
Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages

 , , ,
, , ,
Abstract
1. Introduction
2. Results
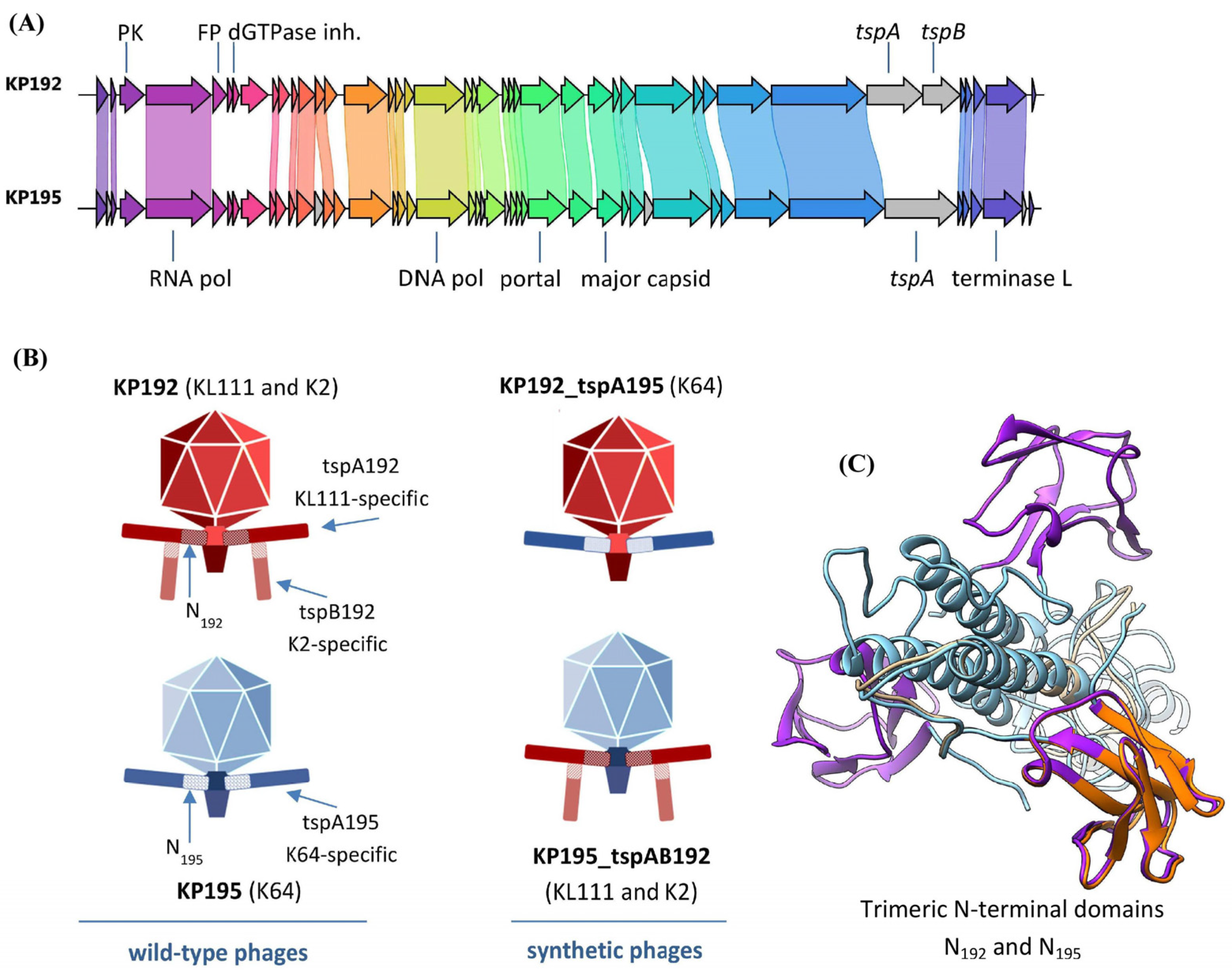
2.1. Bacteriophages KP192 and KP195 Have Different Tailspike Proteins and, Hence, Different Host Ranges
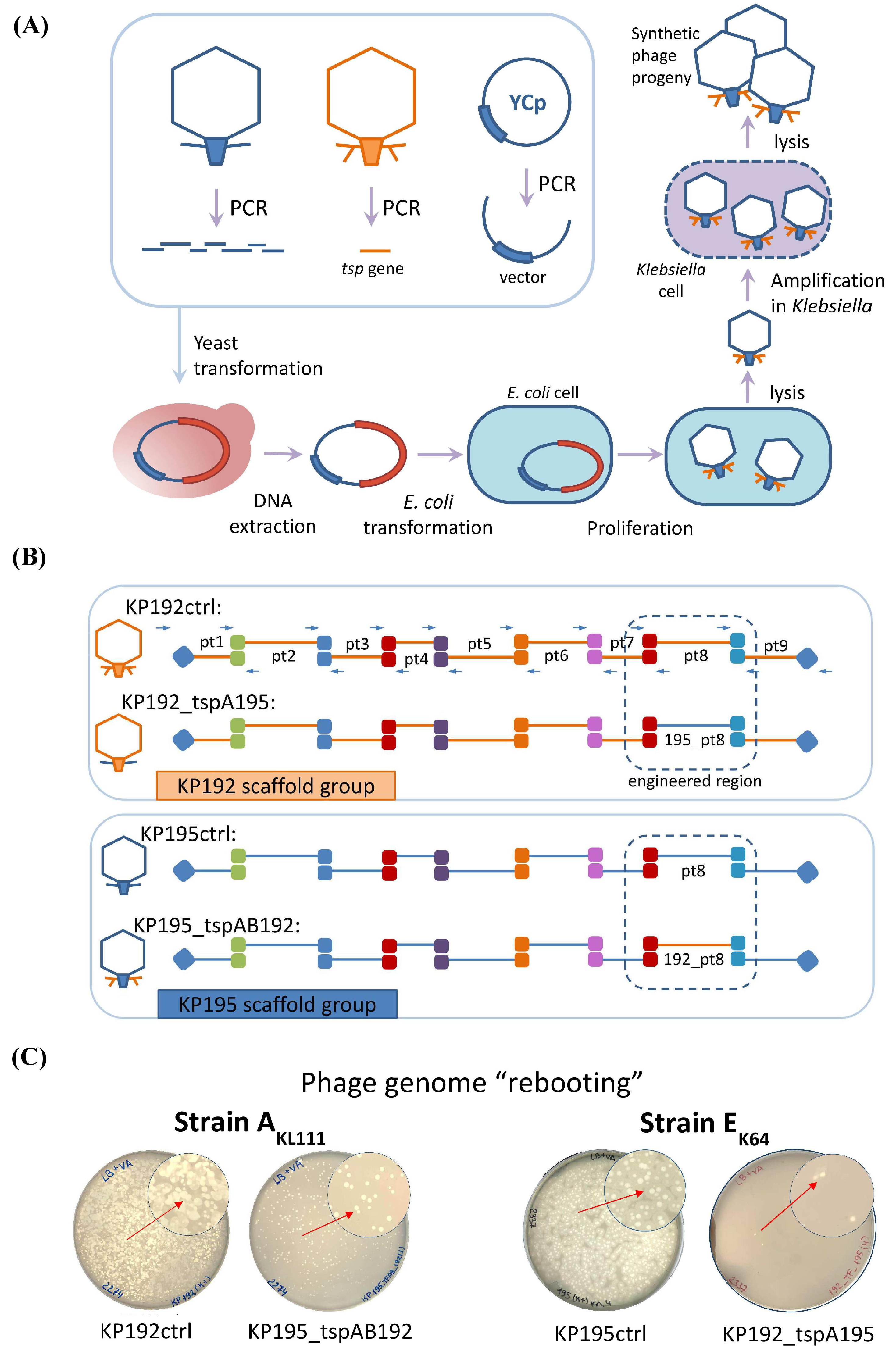
2.2. Design and Assembly of Synthetic Phage Genomes
2.3. “Rebooting” of the Klebsiella Phage Genomes Using E. coli as an Intermediate Host
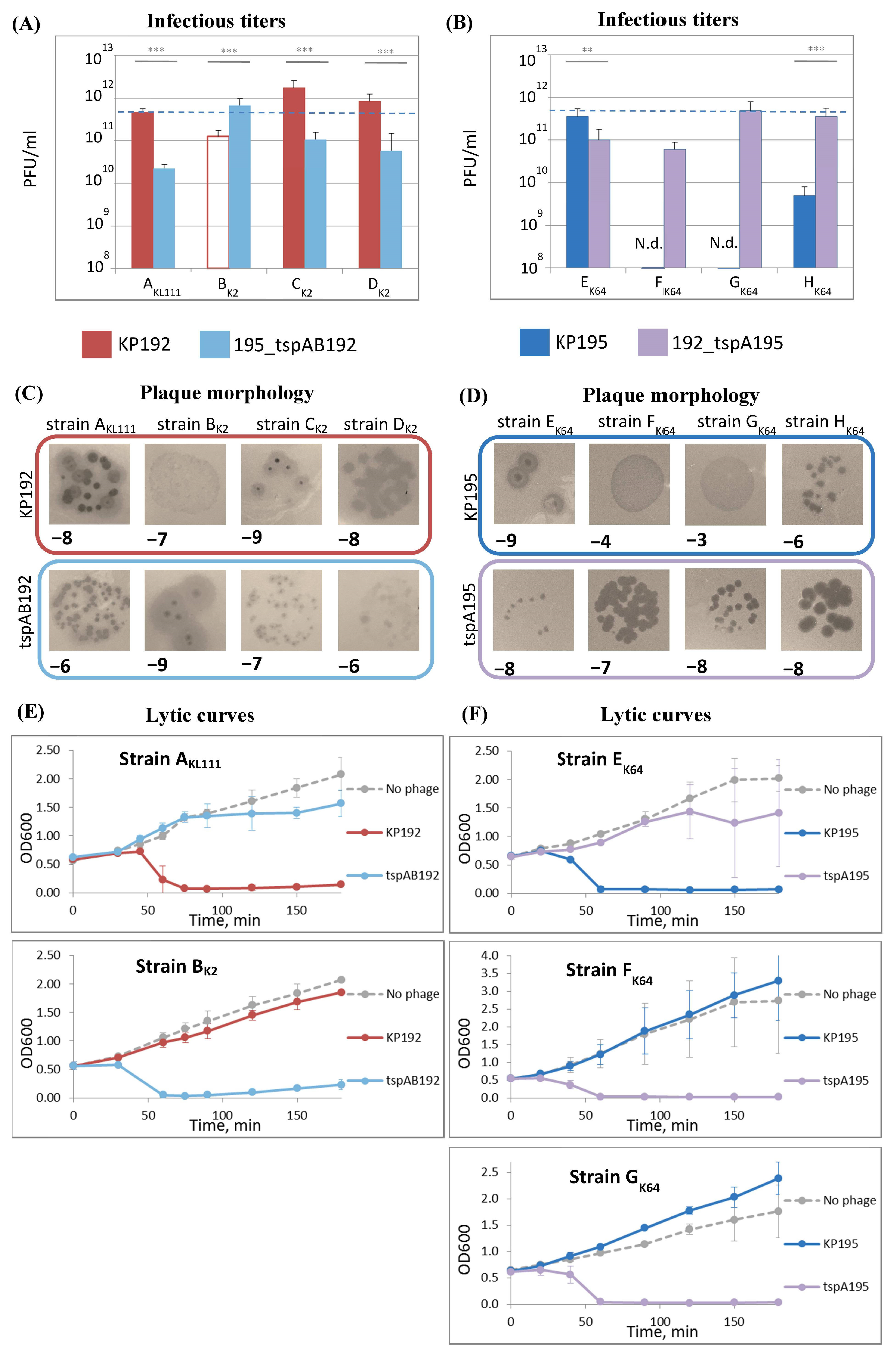
2.4. The Efficiency of Phage Replication Depends on Its Genomic Scaffold and the Klebsiella Strain Used
2.5. Analysis of the Differences Between the Genomes of Phages KP192 and KP195 That Potentially Affect the Efficiency of Phage Reproduction
3. Discussion
4. Materials and Methods
4.1. Phages, Bacterial, and Yeast Strains
4.2. Culturing Conditions
4.3. Determination of the Capsular Type of the AKL111 Strain
4.4. Preparation of DNA Fragments for Assembly of Phage Genomes
4.5. Phage Genome Assembly in Yeast
4.6. Yeast Colony Screening
4.7. Isolation of a Yeast Centromeric Plasmid Containing the Bacteriophage Genome
4.8. Phage Genome “Rebooting”
4.9. Verification of Genome Assembly Accuracy and Genome Sequencing
4.10. Phage Propagation and Purification
4.11. Determination of Infectious Titer of Phage Samples
4.12. Determination of Pseudo-Physical Titer (TiterPP) of Phage Samples
4.13. Determination of the Efficiency of Plating
4.14. Bacterial Killing Assay
4.15. Bioinformatic Analysis of the Differences Between the KP192 and KP195 Genomes
4.16. Protein Structure Modeling and Visualization
4.17. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CEMTC | Collection of Extremophilic Microorganisms and Type Cultures |
| EOP | Efficiency of plating |
| MOI | Multiplicity of infection |
| PCLU | Protein concentration-linked unit |
| PFU | Plaque-forming unit |
| rEOP | Relative efficiency of plating |
| TAR | Transformation-associated recombination cloning |
| titerPP | Pseudo-physical phage titer |
| tsp | Tailspike protein |
Appendix A
Appendix A.1. Validation of Pseudo-Physical Titer Determination of Phage Samples
References
- Strathdee, S.A.; Hatfull, G.F.; Mutalik, V.K.; Schooley, R.T. Phage Therapy: From Biological Mechanisms to Future Directions. Cell 2023, 186, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Watanabe, S.; Miyanaga, K.; Kiga, K.; Sasahara, T.; Aiba, Y.; Tan, X.-E.; Veeranarayanan, S.; Thitiananpakorn, K.; Nguyen, H.M.; et al. A Comprehensive Review on Phage Therapy and Phage-Based Drug Development. Antibiotics 2024, 13, 870. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically Engineered Phages: A Review of Advances over the Last Decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed]
- Dunne, M.; Prokhorov, N.S.; Loessner, M.J.; Leiman, P.G. Reprogramming Bacteriophage Host Range: Design Principles and Strategies for Engineering Receptor Binding Proteins. Curr. Opin. Biotechnol. 2021, 68, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing Phage Therapy through Synthetic Biology and Genome Engineering. Curr. Opin. Biotechnol. 2021, 68, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Du, J.; Dunne, M.; Kilcher, S.; Loessner, M.J. Engineering Therapeutic Phages for Enhanced Antibacterial Efficacy. Curr. Opin. Virol. 2022, 52, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Łobocka, M.; Dąbrowska, K.; Górski, A. Engineered Bacteriophage Therapeutics: Rationale, Challenges and Future. BioDrugs 2021, 35, 255–280. [Google Scholar] [CrossRef] [PubMed]
- Mahler, M.; Costa, A.R.; Van Beljouw, S.P.B.; Fineran, P.C.; Brouns, S.J.J. Approaches for Bacteriophage Genome Engineering. Trends Biotechnol. 2023, 41, 669–685. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Lemire, S.; Pires, D.P.; Lu, T.K. Engineering Modular Viral Scaffolds for Targeted Bacterial Population Editing. Cell Syst. 2015, 1, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.R.; Azeredo, J.; Pires, D.P. Synthetic Biology to Engineer Bacteriophage Genomes. In Bacteriophage Therapy; Azeredo, J., Sillankorva, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2024; Volume 2734, pp. 261–277. ISBN 978-1-0716-3522-3. [Google Scholar]
- Gibson, D.G.; Benders, G.A.; Andrews-Pfannkoch, C.; Denisova, E.A.; Baden-Tillson, H.; Zaveri, J.; Stockwell, T.B.; Brownley, A.; Thomas, D.W.; Algire, M.A.; et al. Complete Chemical Synthesis, Assembly, and Cloning of a Mycoplasma Genitalium Genome. Science 2008, 319, 1215–1220. [Google Scholar] [CrossRef] [PubMed]
- Marinelli, L.J.; Piuri, M.; Swigoňová, Z.; Balachandran, A.; Oldfield, L.M.; Van Kessel, J.C.; Hatfull, G.F. BRED: A Simple and Powerful Tool for Constructing Mutant and Recombinant Bacteriophage Genomes. PLoS ONE 2008, 3, e3957. [Google Scholar] [CrossRef] [PubMed]
- Wetzel, K.S.; Guerrero-Bustamante, C.A.; Dedrick, R.M.; Ko, C.-C.; Freeman, K.G.; Aull, H.G.; Divens, A.M.; Rock, J.M.; Zack, K.M.; Hatfull, G.F. CRISPY-BRED and CRISPY-BRIP: Efficient Bacteriophage Engineering. Sci. Rep. 2021, 11, 6796. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Lemire, S.; Grimon, D.; Dams, D.; Maciejewska, B.; Lu, T.; Drulis-Kawa, Z.; Briers, Y. Engineering the Modular Receptor-Binding Proteins of Klebsiella Phages Switches Their Capsule Serotype Specificity. mBio 2021, 12, e00455-21. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Krishna, R.; Wang, J.; Ahern, W.; Sturmfels, P.; Venkatesh, P.; Kalvet, I.; Lee, G.R.; Morey-Burrows, F.S.; Anishchenko, I.; Humphreys, I.R.; et al. Generalized Biomolecular Modeling and Design with RoseTTAFold All-Atom. Science 2024, 384, eadl2528. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Chipman, P.R.; Kostyuchenko, V.A.; Mesyanzhinov, V.V.; Rossmann, M.G. Three-Dimensional Rearrangement of Proteins in the Tail of Bacteriophage T4 on Infection of Its Host. Cell 2004, 118, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.; Fàbrega-Ferrer, M.; Machón, C.; Conesa, J.J.; Fernández, F.J.; Pérez-Luque, R.; Pérez-Ruiz, M.; Pous, J.; Vega, M.C.; Carrascosa, J.L.; et al. Structures of T7 Bacteriophage Portal and Tail Suggest a Viral DNA Retention and Ejection Mechanism. Nat. Commun. 2019, 10, 3746. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xiao, H.; Wang, L.; Wang, X.; Tan, Z.; Han, Z.; Li, X.; Yang, F.; Liu, Z.; Song, J.; et al. Structural Changes in Bacteriophage T7 upon Receptor-Induced Genome Ejection. Proc. Natl. Acad. Sci. USA 2021, 118, e2102003118. [Google Scholar] [CrossRef] [PubMed]
- Patro, L.P.P.; Sudhakar, K.U.; Rathinavelan, T. K-PAM: A Unified Platform to Distinguish Klebsiella Species K- and O-Antigen Types, Model Antigen Structures and Identify Hypervirulent Strains. Sci. Rep. 2020, 10, 16732. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Zhou, J.; Chen, G.-Q.; Xiu, Z.-L. Efficient Genome Engineering of a Virulent Klebsiella Bacteriophage Using CRISPR-Cas9. J. Virol. Methods 2018, 92, e00534-18. [Google Scholar] [CrossRef] [PubMed]
- Meile, S.; Du, J.; Staubli, S.; Grossmann, S.; Koliwer-Brandl, H.; Piffaretti, P.; Leitner, L.; Matter, C.I.; Baggenstos, J.; Hunold, L.; et al. Engineered Reporter Phages for Detection of Escherichia coli, Enterococcus, and Klebsiella in Urine. Nat. Commun. 2023, 14, 4336. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, S.; Jing, S.; Zeng, Y.; Yang, L.; Mu, Y.; Ding, Z.; Song, Y.; Sun, Y.; Zhang, G.; et al. Data-Driven Engineering of Phages with Tunable Capsule Tropism for Klebsiella Pneumoniae. Adv. Sci. 2024, 11, 2309972. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Huang, X.; Zhao, T.; Zhang, J.; Xiang, Y. Isolation and Characterization of Three Lytic Podo-Bacteriophages with Two Receptor Recognition Modules against Multidrug-Resistant Klebsiella Pneumoniae. bioRxiv 2024. [Google Scholar] [CrossRef]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the Architecture of Depolymerase-Containing Receptor Binding Proteins in Klebsiella Phages. Front. Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Squeglia, F.; Maciejewska, B.; Łątka, A.; Ruggiero, A.; Briers, Y.; Drulis-Kawa, Z.; Berisio, R. Structural and Functional Studies of a Klebsiella Phage Capsule Depolymerase Tailspike: Mechanistic Insights into Capsular Degradation. Structure 2020, 28, 613–624.e4. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Zhang, Z.; Tao, X.; Shi, X.; Lin, P.; Liao, D.; Ma, C.; Cai, X.; Lin, W.; Jiang, X.; et al. Structural and Functional Basis of Bacteriophage K64-ORF41 Depolymerase for Capsular Polysaccharide Degradation of Klebsiella Pneumoniae K64. Int. J. Biol. Macromol. 2024, 265, 130917. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sheng, Y.; Ma, R.; Xu, M.; Liu, F.; Qin, R.; Zhu, M.; Zhu, X.; He, P. Identification of a Depolymerase Specific for K64-Serotype Klebsiella Pneumoniae: Potential Applications in Capsular Typing and Treatment. Antibiotics 2021, 10, 144. [Google Scholar] [CrossRef] [PubMed]
- Solovieva, E.V.; Myakinina, V.P.; Kislichkina, A.A.; Krasilnikova, V.M.; Verevkin, V.V.; Mochalov, V.V.; Lev, A.I.; Fursova, N.K.; Volozhantsev, N.V. Comparative Genome Analysis of Novel Podoviruses Lytic for Hypermucoviscous Klebsiella Pneumoniae of K1, K2, and K57 Capsular Types. Virus Res. 2018, 243, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Larionov, V.; Kouprina, N.; Graves, J.; Resnick, M.A. Highly Selective Isolation of Human DNAs from Rodent–Human Hybrid Cells as Circular Yeast Artificial Chromosomes by Transformation-Associated Recombination Cloning. Proc. Natl. Acad. Sci. USA 1996, 93, 13925–13930. [Google Scholar] [CrossRef] [PubMed]
- Jaschke, P.R.; Lieberman, E.K.; Rodriguez, J.; Sierra, A.; Endy, D. A Fully Decompressed Synthetic Bacteriophage øX174 Genome Assembled and Archived in Yeast. Virology 2012, 434, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J.; Designing, P. Aeruginosa Synthetic Phages with Reduced Genomes. Sci. Rep. 2021, 11, 2164. [Google Scholar] [CrossRef] [PubMed]
- Fournet-Fayard, S.; Joly, B.; Forestier, C. Transformation of Wild Type Klebsiella Pneumoniae with Plasmid DNA by Electroporation. J. Microbiol. Methods 1995, 24, 49–54. [Google Scholar] [CrossRef]
- Abedon, S.T. Bacteriophage Adsorption: Likelihood of Virion Encounter with Bacteria and Other Factors Affecting Rates. Antibiotics 2023, 12, 723. [Google Scholar] [CrossRef] [PubMed]
- Zillig, W.; Fujiki, H.; Blum, W.; Janeković, D.; Schweiger, M.; Rahmsdorf, H.; Ponta, H.; Hirsch-Kauffmann, M. In Vivo and In Vitro Phosphorylation of DNA-Dependent RNA Polymerase of Escherichia Coli by Bacteriophage-T7-Induced Protein Kinase. Proc. Natl. Acad. Sci. USA 1975, 72, 2506–2510. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.S.; Nicholson, A.W. Phosphorylation of Escherichia Coli Translation Initiation Factors by the Bacteriophage T7 Protein Kinase. Biochemistry 1992, 31, 4822–4827. [Google Scholar] [CrossRef] [PubMed]
- Robertson, E.S.; Aggison, L.A.; Nicholson, A.W. Phosphorylation of Elongation Factor G and Ribosomal Protein S6 in Bacteriophage T7-infected Escherichia Coli. Mol. Microbiol. 1994, 11, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Bartolec, T.; Mitosch, K.; Potel, C.; Corona, F.; Yang, A.L.J.; Burtscher, M.L.; Koumoutsi, A.; Becher, I.; Bobonis, J.; Karcher, N.; et al. Pervasive Phosphorylation by Phage T7 Kinase Disarms Bacterial Defenses. bioRxiv 2024. [Google Scholar] [CrossRef]
- Pyra, A.; Brzozowska, E.; Pawlik, K.; Gamian, A.; Dauter, M.; Dauter, Z. Tail Tubular Protein A: A Dual-Function Tail Protein of Klebsiella Pneumoniae Bacteriophage KP32. Sci. Rep. 2017, 7, 2223. [Google Scholar] [CrossRef] [PubMed]
- Brzozowska, E.; Pyra, A.; Pawlik, K.; Janik, M.; Górska, S.; Urbańska, N.; Drulis-Kawa, Z.; Gamian, A. Hydrolytic Activity Determination of Tail Tubular Protein A of Klebsiella Pneumoniae Bacteriophages towards Saccharide Substrates. Sci. Rep. 2017, 7, 18048. [Google Scholar] [CrossRef] [PubMed]
- Ipoutcha, T.; Racharaks, R.; Huttelmaier, S.; Wilson, C.J.; Ozer, E.A.; Hartmann, E.M. A Synthetic Biology Approach to Assemble and Reboot Clinically Relevant Pseudomonas Aeruginosa Tailed Phages. Microbiol. Spectr. 2024, 12, e02897-23. [Google Scholar] [CrossRef] [PubMed]
- Morozova, V.; Babkin, I.; Kozlova, Y.; Baykov, I.; Bokovaya, O.; Tikunov, A.; Ushakova, T.; Bardasheva, A.; Ryabchikova, E.; Zelentsova, E.; et al. Isolation and Characterization of a Novel Klebsiella Pneumoniae N4-like Bacteriophage KP8. Viruses 2019, 11, 1115. [Google Scholar] [CrossRef] [PubMed]
- Gietz, D.; Jean, A.S.; Woods, R.A.; Schiestl, R.H. Improved Method for High Efficiency Transformation of Intact Yeast Cells. Nucl. Acids Res. 1992, 20, 1425. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D. Yeast Transformation by the LiAc/SS Carrier DNA/PEG Method. In Yeast Protocols; Xiao, W., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2014; Volume 1163, pp. 33–44. ISBN 978-1-4939-0798-4. [Google Scholar]
- Morozova, V.V.; Yakubovskij, V.I.; Baykov, I.K.; Kozlova, Y.N.; Tikunov, A.Y.; Babkin, I.V.; Bardasheva, A.V.; Zhirakovskaya, E.V.; Tikunova, N.V. StenM_174: A Novel Podophage That Infects a Wide Range of Stenotrophomonas spp. and Suggests a New Subfamily in the Family Autographiviridae. Viruses 2023, 16, 18. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. CP Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Chapter 2. Bacteriophage λ and Its Vectors. In Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Woodbury, NY, USA, 2001; Volume 1, pp. 2.25–2.106. ISBN 978-0-87969-577-4. [Google Scholar]
- Carroll-Portillo, A.; Coffman, C.N.; Varga, M.G.; Alcock, J.; Singh, S.B.; Lin, H.C. Standard Bacteriophage Purification Procedures Cause Loss in Numbers and Activity. Viruses 2021, 13, 328. [Google Scholar] [CrossRef] [PubMed]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Phages with tspA192 and tspB192 Tailspikes | Phages with tspA195 Tailspikes | |||
|---|---|---|---|---|
| Name | KP192 (WT 1)/ KP192ctrl (synthetic) | KP195_ tspAB192 (synthetic) | KP195 (WT)/ KP195ctrl (synthetic) | KP192_ tspA195 (synthetic) |
| Pictogram |  |  |  |  |
| Capsular specificity | KL111 and K2 | K64 | ||
| Genomic scaffold | KP192 | KP195 | KP195 | KP192 |
| Klebsiella strain used for genome “rebooting” | AKL111 | AKL111 | EK64 | EK64 |
| Plates efficiently on strains | AKL111 CK2 DK2 | BK2 | EK64 | FK64 GK64 HK64 |
| Plates poorly on strains | BK2 | AKL111 CK2 DK2 | FK64 GK64 HK64 | EK64 |
| Product Name | Amino Acid Identity | Locus Tag 1 | Note |
|---|---|---|---|
| Protein kinase | 81% | HOT22_gp03, HOT24_gp04 | The differences are located in two regions |
| Fusion protein | 77% | HOT22_gp05, HOT24_gp06 | The differences are located in the N-terminal region |
| dGTPase inhibitor | 66% | HOT22_gp07, HOT24_gp08 | |
| DNA ligase | 79% | HOT22_gp08, HOT24_gp09 | The differences are located in two regions |
| Nucleotide kinase | 79% | HOT22_gp10, HOT24_gp11 | |
| HNH endonuclease | N/A 2 | HOT24_gp14 | The gene is absent in the KP192 phage |
| Hypothetical protein | 80% | HOT22_gp18, HOT24_gp20 | |
| DNA polymerase | 93% | HOT22_gp19, HOT24_gp21 | The enzyme of the KP195 phage contains an insert near the 520 amino acid residue |
| Hypothetical protein | N/A | HOT24_gp24 | The gene is absent in the KP192 phage |
| Homing endonuclease | N/A | HOT24_gp35 | The gene is absent in the KP192 phage |
| Tailspike protein A | 19% | HOT22_gp35, HOT24_gp41 | |
| Tailspike protein B | N/A | HOT22_gp36 | The gene is absent in the KP195 phage |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baykov, I.K.; Kurchenko, O.M.; Mikhaylova, E.E.; Miroshnikova, A.V.; Morozova, V.V.; Khlebnikova, M.I.; Tikunov, A.Y.; Kozlova, Y.N.; Tikunova, N.V. Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages. Int. J. Mol. Sci. 2025, 26, 6824. https://doi.org/10.3390/ijms26146824
Baykov IK, Kurchenko OM, Mikhaylova EE, Miroshnikova AV, Morozova VV, Khlebnikova MI, Tikunov AY, Kozlova YN, Tikunova NV. Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages. International Journal of Molecular Sciences. 2025; 26(14):6824. https://doi.org/10.3390/ijms26146824
Chicago/Turabian StyleBaykov, Ivan K., Olga M. Kurchenko, Ekaterina E. Mikhaylova, Anna V. Miroshnikova, Vera V. Morozova, Marianna I. Khlebnikova, Artem Yu. Tikunov, Yuliya N. Kozlova, and Nina V. Tikunova. 2025. "Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages" International Journal of Molecular Sciences 26, no. 14: 6824. https://doi.org/10.3390/ijms26146824
APA StyleBaykov, I. K., Kurchenko, O. M., Mikhaylova, E. E., Miroshnikova, A. V., Morozova, V. V., Khlebnikova, M. I., Tikunov, A. Y., Kozlova, Y. N., & Tikunova, N. V. (2025). Replacement of the Genomic Scaffold Improves the Replication Efficiency of Synthetic Klebsiella Phages. International Journal of Molecular Sciences, 26(14), 6824. https://doi.org/10.3390/ijms26146824