFAS structure. J. Comput. Chem. 2022, 43, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Saunders, C.; Khaled, M.B.; Weaver, J.D.; Tantillo, D.J. Prediction of 19F NMR Chemical Shifts for Fluorinated Aromatic Compounds. J. Org. Chem. 2018, 83, 3220–3225. [Google Scholar] [CrossRef] [PubMed]
- Dumon, A.S.; Rzepa, H.S.; Alamillo-Ferrer, C.; Bures, J.; Procter, R.; Sheppard, T.D.; Whiting, A. A computational tool to accurately and quickly predict 19F NMR chemical shifts of molecules with fluorine–carbon and fluorine–boron bonds. Phys. Chem. Chem. Phys. 2022, 24, 20409. [Google Scholar] [CrossRef] [PubMed]
- Ukhanev, S.A.; Fedorov, S.V.; Krivdin, L.B. Computational 19F NMR of trifluoromethyl derivatives of alkenes, pyrimidines, and indenes. Magn. Reson. Chem. 2023, 61, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Fukaya, H.; Ono, T. DFT-GIAO calculations of 19F NMR chemical shifts for perfluoro compounds. J. Comput. Chem. 2004, 25, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Benassi, E. An inexpensive density functional theory-based protocol to predict accurate 19 F-NMR chemical shifts. J. Comput. Chem. 2022, 43, 170–183. [Google Scholar] [CrossRef] [PubMed]
- Saielli, G.; Bini, R.; Bagno, A. Computational 19F NMR. 1. General features. Theor. Chem. Acc. 2012, 131, 1140. [Google Scholar] [CrossRef]
- Saielli, G.; Bini, R.; Bagno, A. Computational 19F NMR. 2. Organic compounds. RSC Adv. 2014, 4, 41605–41611. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Segmented contracted basis sets optimized for nuclear magnetic shielding. J. Chem. Theory Comput. 2015, 11, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Bartlett, R.J. Many-body perturbation theory and coupled cluster theory for electron correlation in molecules. Annu. Rev. Phys. Chem. 1981, 32, 359–401. [Google Scholar] [CrossRef]
- Harding, M.E.; Lenhart, M.; Auer, A.A.; Gauss, J. Quantitative prediction of gas-phase nuclear magnetic shielding constants. J. Chem. Phys. 2008, 128, 244111. [Google Scholar] [CrossRef] [PubMed]
- Chesnut, D.B.; Byrd, E.F.C. The use of locally dense basis sets in correlated NMR chemical shielding calculations. Chem. Phys. 1996, 213, 153–158. [Google Scholar] [CrossRef]
- Rusakova, I.L.; Ukhanev, S.A.; Rusakov, Y.Y. On the relativistic effects on 19F nuclear magnetic resonance chemical shifts in the presence of iodine atoms. J. Fluor. Chem. 2023, 271, 110188. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Nikurashina, Y.A.; Rusakova, I.L. On the utmost importance of the geometry factor of accuracy in the quantum chemical calculations of P NMR chemical shifts: New efficient pecG-n (n = 1, 2) basis sets for the geometry optimization procedure. J. Chem. Phys. 2024, 160, 084109. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L. Getaway from the Geometry Factor Error in the Molecular Property Calculations: Efficient pecG-n (n = 1, 2) Basis Sets for the Geometry Optimization of Molecules Containing Light p Elements. J. Chem. Theory Comput. 2024, 20, 6661–6673. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.C. The Self-consistent Field for Molecules and Solids. In Quantum Theory of Molecules and Solids Volume 4. (Pure & Applied Physics); Slater, J.C., Ed.; McGraw Hill: New York, NY, USA, 1974; Volume 4. [Google Scholar]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B 1992, 45, 13244. [Google Scholar] [CrossRef] [PubMed]
- Kohn, W.; Sham, L.J. Self-Consistent Equations Including Exchange and Correlation Effects. Phys. Rev. 1965, 140, A1133–A1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P. Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys. Rev. 1986, 33, 8822–8824. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed]
- Burke, K.; Perdew, J.P.; Wang, Y. Electronic Density Functional Theory, 1st ed.; Dobson, F.J., Vignale, G., Das, M.P., Eds.; Springer: Boston, MA, USA, 1998; pp. 81–111. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Boese, A.D.; Handy, N.C. New exchange-correlation density functionals: The role of the kinetic-energy density. J. Chem. Phys. 2002, 116, 9559–9569. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, Q.; Muller, R.P.; Goddard, W.A. An extended hybrid density functional (X3LYP) with improved descriptions of nonbond interactions and thermodynamic properties of molecular systems. J. Chem. Phys. 2005, 122, 14105. [Google Scholar] [CrossRef] [PubMed]
- Hoe, W.-M.; Cohen, A.J.; Handy, N.C. Assessment of a new local exchange functional OPTX. Chem. Phys. Lett. 2001, 341, 319–328. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Hamprecht, F.A.; Cohen, A.J.; Tozer, D.J.; Handy, N.C. Development and assessment of new exchange-correlation functionals. J. Chem. Phys. 1998, 109, 6264–6271. [Google Scholar] [CrossRef]
- Wilson, P.J.; Bradley, T.J.; Tozer, D.J. Hybrid exchange-correlation functional determined from thermochemical data and ab initio potentials. J. Chem. Phys. 2001, 115, 9233–9242. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Exchange functionals with improved long-range behavior and adiabatic connection methods without adjustable parameters: The mPW and mPW1PW models. J. Chem. Phys. 1998, 108, 664–675. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ernzerhof, M.; Burke, K.J. Rationale for mixing exact exchange with density functional approximations. Chem. Phys. 1996, 105, 9982–9985. [Google Scholar] [CrossRef]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof exchange-correlation functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V.J. Toward reliable density functional methods without adjustable parameters: The PBE0 model. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Hybrid Functional Based on a Screened Coulomb Potential. J. Chem. Phys. 2003, 118, 8207–8215. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the Density Functional Ladder: Nonempirical Meta–Generalized Gradient Approximation Designed for Molecules and Solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Perdew, J.P.; Ruzsinszky, A.; Tao, J.; Staroverov, V.N.; Scuseria, G.E.; Csonka, G.I. Prescription for the design and selection of density functional approximations: More constraint satisfaction with fewer fits. J. Chem. Phys. 2005, 123, 062201. [Google Scholar] [CrossRef] [PubMed]
- Helgaker, T.; Gauss, J.; Jørgensen, P.; Olsen, J. The prediction of molecular equilibrium structures by the standard electronic wave functions. J. Chem. Phys. 1997, 106, 6430–6440. [Google Scholar] [CrossRef]
- Faber, R.; Sauer, S.P.A. On the discrepancy between theory and experiment for the F–F spin–spin coupling constant of difluoroethyne. Phys. Chem. Chem. Phys. 2012, 14, 16440–16447. [Google Scholar] [CrossRef] [PubMed]
- Temelso, B.; Valeev, E.F.; Sherrill, C.D. A Comparison of One-Particle Basis Set Completeness, Higher-Order Electron Correlation, Relativistic Effects, and Adiabatic Corrections for Spectroscopic Constants of BH, CH+, and NH+. J. Phys. Chem. A 2004, 108, 3068–3075. [Google Scholar] [CrossRef]
- Heckert, M.; Kállay, M.; Tew, D.P.; Klopper, W.; Gauss, J. Basis-set extrapolation techniques for the accurate calculation of molecular equilibrium geometries using coupled-cluster theory. J. Chem. Phys. 2006, 125, 044108. [Google Scholar] [CrossRef] [PubMed]
- Walker, M.; Harvey, A.J.A.; Sen, A.; Dessent, C.E.H. Performance of M06, M06-2X, and M06-HF Density Functionals for Conformationally Flexible Anionic Clusters: M06 Functionals Perform Better than B3LYP for a Model System with Dispersion and Ionic Hydrogen-Bonding Interactions. J. Phys. Chem. A 2013, 117, 12590–12600. [Google Scholar] [CrossRef] [PubMed]
- Tarakeshwar, P.; Kim, K.S.; Kraka, E.; Cremer, D. Structure and stability of fluorine-substituted benzene-argon complexes: The decisive role of exchange-repulsion and dispersion interactions. J. Chem. Phys. 2001, 115, 6018–6029. [Google Scholar] [CrossRef]
- Perdew, J.P.; Schmidt, K. Jacob’s ladder of density functional approximations for the exchange-correlation energy. AIP Conf. Proc. 2001, 577, 1–20. [Google Scholar] [CrossRef]
- Lopez, X.; Ugalde, J.M.; Sarasola, C.; Cossio, F.P. On the accuracy of density functional theory for ion-molecule clusters. A case study of PL+ clusters of the first and second row hydrides. Can. J. Chem. 1996, 74, 1032–1048. [Google Scholar] [CrossRef]
- Schultz, N.E.; Zhao, Y.; Truhlar, D.G. Density Functionals for Inorganometallic and Organometallic Chemistry. J. Phys. Chem. A 2005, 109, 11127–11143. [Google Scholar] [CrossRef] [PubMed]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1371. [Google Scholar] [CrossRef]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- van Mourik, T.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. VIII. Standard and augmented sextuple zeta correlation consistent basis sets for aluminum through argon. Int. J. Quantum Chem. 2000, 76, 205–221. [Google Scholar] [CrossRef]
- Peterson, K.A.; Woon, D.E.; Dunning, T.H. Benchmark calculations with correlated molecular wave functions. IV. The classical barrier height of the H+H2→H2+H reaction. J. Chem. Phys. 1994, 100, 7410–7415. [Google Scholar] [CrossRef]
- Wilson, A.K.; van Mourik, T.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. VI. Sextuple zeta correlation consistent basis sets for boron through neon. J. Mol. Struct Theochem. 1996, 388, 339–349. [Google Scholar] [CrossRef]
- Dill, J.D.; Pople, J.A. Self-consistent molecular orbital methods. XV. Extended Gaussian-type basis sets for lithium, beryllium, and boron. J. Chem. Phys. 1975, 62, 2921–2923. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-Consistent Molecular-Orbital Methods. IX. An Extended Gaussian- Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Francl, M.M.; Pietro, W.J.; Hehre, W.J.; Binkley, J.S.; Gordon, M.S.; DeFrees, D.J.; Pople, J.A. Self-consistent molecular orbital methods. XXIII. A polarizationtype basis set for second-row elements. J. Chem. Phys. 1982, 77, 3654–3665. [Google Scholar] [CrossRef]
- Gordon, M.S.; Binkley, J.S.; Pople, J.A.; Pietro, W.J.; Hehre, W.J. Self-consistent molecular-orbital methods. 22. Small split-valence basis sets for second-row elements. J. Am. Chem. Soc. 1982, 104, 2797–2803. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Hehre, W.J.; Ditchfield, R.; Pople, J.A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. [Google Scholar] [CrossRef]
- Clark, T.; Chandrasekhar, J.; Spitznagel, G.W.; von Ragué Schleyer, P. Efficient diffuse function-augmented basis sets for anion calculations. III. The 3-21+G basis set for first-row elements, Li–F. J. Comput. Chem. 1983, 4, 294–301. [Google Scholar] [CrossRef]
- Spitznagel, G.W.; Clark, T.; von Ragué Schleyer, P.; Hehre, W.J. An evaluation of the performance of diffuse function-augmented basis sets for second row elements, Na–Cl. J. Comput. Chem. 1987, 8, 1109–1116. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- McLean, A.D.; Chandler, G.S. Contracted Gaussian basis sets for molecular calculations. I. Second row atoms, Z = 11–18. J. Chem. Phys. 1980, 72, 5639–5648. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets: Principles. J. Chem. Phys. 2001, 115, 9113–9125. [Google Scholar] [CrossRef]
- Jensen, F. Polarization consistent basis sets. II. Estimating the Kohn-Sham basis set limit. J. Chem. Phys. 2002, 116, 7372–7379. [Google Scholar] [CrossRef]
- Jensen, F.; Helgaker, T. Polarization consistent basis sets. V. The elements Si-Cl. J. Chem. Phys. 2004, 121, 3463–3470. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Polarization Consistent Basis Sets. 4: The Elements He, Li, Be, B, Ne, Na, Mg, Al, and Ar. J. Phys. Chem. A 2007, 111, 11198–11204. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Polarization consistent basis sets. III. The importance of diffuse functions. J. Chem. Phys. 2002, 117, 9234–9240. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Semenov, V.A.; Rusakova, I.L. Quelling the Geometry Factor Effect in Quantum Chemical Calculations of 13C NMR Chemical Shifts with the Aid of the pecG-n (n = 1, 2) Basis Sets. Int. J. Mol. Sci. 2024, 25, 10588. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L. An efficient method for generating property-energy consistent basis sets. New pecJ-n (n = 1, 2) basis sets for high-quality calculations of indirect nuclear spin–spin coupling constants involving 1H, 13C, 15N, and 19F nuclei. Phys. Chem. Chem. Phys. 2021, 23, 14925–14939. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; Jensen, F., Ed.; John Wiley & Sons Ltd.: Chichester, UK, 2007; Chapter 5; pp. 192–231. [Google Scholar]
- Easton, R.E.; Giesen, D.J.; Welch, A.; Cramer, C.J.; Truhlar, D.G. The MIDI! basis set for quantum mechanical calculations of molecular geometries and partial charges. Theor. Chim. Acta 1996, 93, 281–301. [Google Scholar] [CrossRef]
- Boggs, J.E.; Niu, Z. A b Initio Calculation of Amine Out-of-Plane Angles. J. Comput. Chem. 1985, 6, 46–55. [Google Scholar] [CrossRef]
- Harris, R.K.; Becker, E.D.; Cabral de Menezes, S.M.; Goodfellow, R.; Granger, P. NMR Nomenclature: Nuclear Spin Properties and Conventions for Chemical Shifts (IUPAC Recommendations 2001). Solid State Nucl. Magn. Reson. 2002, 22, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.K.; Becker, E.D.; Cabral De Menezes, S.M.; Granger, P.; Hoffman, R.E.; Zilm, K.W. Further Conventions for NMR Shielding and Chemical Shifts (IUPAC Recommendations 2008). Magn. Reson. Chem. 2008, 46, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L. New efficient pecS-n (n = 1, 2) basis sets for quantum chemical calculations of 31P NMR chemical shifts. Phys. Chem. Chem. Phys. 2023, 25, 18728–18741. [Google Scholar] [CrossRef] [PubMed]
- Paschoal, D.; Fonseca Guerra, C.; de Oliveira, M.A.L.; Ramalho, T.C.; Dos Santos, H.F. Predicting Pt-195 NMR Chemical Shift Using New Relativistic All-Electron Basis Set. J. Comput. Chem. 2016, 37, 2360–2373. [Google Scholar] [CrossRef] [PubMed]
- Franca, C.A.; Pis Diez, R.; Jubert, A.H. On the calculation of 15N chemical shifts using linear regression formulae. A performance comparison of different methods. J. Mol. Struct. Theochem. 2008, 856, 1–8. [Google Scholar] [CrossRef]
- Rusakov, Y.Y.; Rusakova, I.L. New pecS-n (n = 1, 2) basis sets for quantum chemical calculations of the NMR chemical shifts of H, C, N, and O nuclei. J. Chem. Phys. 2022, 156, 244112. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Semenov, V.A.; Rusakova, I.L. On the Efficiency of the Density Functional Theory (DFT)-Based Computational Protocol for 1H and 13C Nuclear Magnetic Resonance (NMR) Chemical Shifts of Natural Products: Studying the Accuracy of the pecS-n (n = 1, 2) Basis Sets. Int. J. Mol. Sci. 2023, 24, 14623. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Nikurashina, Y.A.; Rusakova, I.L. Going beyond the routine consideration of solvent effects on 31P NMR shielding constants: A meticulous basis set study and new aug-pecS-n (n = 1 and 2) basis sets for phosphorus atoms. Phys. Chem. Chem. Phys. 2025, 27, 6730. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L. An unusual way of augmenting one-electron basis sets: New aug-pecS-n (n = 1, 2) basis sets for H, C, N, and O atoms for NMR shielding constant calculations that require extra diffuse functions. J. Chem. Phys. 2025, 162, 164111. [Google Scholar] [CrossRef] [PubMed]
- Jensen, F. Basis set convergence of nuclear magnetic shielding constants calculated by density functional methods. J. Chem. Theory Comput. 2008, 4, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Rzepiela, K.; Kaminský, J.; Buczek, A.; Broda, M.A.; Kupka, T. Electron Correlation or Basis Set Quality: How to Obtain Converged and Accurate NMR Shieldings for the Third-Row Elements? Molecules 2022, 27, 8230. [Google Scholar] [CrossRef] [PubMed]
- Rusakov, Y.Y.; Rusakova, I.L. New pecJ-n (n = 1, 2) Basis Sets for Selenium Atom Purposed for the Calculations of NMR Spin–Spin Coupling Constants Involving Selenium. Int. J. Mol. Sci. 2023, 24, 7841. [Google Scholar] [CrossRef] [PubMed]
- Jameson, C.J.; Jameson, A.K.; Burrell, P.M. 19F nuclear magnetic shielding scale from gas phase studies. J. Chem. Phys. 1980, 73, 6013. [Google Scholar] [CrossRef]
- Jameson, C.J.; Jameson, A.K.; Honarbakhsh, J. 19F nuclear magnetic shielding scale from gas phase studies. II. J. Chem. Phys. 1984, 81, 5266. [Google Scholar] [CrossRef]
- Hindermann, D.K.; Cornwell, C.D. Fluorine and Proton NMR Study of Gaseous Hydrogen Fluoride. J. Chem. Phys. 1968, 48, 2017. [Google Scholar] [CrossRef]
- Ruden, T.A.; Lutnæs, O.B.; Helgaker, T. Vibrational corrections to indirect nuclear spin–spin coupling constants calculated by density-functional theory. J. Chem. Phys. 2003, 118, 9572–9581. [Google Scholar] [CrossRef]
- Ruud, K.; Åstrand, P.-O.; Taylor, P.R. Zero-point vibrational effects on proton shieldings: Functional-group contributions from ab initio calculations. J. Am. Chem. Soc. 2001, 123, 4826–4833. [Google Scholar] [CrossRef] [PubMed]
- Mezhenkova, T.V.; Karpova, V.M.; Zonova, Y.V. Formation of polyfluorofluorenes in the reactions of perfluoro-1,1-diphenylalkanes with antimony pentafluoride. J. Fluor. Chem. 2018, 207, 59–66. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. Theochem. 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, J.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Schlegel, H.B.; Scalmani, G.; Barone, V.; Mennucci, B.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Wolinski, K.; Hinton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Stanton, J.F.; Gauss, J.; Cheng, L.; Harding, M.E.; Matthews, D.A.; Szalay, P.G.; Asthana, A.; Auer, A.A.; Bartlett, R.J.; Benedikt, U.; et al. CFOUR, a Quantum Chemical Program Package. Available online: http://www.cfour.de (accessed on 1 June 2025).
- Aidas, K.; Angeli, C.; Bak, K.L.; Bakken, V.; Bast, R.; Boman, L.; Christiansen, O.; Cimiraglia, R.; Coriani, S.; Dahle, P.; et al. The Dalton quantum chemistry program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 269–284. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | pcS-2/pcS-1 | pcS-2 | pcS-3/pcS-2 | pcS-3/pcS-1 | pcS-3 | pcS-4/pcS-3 | Exp. [2] |
|---|---|---|---|---|---|---|---|
| 1 | −76.61 | −76.45 | −75.07 | −71.25 | −78.78 | −77.30 | −78.49 |
| 2 | −244.45 | −257.03 | −261.96 | −255.75 | −267.90 | −266.77 | −275.26 |
| 4 | −111.88 | −106.26 | −109.26 | −111.10 | −111.64 | −110.44 | −110.72 |
| 9 | −84.18 | −80.88 | −85.22 | −89.67 | −85.92 | −84.72 | −83.46 |
| 11 | −167.82 | −198.27 | −196.65 | −184.62 | −205.13 | −203.47 | −214.40 |
| 12 | −39.72 | −32.53 | −33.96 | −36.33 | −31.90 | −30.60 | −26.05 |
| 13 | −118.75 | −127.88 | −126.68 | −121.62 | −129.54 | −128.25 | −131.53 |
| 15 | −64.17 | −56.72 | −43.97 | −55.45 | −38.52 | −36.25 | −32.72 |
| 21 [1] | −206.70 | −203.69 | −209.93 | −208.68 | −213.71 | −212.41 | −208.55 |
| −102.23 | −101.86 | −103.89 | −104.25 | −106.56 | −104.88 | −101.82 | |
| −134.01 | −129.88 | −135.21 | −135.50 | −137.85 | −136.24 | −129.66 | |
| 22 | −134.26 | −135.13 | −137.97 | −135.32 | −141.88 | −140.43 | −143.48 |
| 23 | −67.995 | −61.080 | −59.208 | −61.315 | −59.216 | −57.307 | −56.41 |
| 24 | −66.39 | −64.45 | −59.28 | −54.74 | −62.67 | −60.63 | −63.34 |
| 25 | −5.73 | 7.19 | 13.36 | 0.68 | 46.28 | 24.77 | 30.98 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ukhanev, S.A.; Rusakov, Y.Y.; Rusakova, I.L. A Quest for Effective 19F NMR Spectra Modeling: What Brings a Good Balance Between Accuracy and Computational Cost in Fluorine Chemical Shift Calculations? Int. J. Mol. Sci. 2025, 26, 6930. https://doi.org/10.3390/ijms26146930
Ukhanev SA, Rusakov YY, Rusakova IL. A Quest for Effective 19F NMR Spectra Modeling: What Brings a Good Balance Between Accuracy and Computational Cost in Fluorine Chemical Shift Calculations? International Journal of Molecular Sciences. 2025; 26(14):6930. https://doi.org/10.3390/ijms26146930
Chicago/Turabian StyleUkhanev, Stepan A., Yuriy Yu. Rusakov, and Irina L. Rusakova. 2025. "A Quest for Effective 19F NMR Spectra Modeling: What Brings a Good Balance Between Accuracy and Computational Cost in Fluorine Chemical Shift Calculations?" International Journal of Molecular Sciences 26, no. 14: 6930. https://doi.org/10.3390/ijms26146930
APA StyleUkhanev, S. A., Rusakov, Y. Y., & Rusakova, I. L. (2025). A Quest for Effective 19F NMR Spectra Modeling: What Brings a Good Balance Between Accuracy and Computational Cost in Fluorine Chemical Shift Calculations? International Journal of Molecular Sciences, 26(14), 6930. https://doi.org/10.3390/ijms26146930