
Identification of Genetic Variants Using Next-Generation Sequencing in Pediatric Myelodysplastic Syndrome: From Disease Biology to Clinical Applications
 , , , , and
, , , , and
Abstract
1. Introduction
2. Results
2.1. Clinical and Cytogenetic Characteristics
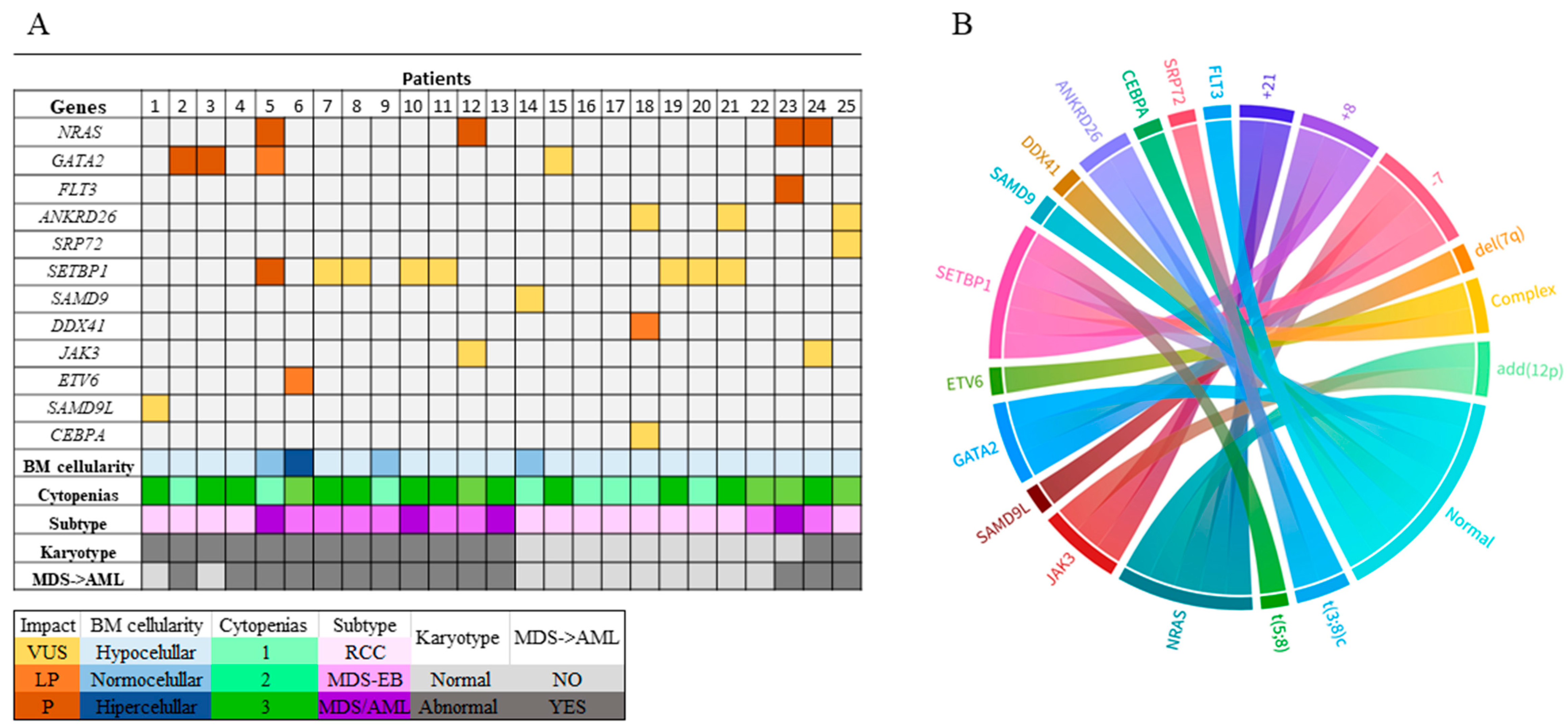
2.2. Genomic Alterations
3. Discussion
4. Material and Methods
4.1. Patients and Controls
4.2. Cytogenetic Analyses
4.3. Next-Generation Sequencing
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chisholm, K.M.; Bohling, S.D. Childhood Myelodysplastic Syndrome. Clin. Lab. Med. 2023, 43, 639–655. [Google Scholar] [CrossRef] [PubMed]
- Arber, D.A.; Orazi, A.; Hasserjian, R.P.; Borowitz, M.J.; Calvo, K.R.; Kvasnicka, H.-M.; Wang, S.A.; Bagg, A.; Barbui, T.; Branford, S.; et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: Integrating morphologic, clinical, and genomic data. Blood 2022, 140, 1200–1228. [Google Scholar] [CrossRef] [PubMed]
- Bernard, E.; Tuechler, H.; Greenberg, P.L.; Hasserjian, R.P.; Ossa, J.E.A.; Nannya, Y.; Devlin, S.M.; Creignou, M.; Pinel, P.; Monnier, L.; et al. Molecular International Prognostic Scoring System for Myelodysplastic Syndromes. NEJM Evid. 2022, 1, EVIDoa2200008. [Google Scholar] [CrossRef] [PubMed]
- Babcock, S.; Calvo, K.R.; Hasserjian, R.P. Pediatric myelodysplastic syndrome. Semin. Diagn. Pathol. 2023, 40, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, J.R.; Ma, J.; Lamprecht, T.; Walsh, M.; Wang, S.; Bryant, V.; Song, G.; Wu, G.; Easton, J.; Kesserwan, C.; et al. The genomic landscape of pediatric myelodysplastic syndromes. Nat Commun. 2017, 8, 1557. [Google Scholar] [CrossRef] [PubMed]
- McCall, D.; Abuasab, T.; Rodriguez-Sevilla, J.J.; Mohamed, S.F.; Patnaik, A.; Devireddy, K.; Arani, N.; Sheikh, I.; Jamshidi, R.; Gibson, A.; et al. Characteristics and outcomes of children, adolescent, and young adult patients with myelodysplastic neoplasms: A single-center retrospective analysis. Leuk Res. 2024, 144, 107563. [Google Scholar] [CrossRef] [PubMed]
- Pastor, V.; Hirabayashi, S.; Karow, A.; Wehrle, J.; Kozyra, E.J.; Nienhold, R.; Ruzaike, G.; Lebrecht, D.; Yoshimi, A.; Niewisch, M.; et al. Mutational landscape in children with myelodysplastic syndromes is distinct from adults: Specific somatic drivers and novel germline variants. Leukemia 2017, 31, 759–762. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Cheng, L.; Peng, Y.; Wang, L.; Zhang, W.; Yin, Y.; Zhang, J.; Wu, X. The role of genetic factors in pediatric myelodysplastic syndromes with different outcomes. BMC Pediatr. 2024, 24, 28. [Google Scholar] [CrossRef] [PubMed]
- Wlodarski, M.W.; Hirabayashi, S.; Pastor, V.; Starý, J.; Hasle, H.; Masetti, R.; Dworzak, M.; Schmugge, M.; Van Den Heuvel-Eibrink, M.; Ussowicz, M.; et al. Prevalence, clinical characteristics, and prognosis of GATA2-related myelodysplastic syndromes in children and adolescents. Blood 2016, 127, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Inam, Z.; Pan, M.; Diab, Y.; Schore, R.; Vatsayan, A.; Cheng, J. Pediatric Myelodysplastic Syndrome. Arch. Pathol. Lab. Med. 2024, 149, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Avagyan, S.; Shimamura, A. Lessons from pediatric MDS: Approaches to germline predisposition to hematologic malignancies. Front. Oncol. 2022, 12, 813149. [Google Scholar] [CrossRef] [PubMed]
- Lovatel, V.L.; Bueno, A.P.; de Kós, E.A.A.; Meyer, L.G.C.; Ferreira, G.M.; Kalonji, M.d.F.; de Mello, F.V.; Milito, C.B.; da Costa, E.S.; Abdelhay, E.; et al. A Novel Constitutional t(3;8)(p26;q21) and ANKRD26 and SRP72 Variants in a Child with Myelodysplastic Neoplasm: Clinical Implications. J. Clin. Med. 2023, 12, 3171. [Google Scholar] [CrossRef] [PubMed]
- Kristan, A.; Debeljak, N. Targeted Next-Generation Sequencing in Rare Diseases. Methods Mol. Biol. 2025, 2866, 45–57. [Google Scholar] [PubMed]
- Alawieh, D.; Cysique-Foinlan, L.; Willekens, C.; Renneville, A. RAS mutations in myeloid malignancies: Revisiting old questions with novel insights and therapeutic perspectives. Blood Cancer J. 2024, 14, 72. [Google Scholar] [CrossRef] [PubMed]
- Pikman, Y.; Stieglitz, E. Targeting the Ras pathway in pediatric hematologic malignancies. Curr. Opin. Pediatr. 2021, 33, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Calvo, K.R.; Hickstein, D.D. The spectrum of GATA2 deficiency syndrome. Blood 2023, 141, 1524–1532. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, S.S.; Erlacher, M.; Wlodarski, M.W. Genetic and Clinical Spectrum of SAMD9 and SAMD9L Syndromes: From Variant Interpretation to Patient Management. Blood 2025, 145, 475–485. [Google Scholar] [CrossRef] [PubMed]
- Cammenga, J. Of gains and losses: SAMD9/SAMD9L and monosomy 7 in myelodysplastic syndrome. Exp. Hematol. 2024, 134, 104217. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Chen, B. Acute myeloid leukemia carrying ETV6 mutations: Biologic and clinical features. Hematology 2018, 9, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Makishima, H.; Saiki, R.; Nannya, Y.; Korotev, S.; Gurnari, C.; Takeda, J.; Momozawa, Y.; Best, S.; Krishnamurthy, P.; Yoshizato, T.; et al. Germ line DDX41 mutations define a unique subtype of myeloid neoplasms. Blood 2023, 141, 534–549. [Google Scholar] [CrossRef] [PubMed]
- Enjeti, A.K.; Walker, N.; Fahey, O.; Johnston, E.; Legge-Wilkinson, H.; Ramsurrun, N.; Sillar, J.; Lincz, L.F.; Ziolkowski, A.; Mossman, D. Certainty in uncertainty: Determining the rate and reasons for reclassification of variants of uncertain significance in haematological malignancies. EJHaem 2024, 5, 957–963. [Google Scholar] [CrossRef]
- Makishima, H. Somatic SETBP1 mutations in myeloid neoplasms. Int. J. Hematol. 2017, 105, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Giudice, V.; Serio, B.; Errichiello, S.; Ferrara, I.; Galdiero, A.; Bertolini, A.; Visconti, R.; De Novellis, D.; Guariglia, R.; Luponio, S.; et al. Subclones with variants of uncertain clinical significance might contribute to ineffective hemopoiesis and leukemia predisposition. Eur. J. Haematol. 2023, 111, 729–741. [Google Scholar] [CrossRef] [PubMed]
- Heredia-Torrejón, M.; Montañez, R.; González-Meneses, A.; Carcavilla, A.; Medina, M.A.; Lechuga-Sancho, A.M. VUS next in rare diseases? Deciphering genetic determinants of biomolecular condensation. Orphanet J. Rare Dis. 2024, 19, 327. [Google Scholar] [CrossRef] [PubMed]
- de Kós, E.A.A.; Lovatel, V.L.; Tavares, R.D.C.B.; Ferreira, G.M.; Gomes, B.; Bueno, A.P.S.; da Costa, E.S.; de Souza Fernandez, T. Outcome of Allogeneic Hematopoietic Stem Cell Transplantation in a Child with Myelodysplastic Neoplasm with Complex Karyotype and ETV6 Variant. Mediterr. J. Hematol. Infect. Dis. 2024, 16, e2024040. [Google Scholar] [CrossRef] [PubMed]
- Niemeyer, C.M.; Strahm, B. Allogeneic Hematopoietic Cell Transplantation in Pediatric MDS Including Refractory Cytopenia of Childhood and in Juvenile Myelomonocytic Leukemia. In The EBMT Handbook: Hematopoietic Cell Transplantation and Cellular Therapies, 8th ed.; Sureda, A., Corbacioglu, S., Greco, R., Kröger, N., Carreras, E., Eds.; Springer: Cham, Switzerland, 2024; Chapter 75. [Google Scholar]
- McGowan-Jordan, J.; Hastings, R.J.; Moore, S. (Eds.) ISCN 2020: An International System for Human Cytogenomic Nomenclature; Karger: Basel, Switzerland, 2020. [Google Scholar]

{kind=link}
| Patient (n.) | Age/ Gender | Karyotype | Subtype | BM Cellularity | Cytopenia | Blast Counts (%) | Evolution from MDS to AML | HSCT | Alive |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 3 months/F | 45,XX,-7[24]/46,XX[2] | RCC | hypocellular | N, T, A | 0% | NO | NO | YES |
| 2 | 10/M | 45,XY,-7[26] | RCC | hypocellular | T | 4.5% | YES | YES | NO |
| 3 | 1/M | 46,XY,del(7)(q22q32)[5]/45,XY,-7[15] | RCC | hypocellular | N, T, A | 2% | NO | YES | NO |
| 4 | 15/M | 45,XY,-7[24] | RCC | hypocellular | N, T, A | 3.8% | YES | NO | NO |
| 5 | 11/F | 45,XX,-7[25]/45,X,del(X)(q23),-7[5] | MDS/AML | normocellular | N | 20–30% | YES | NO | NO |
| 6 * | 3/M | 49,XY,del(3)(q21),del(6)(q21),+der(6)del(6)(q21) | MDS-EB | hypercellular | N, T | 16% | YES | YES | NO |
| +8,+der(12)del(12)(p11)[21] | |||||||||
| 7 | 6/F | 58,XX,+X,+3,+5,+6,+8,+10,+11,+12,+13,+18,+20,+21[5]/ | MDS-EB | hypocellular | N, T, A | 16% | YES | NO | NO |
| 58,XX,idem,dup(1)(q21q31)[14]/46,XX[21] | |||||||||
| 8 | 5/M | 52,XY,+6,+8,+14,+16,+19,+22[3]/46,XY[19] | MDS-EB | hypocellular | N, T, A | 5.8% | YES | NO | NO |
| 9 | 12/F | 46,XX,der(2)t(2;15)(q37;q21)[25] | MDS-EB | normocellular | T | 16% | YES | NO | NO |
| 10 | 5/M | 46,XY,t(5;8)(q32;q22)[23]/46,XY[8] | MDS/AML | hypocellular | N, T, A | 28% | YES | NO | NO |
| 11 | 16/M | 47,XY,+8[30]/46,XY[25] | MDS-EB | hypocellular | N, T, A | 10% | YES | NO | NO |
| 12 | 13/M | 46,XY,add(12)(p12)[19]/47,XY,+8,add(12)(p12)[11] | MDS-EB | hypocellular | N, T | 10% | YES | YES | YES |
| 13 | 1/M | 47,XY,+8,del(11)(q23)[9]/46,XY[13] | MDS/AML | hypocellular | N, T, A | 22% | YES | NO | NO |
| 14 | 17/M | 46,XY[30] | RCC | normocellular | T | 0.3% | NO | NO | YES |
| 15 | 2/F | 46,XX[30] | RCC | hypocellular | N, T, A | 0.5% | NO | NO | YES |
| 16 | 10/F | 46,XX[21] | RCC | hypocellular | T | 0.6% | NO | NO | YES |
| 17 | 10/F | 46,XX[20] | RCC | hypocellular | N | 0% | NO | NO | YES |
| 18 | 5/F | 46,XX[30] | RCC | hypocellular | N | 1% | NO | YES | YES |
| 19 | 1/M | 46,XY[25] | RCC | hypocellular | N, T, A | 2% | NO | NO | YES |
| 20 | 16/M | 46,XY[30] | RCC | hypocellular | T | 1.5% | NO | NO | YES |
| 21 | 7/M | 46,XY[35] | RCC | hypocellular | N, T, A | 1% | NO | NO | YES |
| 22 | 1/M | 46,XY[20] | MDS-EB | hypocellular | N, T | 18% | NO | NO | NO |
| 23 | 16/M | 46,XY[24] | MDS/AML | hypocellular | N, T | 20% | YES | NO | NO |
| 24 | 4/F | 47,XX,+21[5]/47,XX,+8[3]/46,XX[20] | MDS-EB | hypocellular | N, T, A | 15% | YES | NO | NO |
| 25 * | 4/F | 46,XX,t(3;8)(q29;q11)c[26] | RCC | hypocellular | N, T | 5.4% | YES | NO | YES |
| Patient (n.) | Gene | Locus | dbsnp | Impact | Consequence | Genotype | VAF (%) | Protein | Coding |
|---|---|---|---|---|---|---|---|---|---|
| 1 | SAMD9L | chr7: 92761365 | rs1199597457 | VUS | M | A/G | 10.76 | p.Phe1307Ser | c.3920T>C |
| SAMD9L | chr7: 92762622 | - | VUS | M | G/A | 47.80 | p.Ser888Phe | c.2663C>T | |
| 2 | GATA2 | chr3: 128200723 | rs387906634 | p | M | C/T | 45.73 | p.Arg361His | c.1082G>A |
| 3 | GATA2 | chr3: 128205858 | rs1291114301 | P | FI | TC/TC | 100.00 | p.Glu6GlyfsTer179 | c.16_17insG |
| 5 | NRAS | chr1: 115258747 | rs121913237 | P | M | C/T | 50.52 | p.Gly12Asp, p.? | c.35G>A, c.*2043G>A |
| GATA2 | chr3: 128200776 | rs1313081073 | LP | FD | TCTGGCGGCCGA/T | 64.71 | p.Ser340LysfsTer40 | c.1018_1028del TCGGCCGCCAG | |
| SETBP1 | chr18: 42531907 | rs267607042 | P | M | G/A | 48.95 | p.Asp868Asn | c.2602G>A | |
| 6 * | ETV6 | chr12: 12043980 | - | LP | SP | A/G | 3.32 | p.Ter453Trp | c.1359A>G |
| 7 | SETBP1 | chr18: 42532766 | VUS | M | A/C | 4.42 | p.His1154Pro | c.3461A>C | |
| 8 | SETBP1 | chr18: 42532766 | - | VUS | M | A/C | 3.37 | p.His1154Pro | c.3461A>C |
| 10 | SETBP1 | chr18: 42532766 | - | VUS | M | A/C | 2.86 | p.His1154Pro | c.3461A>C |
| 11 | SETBP1 | chr18: 42532766 | VUS | M | A/C | 6.14 | p.His1154Pro | c.3461A>C | |
| 12 | NRAS | chr1: 115256530 | rs1057519834, rs121913254 | P | M | G/T | 47.29 | p.Gln61Lys | c.181C>A |
| JAK3 | chr19: 17953950 | rs55778349 | VUS | M | G/C | 49.75 | p.Pro151Arg | c.452C>G | |
| 14 | SAMD9 | chr7: 92733609 | rs375515095 | VUS | FD | ATT/A | 52.69 | p.Glu600AspfsTer12 | c.1800_1801 delAA |
| 15 | GATA2 | chr3: 128205877 | - | VUS | FI | CCGG/C | 4.05 | c.-3CCGG>G | |
| 18 | DDX41 | chr5: 176943921 | rs1554111842 | LP | FI | T/TACCT | 48.16 | p.Arg10ValfsTer20 | c.25_26 insAGGT |
| ANKRD26 | chr10: 27337805 | rs1216270855, rs561705414 | VUS | FD | CCAT/C | 2.67 | p.Asp579del | c.1736_173 delATG | |
| CEBPA | chr19: 33793023 | rs1289919155 | VUS | FD | CGCC/C | 2.94 | p.Gly104del | c.295_297delGGC | |
| 19 | SETBP1 | chr18: 42532766 | - | VUS | M | A/C | 3.62 | p.His1154Pro | c.3461A>C |
| 20 | SETBP1 | chr18: 42532766 | - | VUS | M | A/C | 3.21 | p.His1154Pro | c.3461A>C |
| 21 | ANKRD26 | chr10: 27389160 | rs779596861 | VUS | M | C/G | 47.18 | p.Glu32Asp | c.96G>C |
| SETBP1 | chr18: 42532766 | - | VUS | M | A/C | 4.51 | p.His1154Pro | c.3461A>C | |
| 23 | NRAS | chr1: 115258747 | rs121913237 | p | M | C/A | 15.35 | p.Gly12Val, p.? | c.35G>T, c.*2043G>T |
| FLT3 | chr13: 28609669 | rs1568403015 | P | FI | A/AG | 8.79 | p.Gly521TrpfsTer5 | c.1559_1560insC | |
| 24 | NRAS | chr1: 115258744 | rs121434596 | P | M | C/T | 5.10 | p.Gly13Asp, p.? | c.38G>A, c.*2046G>A |
| JAK3 | chr19: 17945970 | rs1568403015 | VUS | M | G/A | 38.60 | p.Arg657Trp | c.1969C>T | |
| 25 * | SRP72 | chr4: 57361553 | rs34419325 | VUS | S | A/G | 50.61 | p.Lys557= | c.1671A>G |
| ANKRD26 | chr10: 27353007 | rs12359281 | VUS | M | ATC/GTC | 50.40 | p.Ile425Val | c.1273A>G |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lovatel, V.L.; Ferreira, G.M.; da Silva, B.F.; de Souza Torres, R.; de Cássia Barbosa da Silva Tavares, R.; Bueno, A.P.S.; Abdelhay, E.; de Souza Fernandez, T. Identification of Genetic Variants Using Next-Generation Sequencing in Pediatric Myelodysplastic Syndrome: From Disease Biology to Clinical Applications. Int. J. Mol. Sci. 2025, 26, 6907. https://doi.org/10.3390/ijms26146907
Lovatel VL, Ferreira GM, da Silva BF, de Souza Torres R, de Cássia Barbosa da Silva Tavares R, Bueno APS, Abdelhay E, de Souza Fernandez T. Identification of Genetic Variants Using Next-Generation Sequencing in Pediatric Myelodysplastic Syndrome: From Disease Biology to Clinical Applications. International Journal of Molecular Sciences. 2025; 26(14):6907. https://doi.org/10.3390/ijms26146907
Chicago/Turabian StyleLovatel, Viviane Lamim, Gerson Moura Ferreira, Beatriz Ferreira da Silva, Rayane de Souza Torres, Rita de Cássia Barbosa da Silva Tavares, Ana Paula Silva Bueno, Eliana Abdelhay, and Teresa de Souza Fernandez. 2025. "Identification of Genetic Variants Using Next-Generation Sequencing in Pediatric Myelodysplastic Syndrome: From Disease Biology to Clinical Applications" International Journal of Molecular Sciences 26, no. 14: 6907. https://doi.org/10.3390/ijms26146907
APA StyleLovatel, V. L., Ferreira, G. M., da Silva, B. F., de Souza Torres, R., de Cássia Barbosa da Silva Tavares, R., Bueno, A. P. S., Abdelhay, E., & de Souza Fernandez, T. (2025). Identification of Genetic Variants Using Next-Generation Sequencing in Pediatric Myelodysplastic Syndrome: From Disease Biology to Clinical Applications. International Journal of Molecular Sciences, 26(14), 6907. https://doi.org/10.3390/ijms26146907