
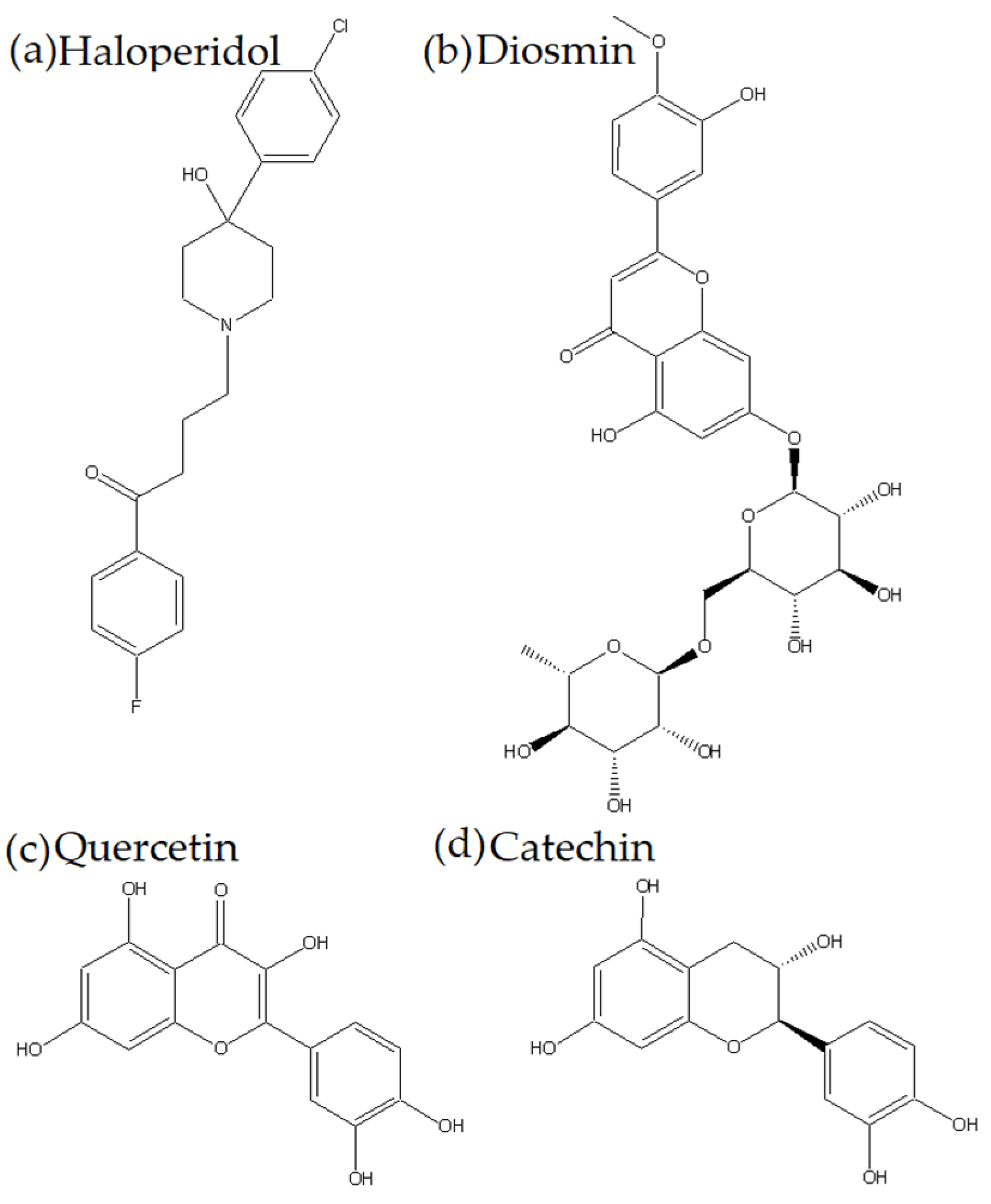
Quercetin, Catechin, and Diosmin as Modulators of Haloperidol–HSA Interactions: A Biophysical and Computational Study

 ,
,  , , ,
, , ,  and
and 
Abstract
1. Introduction
2. Results and Discussion
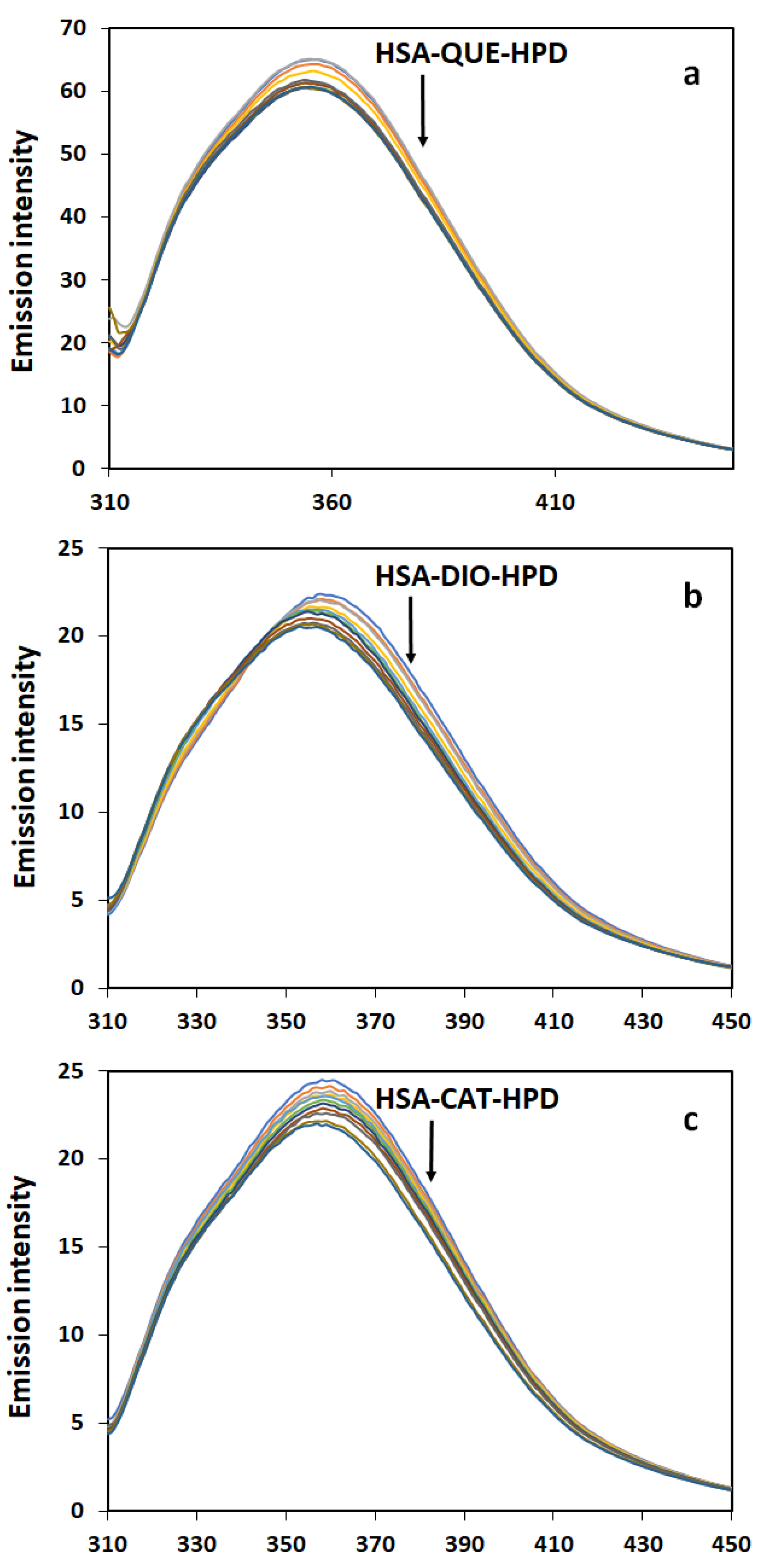
2.1. Fluorescence Quenching Measurements
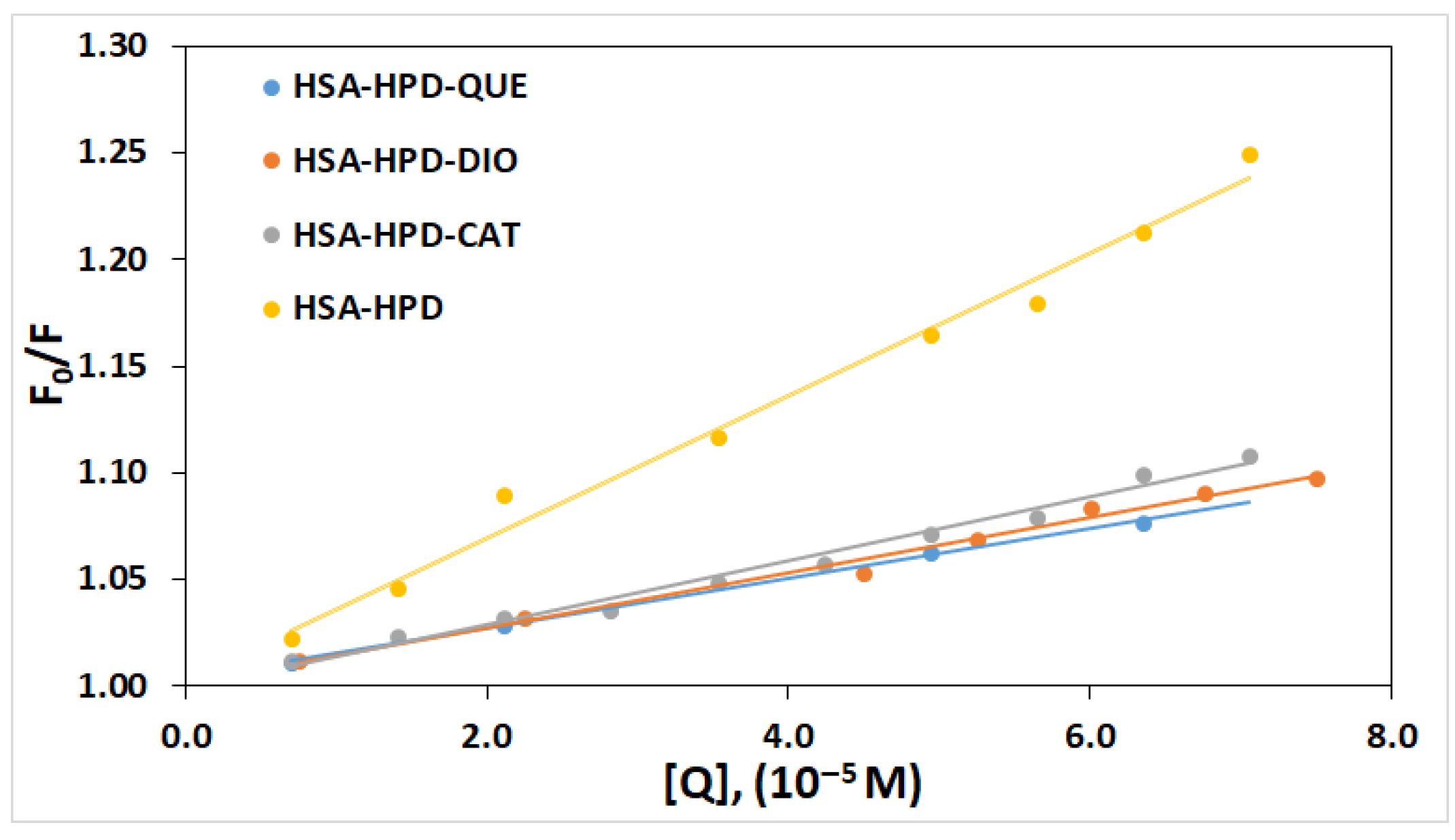
2.2. Quenching Mechanism
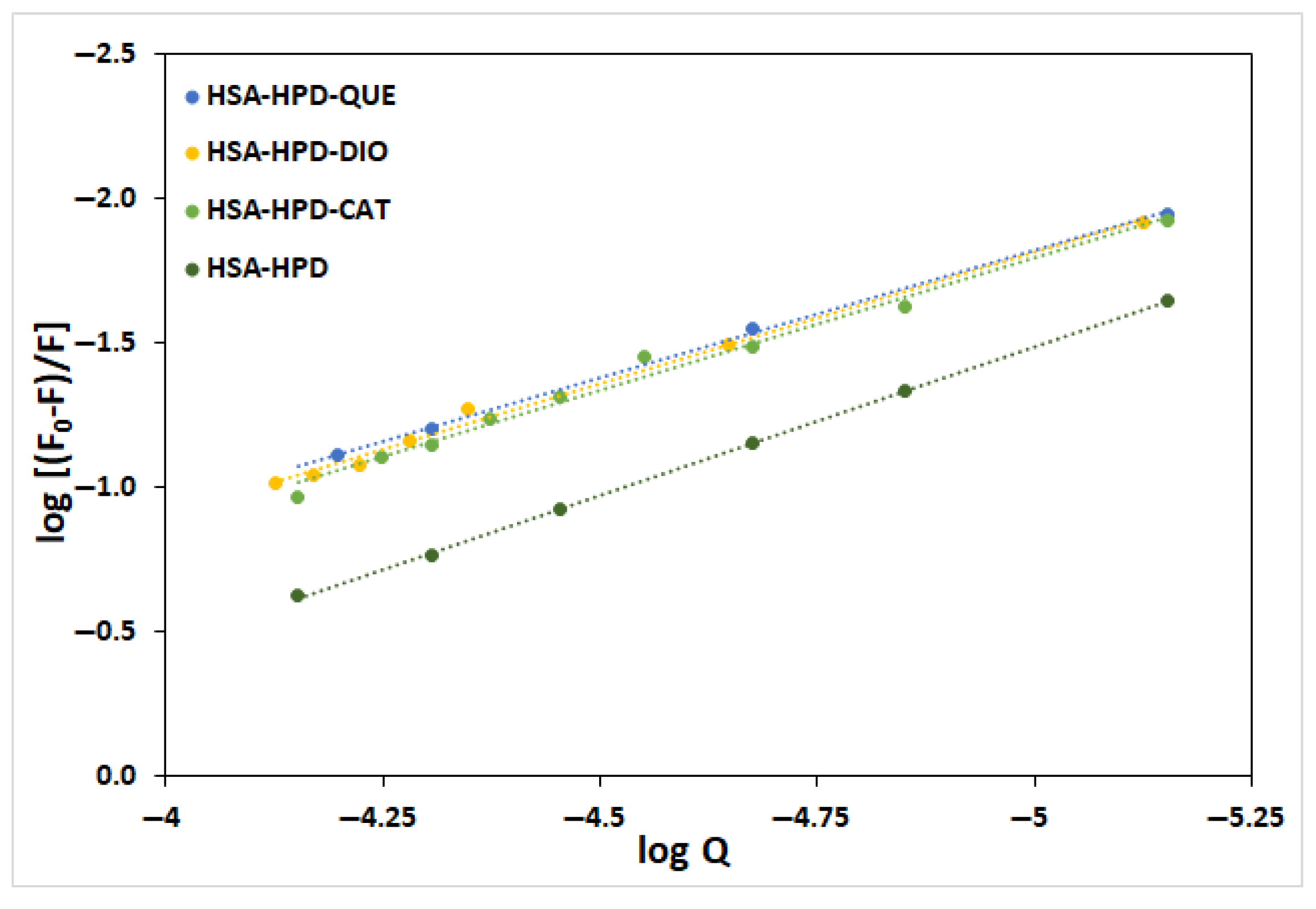
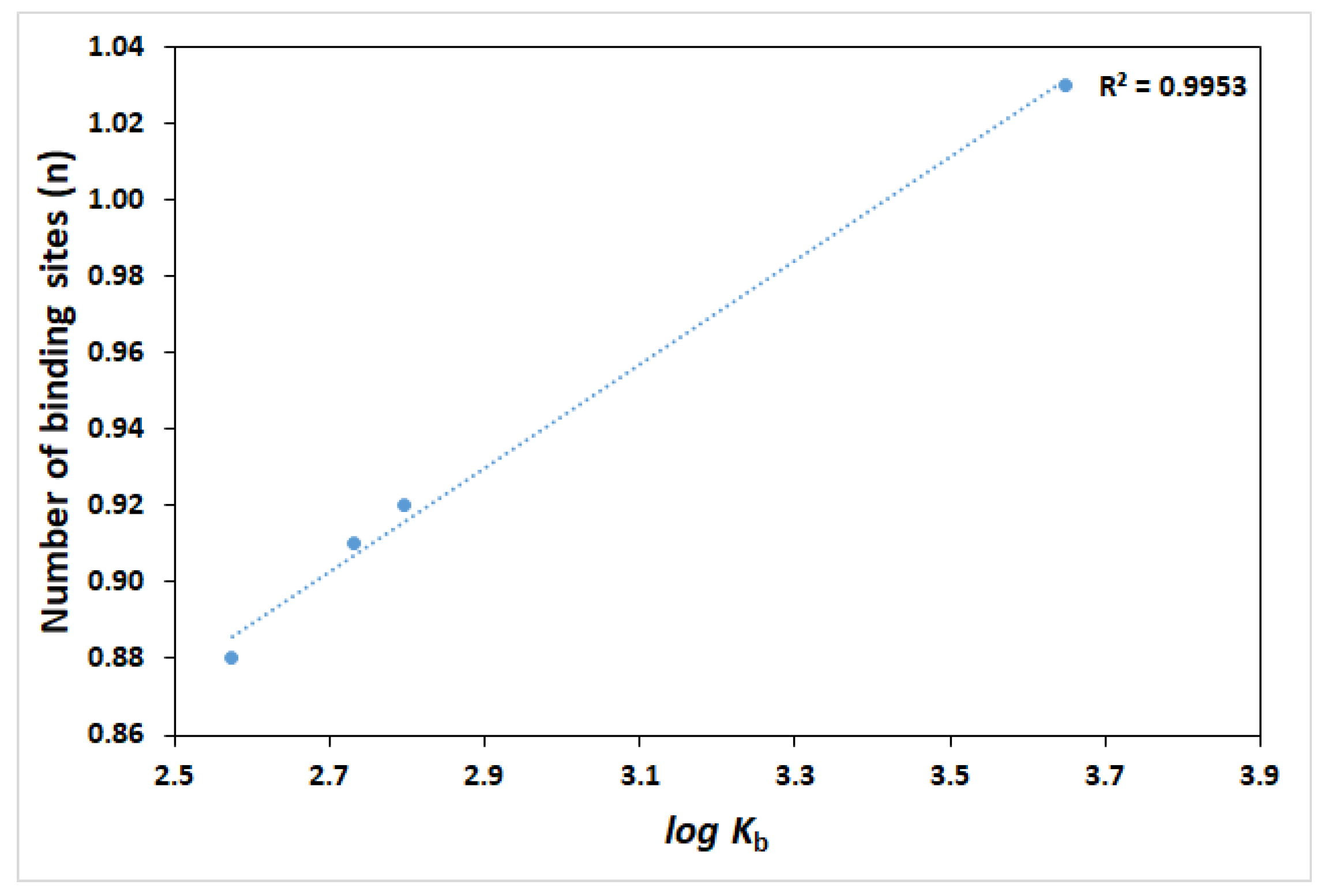
2.3. Binding Constant and Number of Binding Sites
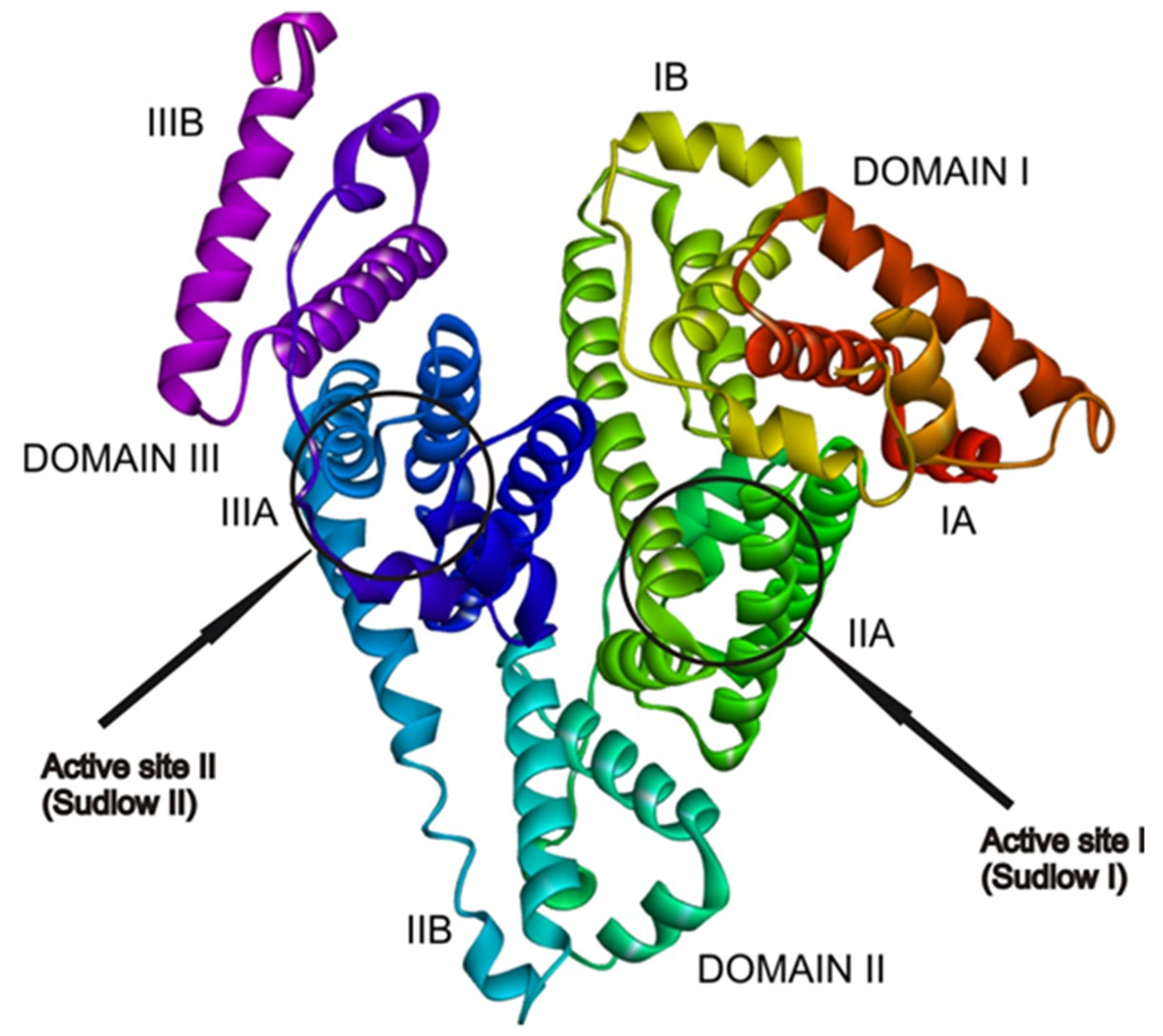
2.4. Docking Analysis
2.5. Molecular Dynamics
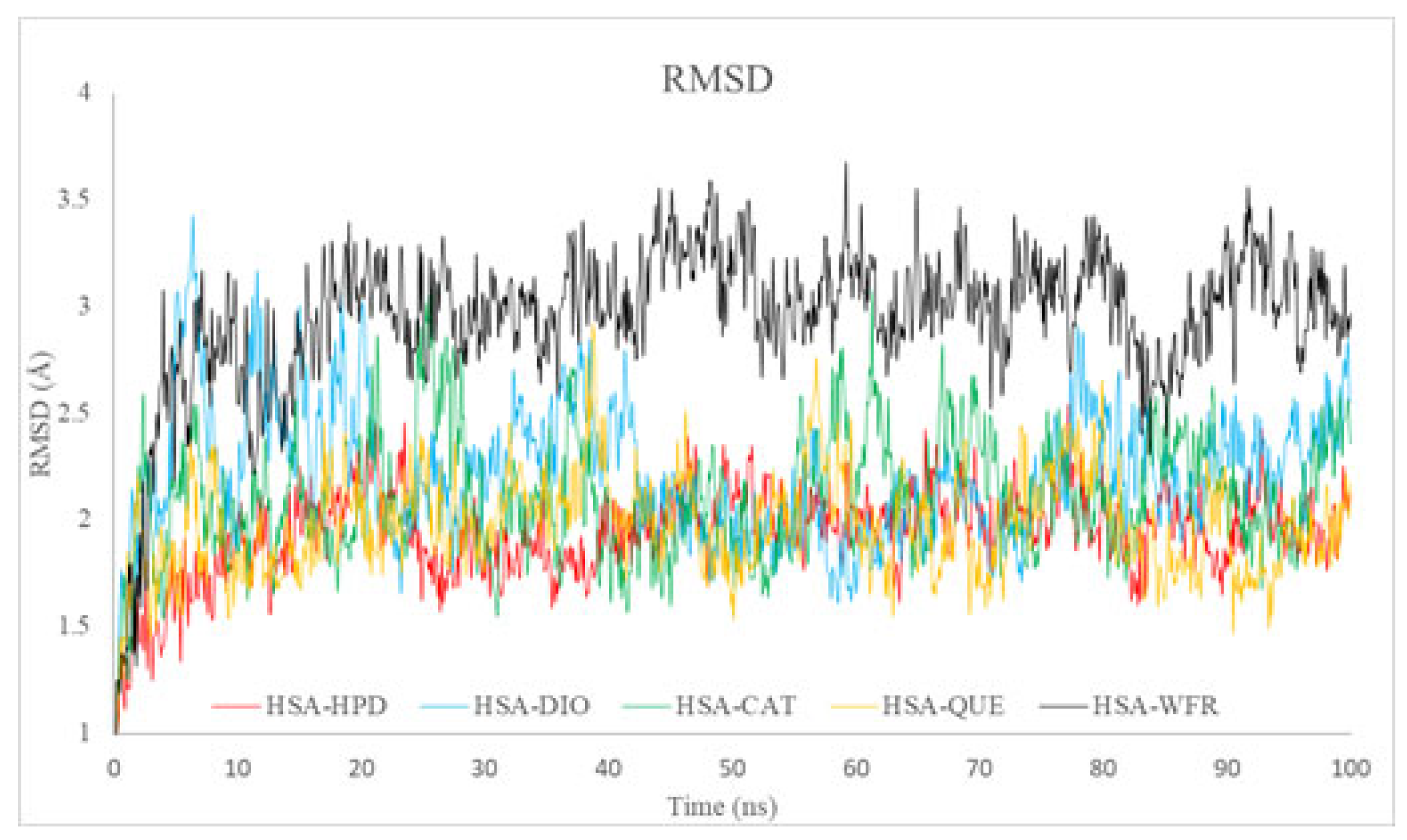
2.5.1. Root Mean Square Deviation Analysis
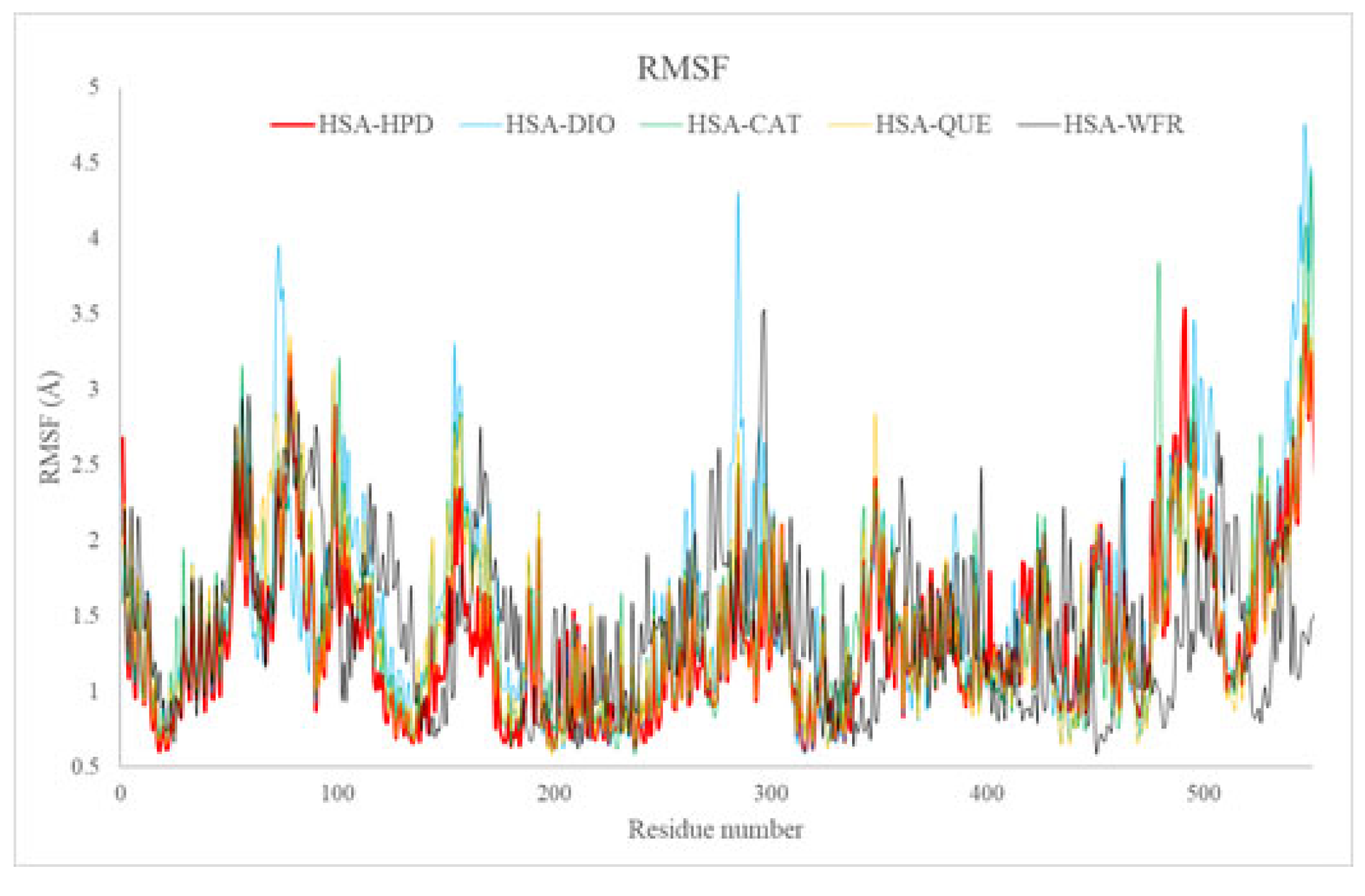
2.5.2. Root Mean Square Fluctuation Analysis
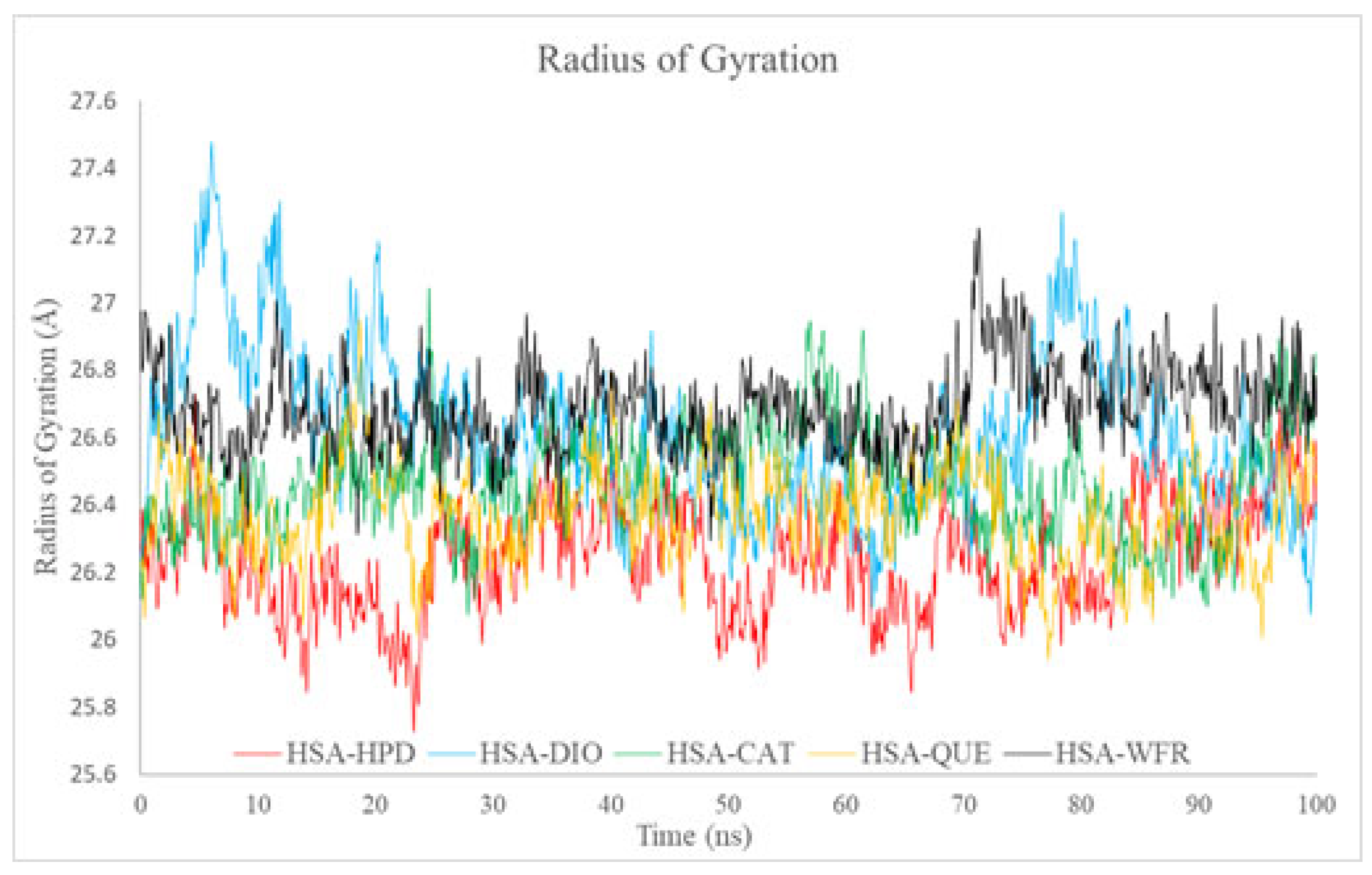
2.5.3. Radius of Gyration Analysis
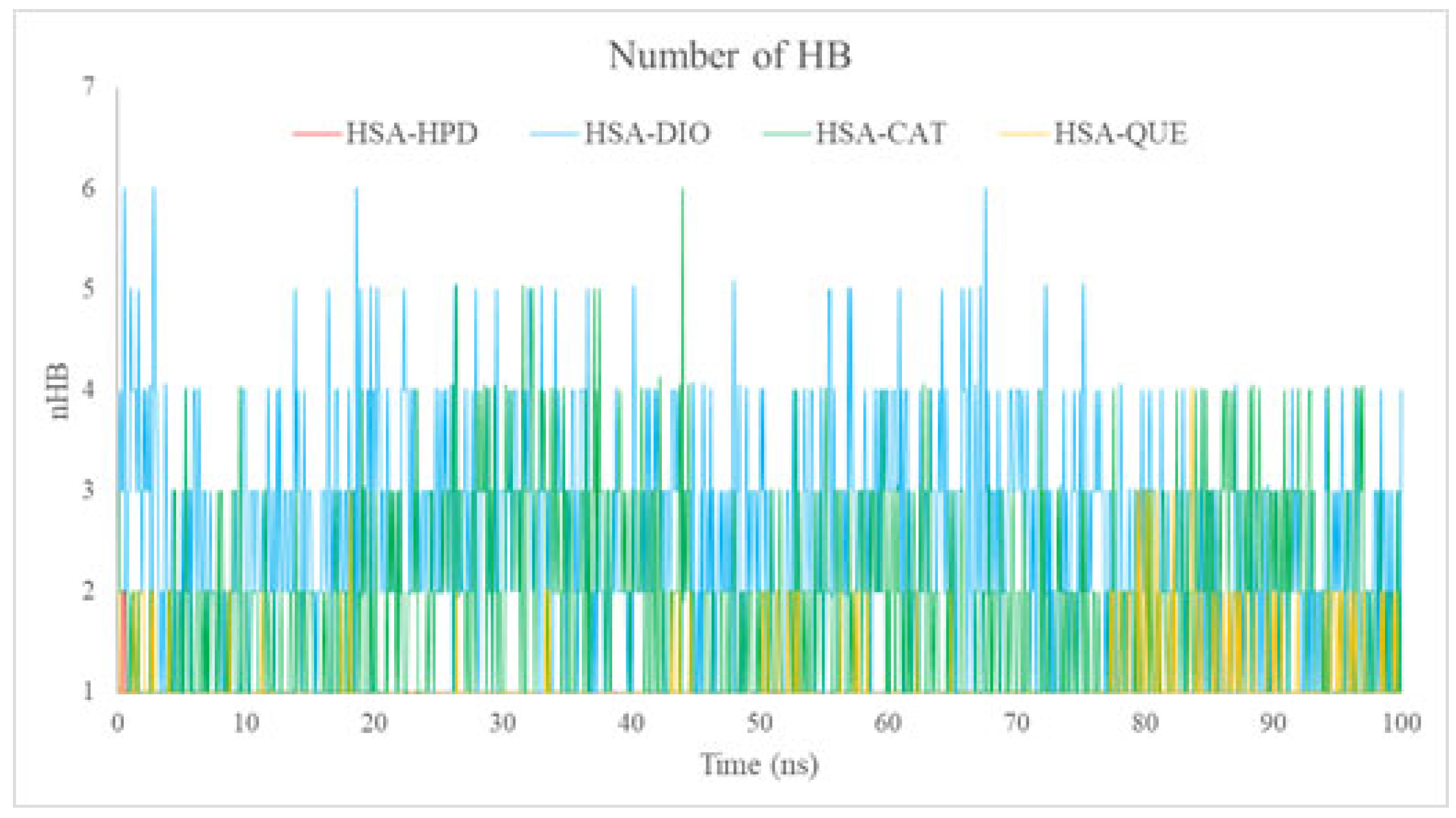
2.5.4. Analysis of the Number of Hydrogen Bonds (nHBs)
2.5.5. Competitive Binding and the Role of Hydrophobic Interactions
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Equipment and Spectral Measurements
3.3. Preparation of Solutions
3.4. Fluorescence Spectra of Haloperidol Binding with HSA in the Absence or Presence of Flavonoids
3.5. Molecular Docking
3.6. Molecular Dynamics Simulations
3.7. Data Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HSA | Human serum albumin |
| HPD | Haloperidol |
| QUE | Quercetin |
| DIO | Diosmin |
| CAT | Catechin |
| MD | Molecular dynamics |
| RMSD | Root Mean Square Deviation |
| RMSF | Root Mean Square Fluctuation |
References
- Maciążek-Jurczyk, M.; Szkudlarek, A.; Chudzik, M.; Pożycka, J.; Sułkowska, A. Alteration of human serum albumin binding properties induced by modifications: A review. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 188, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Rimac, H.; Debeljak, Ž.; Bojić, M.; Miller, L. Displacement of Drugs from Human Serum Albumin: From Molecular Interactions to Clinical Significance. Curr. Med. Chem. 2017, 24, 1930–1947. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Ray, A.; Gopi, A.; Hunter, R. Haloperidol versus risperidone for schizophrenia. Cochrane Database Syst. Rev. 2017, 2017, CD012728. [Google Scholar] [CrossRef]
- Zayed, Y.; Barbarawi, M.; Kheiri, B.; Banifadel, M.; Haykal, T.; Chahine, A.; Rashdan, L.; Aburahma, A.; Bachuwa, G.; Seedahmed, E. Haloperidol for the management of delirium in adult intensive care unit patients: A systematic review and meta-analysis of randomized controlled trials. J. Crit. Care 2019, 50, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Beach, S.R.; Gross, A.F.; Hartney, K.E.; Taylor, J.B.; Rundell, J.R. Intravenous haloperidol: A systematic review of side effects and recommendations for clinical use. Gen. Hosp. Psychiatry 2020, 67, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Williamson, G.; Kay, C.D.; Crozier, A. The Bioavailability, Transport, and Bioactivity of Dietary Flavonoids: A Review from a Historical Perspective. Compr. Rev. Food Sci. Food Saf. 2018, 17, 1054–1112. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Singh, J.P.; Kaur, A.; Singh, N. Phenolic compounds as beneficial phytochemicals in pomegranate (Punica granatum L.) peel: A review. Food Chem. 2018, 261, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, J.M.; Munekata, P.E.S.; Sant’Ana, A.S.; Carvalho, R.B.; Barba, F.J.; Toldra, F.; Mora, L.; Trindade, M.A. Main character-istics of peanut skin and its role for the preservation of meat products. Trends Food Sci. Technol. 2018, 77, 1–10. [Google Scholar] [CrossRef]
- Wen, K.; Fang, X.; Yang, J.; Yao, Y.; Nandakumar, K.S.; Salem, M.L.; Cheng, K. Recent Research on Flavonoids and their Biomedical Applications. Curr. Med. Chem. 2021, 28, 1042–1066. [Google Scholar] [CrossRef] [PubMed]
- Rana, J.N.; Mumtaz, S. Prunin: An Emerging Anticancer Flavonoid. Int. J. Mol. Sci. 2025, 26, 2678. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Surh, Y.-J.; Do, S.-G.; Shin, E.; Shim, K.-S.; Lee, C.-K.; Na, H.-K. Baicalein Inhibits Dextran Sulfate Sodium-induced Mouse Colitis. J. Cancer Prev. 2019, 24, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Zhou, K.; Cheng, R.; Liu, B.; Wang, L.; Xie, H.; Zhang, C. Eupatilin ameliorates dextran sulphate sodium-induced colitis in mice partly through promoting AMPK activation. Phytomedicine 2018, 46, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Liu, P.; Zhong, J.; Hu, Y.; Zhuang, S.; Fan, K.; Liu, Z. Quercetin Attenuates Adhesion Molecule Expression in Intestinal Microvascular Endothelial Cells by Modulating Multiple Pathways. Dig. Dis. Sci. 2018, 63, 3297–3304. [Google Scholar] [CrossRef] [PubMed]
- Lewinska, A.; Adamczyk-Grochala, J.; Kwasniewicz, E.; Deregowska, A.; Wnuk, M. Diosmin-induced senescence, apoptosis and autophagy in breast cancer cells of different p53 status and ERK activity. Toxicol. Lett. 2017, 265, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Gong, W.-Y.; Wu, J.-F.; Liu, B.-J.; Zhang, H.-Y.; Cao, Y.-X.; Sun, J.; Lv, Y.-B.; Wu, X.; Dong, J.-C. Flavonoid components in Scutellaria baicalensis inhibit nicotine-induced proliferation, metastasis and lung cancer-associated inflammation in vitro. Int. J. Oncol. 2014, 44, 1561–1570. [Google Scholar] [CrossRef] [PubMed]
- Rauf, A.; Imran, M.; Khan, I.; Ur-Rehman, M.; Gilani, S.A.; Mehmood, Z.; Mubarak, M.S. Anticancer potential of quercetin: A comprehensive review. Phytother. Res. 2018, 32, 2109–2130. [Google Scholar] [CrossRef] [PubMed]
- Coșarcă, S.; Tanase, C.; Muntean, D.L. Therapeutic Aspects of Catechin and Its Derivatives—An Update. Acta Biol. Marisiensis 2019, 2, 21–29. [Google Scholar] [CrossRef]
- Serra, R.; Ielapi, N.; Bitonti, A.; Candido, S.; Fregola, S.; Gallo, A.; Loria, A.; Muraca, L.; Raimondo, L.; Velcean, L.; et al. Efficacy of a Low-Dose Diosmin Therapy on Improving Symptoms and Quality of Life in Patients with Chronic Venous Disease: Randomized, Double-Blind, Placebo-Controlled Trial. Nutrients 2021, 13, 999. [Google Scholar] [CrossRef] [PubMed]
- Gosslau, A.; Ho, C.-T.; Li, S. The role of rutin and diosmin, two citrus polyhydroxyflavones in disease prevention and treatment. J. Food Bioact. 2019, 5, 43–56. [Google Scholar] [CrossRef]
- Zheng, Y.; Zhang, R.; Shi, W.; Li, L.; Liu, H.; Chen, Z.; Wu, L. Metabolism and pharmacological activities of the natural health-benefiting compound diosmin. Food Funct. 2020, 11, 8472–8492. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.; Zhang, G.; Zhang, J.; Ren, L. Interaction of Flavonoids with Serum Albumin: A Review. Curr. Protein Pept. Sci. 2021, 22, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Sudlow, G.; Birkett, D.J.; Wade, D.N. The characterization of two specific drug binding sites on human serum albumin. Mol. Pharmacol. 1975, 11, 824–832. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Wang, L.; Wang, Y.; Zhou, J.; Ding, J.; Li, W.; Xin, Y.; Fan, S.; Wang, Z.; Wang, Y. Biointeractions of Herbicide Atrazine with Human Serum Albumin: UV-Vis, Fluorescence and Circular Dichroism Approaches. Int. J. Environ. Res. Public Health 2018, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Liu, M.; Guo, Q.; Liu, Y.; Zhao, Y.; Wu, Y.; Sun, B.; Wang, Q.; Liu, J.; Han, J. Investigation of binary and ternary systems of human serum albumin with oxyresveratrol/piceatannol and/or mitoxantrone by multipectroscopy, molecular docking and cytotoxicity evaluation. J. Mol. Liq. 2020, 311, 113364. [Google Scholar] [CrossRef]
- Berić, J.D.; Stojanović, S.D.; Mrkalić, E.M.; Matović, Z.D.; Milovanovic, D.R.; Sovrlic, M.M.; Jelić, R.M. Interaction of haloperidol with human serum albumin and effect of metal ions on the binding. Mon. Für Chem.-Chem. Mon. 2018, 149, 2359–2368. [Google Scholar] [CrossRef]
- Vaneková, Z.; Hubčík, L.; Toca-Herrera, J.L.; Furtműller, P.G.; Valentová, J.; Mučaji, P.; Nagy, M. Study of Interactions between Amlodipine and Quercetin on Human Serum Albumin: Spectroscopic and Modeling Approaches. Molecules 2019, 24, 487. [Google Scholar] [CrossRef] [PubMed]
- López-Yerena, A.; Perez, M.; Vallverdú-Queralt, A.; Escribano-Ferrer, E. Insights into the Binding of Dietary Phenolic Compounds to Human Serum Albumin and Food-Drug Interactions. Pharmaceutics 2020, 12, 1123. [Google Scholar] [CrossRef] [PubMed]
- Barreca, D.; Laganà, G.; Bruno, G.; Magazù, S.; Bellocco, E. Diosmin binding to human serum albumin and its preventive action against degradation due to oxidative injuries. Biochimie 2013, 95, 2042–2049. [Google Scholar] [CrossRef] [PubMed]
- Zhivkova, Z. Studies on drug—Human serum albumin binding: The current state of the matter. Curr. Pharm. Des. 2015, 21, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Lakshmi, P.T.; Krishna, R.; Sakthivel, N. Molecular interaction between human serum albumin (HSA) and phloroglucinol derivative that shows selective anti-proliferative potential. J. Lumin 2017, 192, 990–998. [Google Scholar] [CrossRef]
- Mrkalić, E.; Jelić, R.; Stojanović, S.; Sovrlić, M. Interaction between olanzapine and human serum albumin and effect of metal ions, caffeine and flavonoids on the binding: A spectroscopic study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2021, 249, 119295. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Gan, N.; Sun, Q.; Wu, D.; Gan, R.; Zhang, M.; Tang, P.; Li, H. Study on the interaction of ertugliflozin with human serum albumin in vitro by multispectroscopic methods, molecular docking, and molecular dynamics simulation. Spectrochim. Acta Part A 2019, 219, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shi, S.; Chen, X.; Zhang, W.; Huang, K.; Peng, M. Investigation on the Interaction between Ilaprazole and Bovine Serum Albumin without or with Different C-Ring Flavonoids from the Viewpoint of Food–Drug Interference. J. Agric. Food Chem. 2011, 59, 8499–8506. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Trynda-Lemiesz, L. Paclitaxel–HSA interaction. Binding sites on HSA molecule. Bioorganic Med. Chem. 2004, 12, 3269–3275. [Google Scholar] [CrossRef] [PubMed]
- Kragh-Hansen, U.; Chuang, V.T.G.; Otagiri, M. Practical aspects of the ligand-binding and enzymatic properties of human serum albumin. Biol. Pharm. Bull. 2002, 25, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A new mixing of Hartree–Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Fox, Gaussian 09. 2009. Available online: http://gaussian.com/ (accessed on 1 July 2025.).
- Petitpas, I.; Petersen, C.E.; Ha, C.-E.; Bhattacharya, A.A.; Zunszain, P.A.; Ghuman, J.; Bhagavan, N.V.; Curry, S. Structural basis of albumin–thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc. Natl. Acad. Sci. USA 2003, 100, 6440–6445. [Google Scholar] [CrossRef] [PubMed]
- BIOVIA, Dassault Systèmes, Discovery Studio Modeling Environment. 2017. Available online: https://www.3ds.com/products/biovia/discovery-studio (accessed on 12 July 2025).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.Y.; Berryman, J.T.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E., III; Cisneros, G.A.; Cruzeiro, V.W.D.; et al. Amber 2022; University of California: San Francisco, CA, USA, 2022; Available online: https://ambermd.org/doc12/Amber22.pdf (accessed on 1 October 2021).
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy, 3rd ed.; Springer Science & Business Media: New York, NY, USA, 2006. [Google Scholar] [CrossRef]
- Sharma, A.; Schulman, S.G. Introduction to Fluorescence Spectroscopy; John Willey & Sons: New York, NY, USA, 1999; Available online: https://www.scirp.org/reference/referencespapers?referenceid=1511350 (accessed on 1 January 2021).
- Marjanović, J.S.; Milanović, Ž.; Divac, V.M.; Kosanić, M.M.; Petković, M.R.; Kostić, M.D. Molecular modeling studies, in vitro antioxidant and antimicrobial assay and BSA affinity of novel benzyl-amine derived scaffolds as CYP51B inhibitors. Mol. Divers. 2024, 28. [Google Scholar] [CrossRef] [PubMed]
- Dragojević, J.S.; Milanović, Ž.; Matić, J.; Divac, V.M.; Kosanić, M.; Milivojević, D.; Petković, M.; Singh, F.V.; Kostić, M.D. Dual activity of newly synthesized Zn(II) and Cu(II) schiff base complexes as a potential solution for global challenges in the fight against priority microorganisms. J. Mol. Struct. 2025, 1335, 142052. [Google Scholar] [CrossRef]
- Varshney, A.; Sen, P.; Ahmad, E.; Rehan, M.; Subbarao, N.; Khan, R.H. Ligand binding strategies of human serum albumin: How can the cargo be utilized? Chirality 2010, 22, 77–87. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | KSV/dm3 mol−1 | Kq/dm3 mol−1 s | R2 * | Kb/dm3 mol−1 | n | R2 |
|---|---|---|---|---|---|---|
| HSA-HPD | 3.35 × 103 | 3.37 × 1011 | 0.9869 | 4.45 × 103 | 1.03 | 0.9997 |
| HSA-QUE-HPD | 1.18 × 103 | 1.18 × 1011 | 0.9988 | 3.75 × 102 | 0.88 | 0.9992 |
| HSA-DIO-HPD | 1.30 × 103 | 1.30 × 1011 | 0.9881 | 5.40 × 102 | 0.91 | 0.9933 |
| HSA-CAT-HPD | 1.50 × 103 | 1.50 × 1011 | 0.9841 | 6.24 × 102 | 0.92 | 0.9861 |
| HSA-Ligand Complex | ΔGbind kJ/mol | Ki (nM) | ΔGinter kJ/mol | ΔGvdw+hbond+desolv kJ/mol | ΔGelec kJ/mol | ΔGtotal kJ/mol | ΔGtor kJ/mol |
|---|---|---|---|---|---|---|---|
| HSA-HPD | −40.7 | 75.6 | −43.2 | −42.8 | −0.4 | −6.2 | 8.7 |
| HSA-QUE-HPD | −37.0 | 325.6 | −38.2 | −37.9 | −0.2 | −7.6 | 8.7 |
| HSA-DIO-HPD | −37.2 | 298.4 | −41.5 | −41.4 | −0.1 | −4.5 | 8.7 |
| HSA-CAT-HPD | −38.0 | 221.5 | −42.5 | −42.0 | −0.5 | −4.3 | 8.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrušić, A.; Mrkalić, E.; Jelić, R.; Kočović, A.; Milosavljević, M.; Antonijević, M.; Sovrlić, M. Quercetin, Catechin, and Diosmin as Modulators of Haloperidol–HSA Interactions: A Biophysical and Computational Study. Int. J. Mol. Sci. 2025, 26, 6834. https://doi.org/10.3390/ijms26146834
Petrušić A, Mrkalić E, Jelić R, Kočović A, Milosavljević M, Antonijević M, Sovrlić M. Quercetin, Catechin, and Diosmin as Modulators of Haloperidol–HSA Interactions: A Biophysical and Computational Study. International Journal of Molecular Sciences. 2025; 26(14):6834. https://doi.org/10.3390/ijms26146834
Chicago/Turabian StylePetrušić, Aleksandar, Emina Mrkalić, Ratomir Jelić, Aleksandar Kočović, Miloš Milosavljević, Marko Antonijević, and Miroslav Sovrlić. 2025. "Quercetin, Catechin, and Diosmin as Modulators of Haloperidol–HSA Interactions: A Biophysical and Computational Study" International Journal of Molecular Sciences 26, no. 14: 6834. https://doi.org/10.3390/ijms26146834
APA StylePetrušić, A., Mrkalić, E., Jelić, R., Kočović, A., Milosavljević, M., Antonijević, M., & Sovrlić, M. (2025). Quercetin, Catechin, and Diosmin as Modulators of Haloperidol–HSA Interactions: A Biophysical and Computational Study. International Journal of Molecular Sciences, 26(14), 6834. https://doi.org/10.3390/ijms26146834