
The Therapeutic Potential of Phytochemicals Unlocks New Avenues in the Management of Rheumatoid Arthritis

 , ,
, ,
Abstract

1. Introduction
1.1. Epidemiological Overview and Global Prevalence
1.2. Risk Factors
1.3. Genetic Factors
1.4. Epigenetic Factors
1.5. Environmental Factors
1.6. Lifestyle Factors (Nutrition and Gut Microbiota)
1.7. Personal Factors
1.8. Mechanisms and (Pato)Etiology of Rheumatoid Arthritis Initiation, Development, and Progression
2. Effector Cells Involved in Rheumatoid Arthritis Pathology
2.1. Cytokines and the Impact on Effector Cells
2.2. The Role of Metalloproteinases
2.3. The Role of Angiogenesis
2.4. The Role of Free Radicals
3. Current Rheumatoid Arthritis Drug Treatment
3.1. Conventional Synthetic DMARDs (csDMARDs)
3.2. Methotrexate
3.3. Leflunomide
3.4. Sulfasalazine
3.5. Biologic DMARDs (bDMARDs)
3.6. Tumor Necrosis Factor-Alpha Inhibitors (TNFis)
3.7. Interleukin-1 Inhibitor
3.8. Interleukin-6 Receptor Inhibitor
3.9. Anti-CD20 Antibody
3.10. Targeted Synthetic DMARDs (tsDMARDs)
3.11. Mesenchymal Stem Cells (MSCs)
4. In Vitro Studies Using Plant-Derived Natural Products for the Management of Rheumatoid Arthritis and Signaling Pathways

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Dose, µM | Cell Line | Targets | Main Findings | Modulated Pathway | Reference |
|---|---|---|---|---|---|---|
| Curcumin | 50 | MH7A | TNF-α, IL-6, IL-17 | Inhibition of migration, invasion, and inflammation | PI3K/AKT | [296] |
| Emodin | 15 | L929 | IL-6, IL-1β, COX-2 | Inhibition of inflammation | NF-κB | [297] |
| Ginsenoside compound K | 30 | Isolated FLS | FLUT1, HK2, PKM1, PKM2 | Inhibition of glycolysis | NF-κB | [298] |
| Glytabastan B | 3 and 6 | SW982 | TNF-α, IL-6, IL-8, COX-2, MMP-1 | Inhibition of inflammation and invasion | MAPK, PI3K/AKT, NF-κB | [299] |
| Isobavachalcone | 20 | MH7A | TNF-α, MAPK13, EGFR, PTGS2, MMP-3 | Inhibition of migration, invasion, and inflammation | PI3K/AKT, JAK/STAT | [300] |
| Kaempferol | 10 | HFLS-RA | IL-1β, MMP-2 and -9, N-cadherin, vimentin | Inhibition of inflammation and abnormal proliferation | MAPK | [301] |
| Leocarpinolide B | 20 | SW982 | IL-6, IL-8, IL-1β | Inhibition of proliferation, migration, invasion, and inflammation | NF-κB | [302] |
| Magnoflorine | 10 | MH7A | iNOS; COX-2; IL-6; IL-8; MMP-1, -2, -3, -9, and -13 | Inhibition of proliferation, migration, and invasion | PI3K/AKT, NF-κB, Nrf-2, | [291] |
| Nimbolide | 1 | HIG-82 | MMP-2, IL-6, iNOS, COX-2 | Reduction in inflammation | MAPK, NF-κB, Nrf-2 | [303] |
| Quercetin | 1.5 | L929, HEK293T, MH7A | COX-2, iNOS, IL-6, IL-1β | Reduction in cell apoptosis and improvement in cell injury | NF-κB | [159] |
| Sappanone A | 40 | HFLS-RA | TNF-α, IL-1β, IL-6, IL-10, IL-17A | Inhibition of inflammation | JAK2/STAT3, PI3K/AKT, NF-κB | [304] |
| Shikonin | 1 × 10−7 | MH7A | VEGF, VEGFR2, TNF-α, IL-1β, PDGF, TGF-β | Inhibition of migration, invasion, and adhesion | MAPK (ERK1/2, JNK, p38) | [305] |
| Scopoletin | 30 | HFLS-RA | IL-1β, TNF-α, MMP-3, MMP-9, COX-2, Bcl-2 | Inhibition of proliferation, migration, and invasion | NF-κB | [306] |
| Suberosin | 5 | RA-FLS | IL-6, IL-1β, TNF-α, IL-8, MMP-1, MMP-3, MMP-9, MMP-13 | Inhibition of inflammation | JAK/STAT | [307] |
| Tectoridin | 50 | HFLS-RA | IL-1β, IL-6, COX-2, iNOS | Inhibition of inflammation | MAPK (ERK1/2, JNK, p38) | [308] |
| Umbelliferone | 20 | HFLS-RA | IL-1β, TNF-α, MMP-3, MMP-9, COX-2, Bcl-2 | Inhibition of proliferation, migration, and invasion | NF-κB | [306] |
| Wilforine | 0.4 | Isolated FLS | IL-1β, IL-6, TNF-α, CCND1, GSK-3β, c-Myc, MMP-3 | Inhibition of inflammation and abnormal proliferation | Wnt11/β-catenin | [309] |
5. In Vivo Studies Using Plant-Derived Natural Products for the Management of Rheumatoid Arthritis
5.1. Collagen-Induced Arthritis Model
5.2. Collagen Antibody-Induced Arthritis Model
5.3. Adjuvant Induced Arthritis Model
5.4. Pristane-Induced Arthritis Model
6. Human Clinical Trials Involving Phytochemicals for RA Treatment
7. Conclusions and Future Perspectives
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ACPAs | Anti-citrullinated protein antibodies |
| ADAMTS | A Disintegrin and Metalloproteinase with Thrombospondin Motifs |
| Anti-CarP | Anti-carbamylated protein antibodies |
| CCR6 | Chemokine receptor 6 |
| cDMARDs | Conventional disease-modifying anti-rheumatic drugs |
| COX-2 | Cyclooxygenase-2 |
| CRP | C-creative protein |
| CTLA-4 | T-lymphocyte-associated protein 4 |
| CXCL12 | Chemokine C-X-C motif ligand 12 |
| DALYs | Disability-adjusted life years |
| DNMT | DNA methyltransferases |
| ECM | Extracellular matrix |
| ESR | Erythrocyte sedimentation rate |
| FGFs | Fibroblast growth factors |
| FLS | Fibroblast-like synoviocytes |
| GC | Glucocorticoids |
| GM-CSF | Granulocyte–macrophage colony-stimulating factor |
| HB-EGF | Heparin-binding EGF-like growth factor |
| HDAC | Histone deacetylases |
| HLA-DR | Human leukocyte antigen D-related |
| IgG | Immunoglobulin G |
| IL | Interleukin |
| IRF-5 | Interferon regulatory factor 5 |
| JAK/STAT | Janus-activated kinase signal transduction and activator of transcription |
| MAPK | Mitogen-activated protein kinase |
| M-CSF | Macrophage colony-stimulating factor |
| MDC | Myeloid dendritic cells |
| MHC | Major histocompatibility complex |
| MMPs | Matrix metalloproteinases |
| MSCs | Mesenchymal stem cells |
| MTX | Methotrexate |
| NF-κB | Nuclear factor kappa B |
| NF-κβ | Nuclear factor kappa-B |
| NO | Nitric oxide |
| NSAIDs | Non-steroidal anti-inflammatory drugs |
| OPG | Osteoprotegerin |
| PADI4 | Peptidyl arginine deiminase, type IV enzyme |
| PBMCs | Peripheral blood mononuclear cells |
| PDC | Plasmacytoid dendritic cells |
| PI3/AKT | Phosphatidylinositol 3 kinase-AKT |
| PlGF | Placenta growth factor |
| RA | Rheumatoid arthritis |
| RANKL | Receptor activator of nuclear factor-B ligand |
| RF | Rheumatoid factor |
| ROS | Reactive oxygen species |
| SAM | S-adenosine methionine |
| SLE | Systemic lupus erythematosus |
| SNPs | Single nucleotide polymorphisms |
| SYK/BTK | Spleen tyrosine kinase)/Bruton’s tyrosine kinase |
| TBX5 | T-box transcription factor 5 |
| Tfh | T follicular helper |
| TGF-β | Transforming growth factor beta |
| TIMPs | Tissue inhibitors of metalloproteinases |
| TNF-α | Tumor necrosis factor alpha |
| TRAF1 | Tumors necrosis factor receptor-associated factor 1 |
| Treg | Regulatory T cells |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| VEGF | Vascular endothelial growth factor |
| Wnt/β-catenin | Wingless/integrated |
References
- Damerau, A.; Gaber, T. Modeling Rheumatoid Arthritis In Vitro: From Experimental Feasibility to Physiological Proximity. Int. J. Mol. Sci. 2020, 21, 7916. [Google Scholar] [CrossRef] [PubMed]
- Kour, G.; Choudhary, R.; Anjum, S.; Bhagat, A.; Bajaj, B.K.; Ahmed, Z. Phytochemicals Targeting JAK/STAT Pathway in the Treatment of Rheumatoid Arthritis: Is There a Future? Biochem. Pharmacol. 2022, 197, 114929. [Google Scholar] [CrossRef] [PubMed]
- Hosseinikhah, S.; Barani, M.; Rahdar, A.; Madry, H.; Arshad, R.; Mohammadzadeh, V.; Cucchiarini, M. Nanomaterials for the Diagnosis and Treatment of Inflammatory Arthritis. Int. J. Mol. Sci. 2021, 22, 3092. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Aletaha, D.; Barton, A.; Burmester, G.R.; Emery, P.; Firestein, G.S.; Kavanaugh, A.; McInnes, I.B.; Solomon, D.H.; Strand, V.; et al. Rheumatoid Arthritis. Nat. Rev. Dis. Primers 2018, 4, 18001. [Google Scholar] [CrossRef] [PubMed]
- Jannat, A.; John, P.; Bhatti, A.; Hayat, M.Q. Tomorou Attenuates Progression of Rheumatoid Arthritis through Alteration in ULK-1 Independent Autophagy Pathway in Collagen Induced Arthritis Mice Model. Cell Death Discov. 2019, 5, 142. [Google Scholar] [CrossRef] [PubMed]
- Kour, G.; Haq, S.A.; Bajaj, B.K.; Gupta, P.N.; Ahmed, Z. Phytochemical Add-on Therapy to DMARDs Therapy in Rheumatoid Arthritis: In Vitro and in Vivo Bases, Clinical Evidence and Future Trends. Pharmacol. Res. 2021, 169, 105618. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Wang, C.; Huo, H.; Xu, C.; Sun, H.; Wang, X.; Wang, L.; Li, L. Prodrug-Based Nanomedicines for Rheumatoid Arthritis. Discov. Nano 2024, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Dudics, S.; Langan, D.; Meka, R.R.; Venkatesha, S.H.; Berman, B.M.; Che, C.-T.; Moudgil, K.D. Natural Products for the Treatment of Autoimmune Arthritis: Their Mechanisms of Action, Targeted Delivery, and Interplay with the Host Microbiome. Int. J. Mol. Sci. 2018, 19, 2508. [Google Scholar] [CrossRef] [PubMed]
- Juarez, M.; Bang, H.; Hammar, F.; Reimer, U.; Dyke, B.; Sahbudin, I.; Buckley, C.D.; Fisher, B.; Filer, A.; Raza, K. Identification of Novel Antiacetylated Vimentin Antibodies in Patients with Early Inflammatory Arthritis. Ann. Rheum. Dis. 2016, 75, 1099–1107. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.L.; Wolfe, F.; Huizinga, T.W. Rheumatoid Arthritis. Lancet 2010, 376, 1094–1108. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Knevel, R.; Suwannalai, P.; Van Der Linden, M.P.; Janssen, G.M.C.; Van Veelen, P.A.; Levarht, N.E.W.; Van Der Helm-van Mil, A.H.M.; Cerami, A.; Huizinga, T.W.J.; et al. Autoantibodies Recognizing Carbamylated Proteins Are Present in Sera of Patients with Rheumatoid Arthritis and Predict Joint Damage. Proc. Natl. Acad. Sci. USA 2011, 108, 17372–17377. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, G.R.; Jothi, G.; Mohana, T.; Vasconcelos, A.B.S.; Montalvão, M.M.; Hariharan, G.; Sridharan, G.; Kumar, P.M.; Gurgel, R.Q.; Li, H.-B.; et al. Anti-Inflammatory Natural Products as Potential Therapeutic Agents of Rheumatoid Arthritis: A Systematic Review. Phytomedicine 2021, 93, 153766. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Huang, S. Nanomedicine Is More than a Supporting Role in Rheumatoid Arthritis Therapy. J. Control. Release 2023, 356, 142–161. [Google Scholar] [CrossRef] [PubMed]
- Finckh, A.; Gilbert, B.; Hodkinson, B.; Bae, S.-C.; Thomas, R.; Deane, K.D.; Alpizar-Rodriguez, D.; Lauper, K. Global Epidemiology of Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2022, 18, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Black, R.J.; Cross, M.; Haile, L.M.; Culbreth, G.T.; Steinmetz, J.D.; Hagins, H.; Kopec, J.A.; Brooks, P.M.; Woolf, A.D.; Ong, K.L.; et al. Global, Regional, and National Burden of Rheumatoid Arthritis, 1990–2020, and Projections to 2050: A Systematic Analysis of the Global Burden of Disease Study 2021. Lancet Rheumatol. 2023, 5, e594–e610. [Google Scholar] [CrossRef] [PubMed]
- Globaldata. Available online: https://www.globaldata.com/store/report/epicast-report-rheumatoid-arthritis-epidemiology-forecast-to-2025/ (accessed on 26 June 2025).
- Persistence Market Research. Available online: https://www.persistencemarketresearch.com/market-research/rheumatoid-arthritis-market.asp (accessed on 26 June 2025).
- Biospace. Available online: https://www.biospace.com/press-releases/rheumatoid-arthritis-market-size-to-reach-us-34-7-billion-by-2035-impelled-by-obesity-and-lifestyle-changes (accessed on 26 June 2025).
- Bahuguna, R.; Awasthi, R. Unlocking New Dimensions in Rheumatoid Arthritis Therapy: Harnessing the Power of Lipid Based Vesicles beyond Traditional Therapies. J. Drug Deliv. Sci. Technol. 2023, 89, 105106. [Google Scholar] [CrossRef]
- Ding, Q.; Hu, W.; Wang, R.; Yang, Q.; Zhu, M.; Li, M.; Cai, J.; Rose, P.; Mao, J.; Zhu, Y.Z. Signaling Pathways in Rheumatoid Arthritis: Implications for Targeted Therapy. Signal Transduct. Target. Ther. 2023, 8, 68. [Google Scholar] [CrossRef] [PubMed]
- Galloway, J.; Capron, J.-P.; De Leonardis, F.; Fakhouri, W.; Rose, A.; Kouris, I.; Burke, T. The Impact of Disease Severity and Duration on Cost, Early Retirement and Ability to Work in Rheumatoid Arthritis in Europe: An Economic Modelling Study. Rheumatol. Adv. Pract. 2020, 4, rkaa041. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.D.; Adeyemo, A.; O’Leary, M.E.; Bottaro, A. Animal Models of Rheumatoid Pain: Experimental Systems and Insights. Arthritis Res. Ther. 2017, 19, 146. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Li, J.; Agarwal, S.K. Economic and Humanistic Burden of Rheumatoid Arthritis: Results From the US National Survey Data 2018–2020. ACR Open Rheumatol. 2024, 6, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Akram, M.; Daniyal, M.; Sultana, S.; Owais, A.; Akhtar, N.; Zahid, R.; Said, F.; Bouyahya, A.; Ponomarev, E.; Ali Shariat, M.; et al. Traditional and Modern Management Strategies for Rheumatoid Arthritis. Clin. Chim. Acta 2021, 512, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Patidar, V.; Shah, S.; Kumar, R.; Singh, P.K.; Singh, S.B.; Khatri, D.K. A Molecular Insight of Inflammatory Cascades in Rheumatoid Arthritis and Anti-Arthritic Potential of Phytoconstituents. Mol. Biol. Rep. 2022, 49, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tao, T.; Yao, H.; Zheng, H.; Wang, F.; Gao, Y. Mechanism of Action of Quercetin in Rheumatoid Arthritis Models: Meta-Analysis and Systematic Review of Animal Studies. Inflammopharmacol 2023, 31, 1629–1645. [Google Scholar] [CrossRef] [PubMed]
- Balendran, T.; Lim, K.; Hamilton, J.A.; Achuthan, A.A. Targeting Transcription Factors for Therapeutic Benefit in Rheumatoid Arthritis. Front. Immunol. 2023, 14, 1196931. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.; Bai, L.; Yang, Y.; Huang, J. Dysregulation of lncRNAs in Rheumatoid Arthritis: Biomarkers, Pathogenesis and Potential Therapeutic Targets. Front. Pharmacol. 2021, 12, 652751. [Google Scholar] [CrossRef] [PubMed]
- George, G.; Shyni, G.L.; Raghu, K.G. Current and Novel Therapeutic Targets in the Treatment of Rheumatoid Arthritis. Inflammopharmacol 2020, 28, 1457–1476. [Google Scholar] [CrossRef] [PubMed]
- Luo, T.-T.; Wu, Y.-J.; Yin, Q.; Chen, W.-G.; Zuo, J. The Involvement of Glucose and Lipid Metabolism Alteration in Rheumatoid Arthritis and Its Clinical Implication. J. Inflamm. Res. 2023, 16, 1837–1852. [Google Scholar] [CrossRef] [PubMed]
- Németh, T.; Nagy, G.; Pap, T. Synovial Fibroblasts as Potential Drug Targets in Rheumatoid Arthritis, Where Do We Stand and Where Shall We Go? Ann. Rheum. Dis. 2022, 81, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Mueller, A.-L.; Payandeh, Z.; Mohammadkhani, N.; Mubarak, S.M.H.; Zakeri, A.; Alagheband Bahrami, A.; Brockmueller, A.; Shakibaei, M. Recent Advances in Understanding the Pathogenesis of Rheumatoid Arthritis: New Treatment Strategies. Cells 2021, 10, 3017. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.D.; Leung, S.H.; Viatte, S. Genetics of Rheumatoid Arthritis. Best Pract. Res. Clin. Rheumatol. 2024, 38, 101968. [Google Scholar] [CrossRef] [PubMed]
- Källberg, H.; Padyukov, L.; Plenge, R.M.; Rönnelid, J.; Gregersen, P.K.; Van Der Helm-van Mil, A.H.M.; Toes, R.E.M.; Huizinga, T.W.; Klareskog, L.; Alfredsson, L. Gene-Gene and Gene-Environment Interactions Involving HLA-DRB1, PTPN22, and Smoking in Two Subsets of Rheumatoid Arthritis. Am. J. Hum. Genet. 2007, 80, 867–875. [Google Scholar] [CrossRef] [PubMed]
- Kokkonen, H.; Johansson, M.; Innala, L.; Jidell, E.; Rantapää-Dahlqvist, S. The PTPN221858C/T Polymorphism Is Associated with Anti-Cyclic Citrullinated Peptide Antibody-Positive Early Rheumatoid Arthritis in Northern Sweden. Arthritis Res. Ther. 2007, 9, R56. [Google Scholar] [CrossRef] [PubMed]
- Van Der Helm-van Mil, A.H.M.; Verpoort, K.N.; Breedveld, F.C.; Huizinga, T.W.J.; Toes, R.E.M.; De Vries, R.R.P. The HLA–DRB1 Shared Epitope Alleles Are Primarily a Risk Factor for Anti–Cyclic Citrullinated Peptide Antibodies and Are Not an Independent Risk Factor for Development of Rheumatoid Arthritis. Arthritis Rheum. 2006, 54, 1117–1121. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, W.; Zhang, X.; Zhang, X.; Jiang, L.; Guo, Y.; Wang, X. Association between Polymorphism in TRAF1/C5 Gene and Risk of Rheumatoid Arthritis: A Meta-Analysis. Mol. Biol. Rep. 2014, 41, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.; Padyukov, L.; Kurreeman, F.A.S.; Liljedahl, U.; Wiman, A.; Alfredsson, L.; Toes, R.; Rönnelid, J.; Klareskog, L.; Huizinga, T.W.J.; et al. Association of a Haplotype in the Promoter Region of the Interferon Regulatory Factor 5 Gene with Rheumatoid Arthritis. Arthritis Rheum. 2007, 56, 2202–2210. [Google Scholar] [CrossRef] [PubMed]
- Silman, A.J.; Pearson, J.E. Epidemiology and Genetics of Rheumatoid Arthritis. Arthritis Res. 2002, 4, S265. [Google Scholar] [CrossRef] [PubMed]
- Kapitány, A.; Zilahi, E.; Szántó, S.; Szücs, G.; Szabó, Z.; Végvári, A.; Rass, P.; Sipka, S.; Szegedi, G.; Szekanecz, Z. Association of Rheumatoid Arthritis with HLA-DR1 and HLA-DR4 in Hungary. Ann. N.Y. Acad. Sci. 2005, 1051, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Becart, S.; Whittington, K.B.; Prislovsky, A.; Rao, N.L.; Rosloniec, E.F. The Role of Posttranslational Modifications in Generating Neo-Epitopes That Bind to Rheumatoid Arthritis-Associated HLA-DR Alleles and Promote Autoimmune T Cell Responses. PLoS ONE 2021, 16, e0245541. [Google Scholar] [CrossRef] [PubMed]
- Van Der Helm-van Mil, A.H.M.; Huizinga, T.W.J.; Schreuder, G.M.T.; Breedveld, F.C.; De Vries, R.R.P.; Toes, R.E.M. An Independent Role of Protective HLA Class II Alleles in Rheumatoid Arthritis Severity and Susceptibility. Arthritis Rheum. 2005, 52, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, S.B.; Sensoy, O.; Yan, Z.; Gadi, V.K.; Richardson, M.L.; Nelson, J.L. Immunogenicity of a Rheumatoid Arthritis Protective Sequence When Acquired through Microchimerism. Proc. Natl. Acad. Sci. USA 2019, 116, 19600–19608. [Google Scholar] [CrossRef] [PubMed]
- Okada, E.; Daimon, K.; Fujiwara, H.; Nishiwaki, Y.; Nojiri, K.; Watanabe, M.; Katoh, H.; Shimizu, K.; Ishihama, H.; Fujita, N.; et al. Twenty-Year Longitudinal Follow-up MRI Study of Asymptomatic Volunteers: The Impact of Cervical Alignment on Disk Degeneration. Clin. Spine Surg. A Spine Publ. 2018, 31, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Holoshitz, J. The Rheumatoid Arthritis HLA–DRB1 Shared Epitope. Curr. Opin. Rheumatol. 2010, 22, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Massey, J.; Bowes, J.; Duffus, K.; arcOGEN Consortium; Eyre, S.; Barton, A.; Worthington, J. Replication of Associations of Genetic Loci Outside the HLA Region with Susceptibility to Anti-Cyclic Citrullinated Peptide-Negative Rheumatoid Arthritis. Arthritis Rheumatol. 2016, 68, 1603–1613. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Bowes, J.; Lunt, M.; Eyre, S.; Barton, A.; Worthington, J. Genetic Markers of Rheumatoid Arthritis Susceptibility in Anti-Citrullinated Peptide Antibody Negative Patients. Ann. Rheum. Dis. 2012, 71, 1984–1990. [Google Scholar] [CrossRef] [PubMed]
- Krabben, A.; Huizinga, T.W.J.; Mil, A.H.M. Biomarkers for Radiographic Progression in Rheumatoid Arthritis. Curr. Pharm. Des. 2014, 21, 147–169. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.C.; Espéli, M.; Anderson, C.A.; Linterman, M.A.; Pocock, J.M.; Williams, N.J.; Roberts, R.; Viatte, S.; Fu, B.; Peshu, N.; et al. Human SNP Links Differential Outcomes in Inflammatory and Infectious Disease to a FOXO3-Regulated Pathway. Cell 2013, 155, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.-S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five Amino Acids in Three HLA Proteins Explain Most of the Association between MHC and Seropositive Rheumatoid Arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; González-Gay, M.A.; Rantapää-Dahlqvist, S.; et al. Fine Mapping Seronegative and Seropositive Rheumatoid Arthritis to Shared and Distinct HLA Alleles by Adjusting for the Effects of Heterogeneity. Am. J. Hum. Genet. 2014, 94, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Ishigaki, K.; Sakaue, S.; Terao, C.; Luo, Y.; Sonehara, K.; Yamaguchi, K.; Amariuta, T.; Too, C.L.; Laufer, V.A.; Scott, I.C.; et al. Multi-Ancestry Genome-Wide Association Analyses Identify Novel Genetic Mechanisms in Rheumatoid Arthritis. Nat. Genet. 2022, 54, 1640–1651. [Google Scholar] [CrossRef] [PubMed]
- Hill, R.J.; Zozulya, S.; Lu, Y.-L.; Ward, K.; Gishizky, M.; Jallal, B. The Lymphoid Protein Tyrosine Phosphatase Lyp Interacts with the Adaptor Molecule Grb2 and Functions as a Negative Regulator of T-Cell Activation. Exp. Hematol. 2002, 30, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; Peterson, E.J. Tyrosine Phosphatase PTPN22: Multifunctional Regulator of Immune Signaling, Development, and Disease. Annu. Rev. Immunol. 2014, 32, 83–119. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Liu, W.H.; Delacroix, L.; Wu, S.; Vasile, S.; Dahl, R.; Yang, L.; Musumeci, L.; Francis, D.; Landskron, J.; et al. LYP Inhibits T-Cell Activation When Dissociated from CSK. Nat. Chem. Biol. 2012, 8, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Arechiga, A.F.; Habib, T.; He, Y.; Zhang, X.; Zhang, Z.-Y.; Funk, A.; Buckner, J.H. Cutting Edge: The PTPN22 Allelic Variant Associated with Autoimmunity Impairs B Cell Signaling. J. Immunol. 2009, 182, 3343–3347. [Google Scholar] [CrossRef] [PubMed]
- Gardette, A.; Marzaioli, V.; Bedouhene, S.; Hayem, G.; Hurtado-Nedelec, M.; He, Y.; Dang, P.M.-C.; Dieudé, P.; Zhang, Z.-Y.; Marie, J.-C.; et al. The Protein Tyrosine Phosphatase Lyp/PTPN22 Drives TNFα-Induced Priming of Superoxide Anions Production by Neutrophils and Arthritis. Free Radic. Biol. Med. 2025, 228, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A. Epigenetic Changes in the Pathogenesis of Rheumatoid Arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, D.; Teng, D.; Zhou, Y.; Zhang, L.; Zhong, Z.; Yang, G.-J. Epigenetic Regulation in the Pathogenesis of Rheumatoid Arthritis. Front. Immunol. 2022, 13, 859400. [Google Scholar] [CrossRef] [PubMed]
- Karouzakis, E.; Trenkmann, M.; Gay, R.E.; Michel, B.A.; Gay, S.; Neidhart, M. Epigenome Analysis Reveals TBX5 as a Novel Transcription Factor Involved in the Activation of Rheumatoid Arthritis Synovial Fibroblasts. J. Immunol. 2014, 193, 4945–4951. [Google Scholar] [CrossRef] [PubMed]
- Li, X.-F.; Wu, S.; Yan, Q.; Wu, Y.-Y.; Chen, H.; Yin, S.-Q.; Chen, X.; Wang, H.; Li, J. PTEN Methylation Promotes Inflammation and Activation of Fibroblast-Like Synoviocytes in Rheumatoid Arthritis. Front. Pharmacol. 2021, 12, 700373. [Google Scholar] [CrossRef] [PubMed]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA Methylome Signature in Rheumatoid Arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Liebold, I.; Grützkau, A.; Göckeritz, A.; Gerl, V.; Lindquist, R.; Feist, E.; Zänker, M.; Häupl, T.; Poddubnyy, D.; Zernicke, J.; et al. Peripheral Blood Mononuclear Cells Are Hypomethylated in Active Rheumatoid Arthritis and Methylation Correlates with Disease Activity. Rheumatology 2021, 60, 1984–1995. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wu, L.-F.; Mo, X.-B.; Lu, X.; Tang, H.; Zhu, X.-W.; Xia, W.; Guo, Y.-F.; Wang, M.-J.; Zeng, K.-Q.; et al. Rheumatoid Arthritis–Associated DNA Methylation Sites in Peripheral Blood Mononuclear Cells. Ann. Rheum. Dis. 2019, 78, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Julià, A.; Absher, D.; López-Lasanta, M.; Palau, N.; Pluma, A.; Waite Jones, L.; Glossop, J.R.; Farrell, W.E.; Myers, R.M.; Marsal, S. Epigenome-Wide Association Study of Rheumatoid Arthritis Identifies Differentially Methylated Loci in B Cells. Hum. Mol. Genet. 2017, 26, 2803–2811. [Google Scholar] [CrossRef] [PubMed]
- Lev Maor, G.; Yearim, A.; Ast, G. The Alternative Role of DNA Methylation in Splicing Regulation. Trends Genet. 2015, 31, 274–280. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, A.P.; Kennedy, A.; Penn, H.; Read, J.E.; Amjadi, P.; Green, P.; Syed, K.; Manka, S.W.; Brennan, F.M.; Gregory, B.; et al. Treg Cell Function in Rheumatoid Arthritis Is Compromised by CTLA-4 Promoter Methylation Resulting in a Failure to Activate the Indoleamine 2,3-Dioxygenase Pathway. Arthritis Rheumatol. 2014, 66, 2344–2354. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, A.; Feldmann, M.; Oppermann, U. Towards an Understanding of the Role of DNA Methylation in Rheumatoid Arthritis: Therapeutic and Diagnostic Implications. Ther. Adv. Musculoskelet. 2015, 7, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, M.; Lv, X.; Song, L.; Zhang, D.; He, Y.; Wang, M.; Zhao, X.; Yuan, X.; Shi, G.; et al. Reduced Activity of HDAC3 and Increased Acetylation of Histones H3 in Peripheral Blood Mononuclear Cells of Patients with Rheumatoid Arthritis. J. Immunol. Res. 2018, 2018, 7313515. [Google Scholar] [CrossRef] [PubMed]
- Göschl, L.; Preglej, T.; Boucheron, N.; Saferding, V.; Müller, L.; Platzer, A.; Hirahara, K.; Shih, H.-Y.; Backlund, J.; Matthias, P.; et al. Histone Deacetylase 1 (HDAC1): A Key Player of T Cell-Mediated Arthritis. J. Autoimmun. 2020, 108, 102379. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Lee, S.W.; Lee, S.Y.; Hong, K.W.; Bae, S.S.; Kim, K.; Kim, C.D. SIRT1/Adenosine Monophosphate-Activated Protein Kinase α Signaling Enhances Macrophage Polarization to an Anti-Inflammatory Phenotype in Rheumatoid Arthritis. Front. Immunol. 2017, 8, 1135. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Hu, W.; Wang, R.; Li, Z.; Yu, Y.; Zhuo, Y.; Zhang, Y.; Wang, Z.; Qiu, Y.; Chen, K.; et al. Sp1 S-Sulfhydration Induced by Hydrogen Sulfide Inhibits Inflammation via HDAC6/MyD88/NF-κB Signaling Pathway in Adjuvant-Induced Arthritis. Antioxidants 2022, 11, 732. [Google Scholar] [CrossRef] [PubMed]
- Mu, N.; Gu, J.; Huang, T.; Zhang, C.; Shu, Z.; Li, M.; Hao, Q.; Li, W.; Zhang, W.; Zhao, J.; et al. A Novel NF-κB/YY1/microRNA-10a Regulatory Circuit in Fibroblast-like Synoviocytes Regulates Inflammation in Rheumatoid Arthritis. Sci. Rep. 2016, 6, 20059. [Google Scholar] [CrossRef] [PubMed]
- Stanczyk, J.; Ospelt, C.; Karouzakis, E.; Filer, A.; Raza, K.; Kolling, C.; Gay, R.; Buckley, C.D.; Tak, P.P.; Gay, S.; et al. Altered Expression of microRNA-203 in Rheumatoid Arthritis Synovial Fibroblasts and Its Role in Fibroblast Activation. Arthritis Rheum. 2011, 63, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Gantier, M.P.; Stunden, H.J.; McCoy, C.E.; Behlke, M.A.; Wang, D.; Kaparakis-Liaskos, M.; Sarvestani, S.T.; Yang, Y.H.; Xu, D.; Corr, S.C.; et al. A miR-19 Regulon That Controls NF-κB Signaling. Nucleic Acids Res. 2012, 40, 8048–8058. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Demoruelle, M.K.; Kuhn, K.A.; Buckner, J.H.; Robinson, W.H.; Okamoto, Y.; Norris, J.M.; Deane, K.D. Rheumatoid Arthritis and the Mucosal Origins Hypothesis: Protection Turns to Destruction. Nat. Rev. Rheumatol. 2018, 14, 542–557. [Google Scholar] [CrossRef] [PubMed]
- Willis, V.C.; Demoruelle, M.K.; Derber, L.A.; Chartier-Logan, C.J.; Parish, M.C.; Pedraza, I.F.; Weisman, M.H.; Norris, J.M.; Holers, V.M.; Deane, K.D. Sputum Autoantibodies in Patients with Established Rheumatoid Arthritis and Subjects at Risk of Future Clinically Apparent Disease. Arthritis Rheum. 2013, 65, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Bowers, E.; Lahey, L.J.; Sokolove, J.; Purmalek, M.; Seto, N.L.; Weisman, M.H.; Norris, J.M.; Kaplan, M.J.; Holers, V.M.; et al. Antibody Responses to Citrullinated and Noncitrullinated Antigens in the Sputum of Subjects With Rheumatoid Arthritis and Subjects at Risk for Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Reynisdottir, G.; Karimi, R.; Joshua, V.; Olsen, H.; Hensvold, A.H.; Harju, A.; Engström, M.; Grunewald, J.; Nyren, S.; Eklund, A.; et al. Structural Changes and Antibody Enrichment in the Lungs Are Early Features of Anti-Citrullinated Protein Antibody-Positive Rheumatoid Arthritis. Arthritis Rheumatol. 2014, 66, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A Gene-Environment Interaction between Smoking and Shared Epitope Genes in HLA-DR Provides a High Risk of Seropositive Rheumatoid Arthritis. Arthritis Rheum. 2004, 50, 3085–3092. [Google Scholar] [CrossRef] [PubMed]
- Linn-Rasker, S.P.; Van Der Helm-van Mil, A.H.M.; Van Gaalen, F.A.; Kloppenburg, M.; De Vries, R.R.P.; Le Cessie, S.; Breedveld, F.C.; Toes, R.E.M.; Huizinga, T.W.J. Smoking Is a Risk Factor for Anti-CCP Antibodies Only in Rheumatoid Arthritis Patients Who Carry HLA-DRB1 Shared Epitope Alleles. Ann. Rheum. Dis. 2006, 65, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Källberg, H.; Ding, B.; Padyukov, L.; Bengtsson, C.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Smoking Is a Major Preventable Risk Factor for Rheumatoid Arthritis: Estimations of Risks after Various Exposures to Cigarette Smoke. Ann. Rheum. Dis. 2011, 70, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Klareskog, L.; Alfredsson, L. Exposure to Passive Smoking and Rheumatoid Arthritis Risk: Results from the Swedish EIRA Study. Ann. Rheum. Dis. 2018, 77, 970–972. [Google Scholar] [CrossRef] [PubMed]
- Svendsen, A.J.; Gervin, K.; Lyle, R.; Christiansen, L.; Kyvik, K.; Junker, P.; Nielsen, C.; Houen, G.; Tan, Q. Differentially Methylated DNA Regions in Monozygotic Twin Pairs Discordant for Rheumatoid Arthritis: An Epigenome-Wide Study. Front. Immunol. 2016, 7, 510. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, E. Smoking, Gender and Rheumatoid Arthritis–Epidemiological Clues to Etiology. Jt. Bone Spine 2003, 70, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Xie, W.; Wang, C.; Zhu, Y.; Zhong, D. Causal Relationships Between Environmental Exposures, Iron Metabolism, Hematuria Markers, and Rheumatoid Arthritis: An Investigation Using Mendelian Randomization. Biomedicines 2025, 13, 513. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Y.; Hu, S.; Wu, Y. Causal Relationships between Air Pollution and Common Autoimmune Diseases: A Two-Sample Mendelian Randomization Study. Sci. Rep. 2025, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-X.; Feng, B.-B.; Ruo-Wei, M.; Zhu, L.; Liu, Y.-Y.; Zuo, Y.-Y.; Pan, H.-F.; Wu, G.-C. Long-Term Air Pollution Exposure and Cardiovascular Disease Progression in Rheumatoid Arthritis: A Prospective Cohort Study Using Multi-State Model Analysis. Ecotoxicol. Environ. Saf. 2025, 296, 118187. [Google Scholar] [CrossRef] [PubMed]
- Alsaber, A.; Pan, J.; Al-Herz, A.; Alkandary, D.; Al-Hurban, A.; Setiya, P.; on Behalf of the Krrd Group. Influence of Ambient Air Pollution on Rheumatoid Arthritis Disease Activity Score Index. Int. J. Environ. Res. Public Health 2020, 17, 416. [Google Scholar] [CrossRef] [PubMed]
- Tu, L.; Wei, F.; Song, Y.; Huang, H.; Qing, L.; Luo, X.; Liu, Y.; Chen, H. Development and Internal Validation of a Prediction Model for Rheumatoid Arthritis: A Case-Control Study. Sci. Rep. 2025, 15, 16620. [Google Scholar] [CrossRef] [PubMed]
- Tschernig, T.; Pabst, R. Bronchus-Associated Lymphoid Tissue (BALT) Is Not Present in the Normal Adult Lung but in Different Diseases. Pathobiology 2000, 68, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Adami, G.; Viapiana, O.; Rossini, M.; Orsolini, G.; Bertoldo, E.; Giollo, A.; Gatti, D.; Fassio, A. Association between Environmental Air Pollution and Rheumatoid Arthritis Flares. Rheumatology 2021, 60, 4591–4597. [Google Scholar] [CrossRef] [PubMed]
- Too, C.L.; Muhamad, N.A.; Ilar, A.; Padyukov, L.; Alfredsson, L.; Klareskog, L.; Murad, S.; Bengtsson, C. Occupational Exposure to Textile Dust Increases the Risk of Rheumatoid Arthritis: Results from a Malaysian Population-Based Case–Control Study. Ann. Rheum. Dis. 2016, 75, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L. Silica Exposure among Male Current Smokers Is Associated with a High Risk of Developing ACPA-Positive Rheumatoid Arthritis. Ann. Rheum. Dis. 2010, 69, 1072–1076. [Google Scholar] [CrossRef] [PubMed]
- Blanc, P.; Andersson, L.; Bryngelsson, I.-L. Risk of Rheumatoid Arthritis in a Cohort of Silica-Exposed Swedish Foundry Workers. Eur. Respir. J. 2016, 48, 389. [Google Scholar]
- Mehri, F.; Jenabi, E.; Bashirian, S.; Shahna, F.G.; Khazaei, S. The Association Between Occupational Exposure to Silica and Risk of Developing Rheumatoid Arthritis: A Meta-Analysis. Saf. Health Work 2020, 11, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Rodolfi, S.; Selmi, C. Environmental Factors and Rheumatic Diseases. Best Pract. Res. Clin. Rheumatol. 2025, 39, 102053. [Google Scholar] [CrossRef] [PubMed]
- Alaya, Z.; Braham, M.; Aissa, S.; Kalboussi, H.; Bouajina, E. A Case of Caplan Syndrome in a Recently Diagnosed Patient with Silicosis: A Case Report. Radiol. Case Rep. 2018, 13, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Ilar, A.; Klareskog, L.; Saevarsdottir, S.; Wiebert, P.; Askling, J.; Gustavsson, P.; Alfredsson, L. Occupational Exposure to Asbestos and Silica and Risk of Developing Rheumatoid Arthritis: Findings from a Swedish Population-Based Case-Control Study. RMD Open 2019, 5, e000978. [Google Scholar] [CrossRef] [PubMed]
- Joo, S.H.; Lee, J.; Hutchinson, D.; Song, Y.W. Prevalence of Rheumatoid Arthritis in Relation to Serum Cadmium Concentrations: Cross-Sectional Study Using Korean National Health and Nutrition Examination Survey (KNHANES) Data. BMJ Open 2019, 9, e023233. [Google Scholar] [CrossRef] [PubMed]
- on Behalf of the EIRA Study Group; Johansson, K.; Askling, J.; Alfredsson, L.; Di Giuseppe, D. Mediterranean Diet and Risk of Rheumatoid Arthritis: A Population-Based Case-Control Study. Arthritis Res. Ther. 2018, 20, 175. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Li, J.; Gan, Y.; Liu, J.; Zhao, X.; Chen, J.; Zhang, R.; Zhong, Y.; Chen, X.; Wu, L.; et al. Red Meat Intake Is Associated with Early Onset of Rheumatoid Arthritis: A Cross-Sectional Study. Sci. Rep. 2021, 11, 5681. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.W.; Demoruelle, M.K.; Deane, K.D.; Weisman, M.H.; Buckner, J.H.; Gregersen, P.K.; Mikuls, T.R.; O’Dell, J.R.; Keating, R.M.; Fingerlin, T.E.; et al. Omega-3 Fatty Acids Are Associated with a Lower Prevalence of Autoantibodies in Shared Epitope-Positive Subjects at Risk for Rheumatoid Arthritis. Ann. Rheum. Dis. 2017, 76, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Gan, R.W.; Young, K.A.; Zerbe, G.O.; Demoruelle, M.K.; Weisman, M.H.; Buckner, J.H.; Gregersen, P.K.; Mikuls, T.R.; O’Dell, J.R.; Keating, R.M.; et al. Lower Omega-3 Fatty Acids Are Associated with the Presence of Anti-Cyclic Citrullinated Peptide Autoantibodies in a Population at Risk for Future Rheumatoid Arthritis: A Nested Case-Control Study. Rheumatology 2016, 55, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Costenbader, K.H.; Cook, N.R.; Lee, I.; Hahn, J.; Walter, J.; Bubes, V.; Kotler, G.; Yang, N.; Friedman, S.; Alexander, E.K.; et al. Vitamin D and Marine n-3 Fatty Acids for Autoimmune Disease Prevention: Outcomes Two Years After Completion of a Double-Blind, Placebo-Controlled Trial. Arthritis Rheumatol. 2024, 76, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Karlson, E.W.; Mandl, L.A.; Aweh, G.N.; Grodstein, F. Coffee Consumption and Risk of Rheumatoid Arthritis. Arthritis Rheum. 2003, 48, 3055–3060. [Google Scholar] [CrossRef] [PubMed]
- Pattison, D.J.; Symmons, D.P.M.; Lunt, M.; Welch, A.; Luben, R.; Bingham, S.A.; Khaw, K.; Day, N.E.; Silman, A.J. Dietary Risk Factors for the Development of Inflammatory Polyarthritis: Evidence for a Role of High Level of Red Meat Consumption. Arthritis Rheum. 2004, 50, 3804–3812. [Google Scholar] [CrossRef] [PubMed]
- DeChristopher, L.R.; Uribarri, J.; Tucker, K.L. Intake of High-Fructose Corn Syrup Sweetened Soft Drinks, Fruit Drinks and Apple Juice Is Associated with Prevalent Arthritis in US Adults, Aged 20–30 Years. Nutr. Diabetes 2016, 6, e199. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Costenbader, K.H.; Gao, X.; Al-Daabil, M.; Sparks, J.A.; Solomon, D.H.; Hu, F.B.; Karlson, E.W.; Lu, B. Sugar-Sweetened Soda Consumption and Risk of Developing Rheumatoid Arthritis in Women. Am. J. Clin. Nutr. 2014, 100, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Radu, A.-F.; Bungau, S.G. Management of Rheumatoid Arthritis: An Overview. Cells 2021, 10, 2857. [Google Scholar] [CrossRef] [PubMed]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans—Induced Hypercitrullination Links Periodontal Infection to Autoimmunity in Rheumatoid Arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef] [PubMed]
- Alpizar-Rodriguez, D.; Lesker, T.R.; Gronow, A.; Gilbert, B.; Raemy, E.; Lamacchia, C.; Gabay, C.; Finckh, A.; Strowig, T. Prevotella Copri in Individuals at Risk for Rheumatoid Arthritis. Ann. Rheum. Dis. 2019, 78, 590–593. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, D.; Jia, H.; Feng, Q.; Wang, D.; Liang, D.; Wu, X.; Li, J.; Tang, L.; Li, Y.; et al. The Oral and Gut Microbiomes Are Perturbed in Rheumatoid Arthritis and Partly Normalized after Treatment. Nat. Med. 2015, 21, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of Intestinal Prevotella Copri Correlates with Enhanced Susceptibility to Arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wright, K.; Davis, J.M.; Jeraldo, P.; Marietta, E.V.; Murray, J.; Nelson, H.; Matteson, E.L.; Taneja, V. An Expansion of Rare Lineage Intestinal Microbes Characterizes Rheumatoid Arthritis. Genome Med. 2016, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, C.; Malspeis, S.; Orellana, C.; Sparks, J.A.; Costenbader, K.H.; Karlson, E.W. Association Between Menopausal Factors and the Risk of Seronegative and Seropositive Rheumatoid Arthritis: Results from the Nurses’ Health Studies. Arthritis Care Res. 2017, 69, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- Beydoun, H.A.; el-Amin, R.; McNeal, M.; Perry, C.; Archer, D.F. Reproductive History and Postmenopausal Rheumatoid Arthritis among Women 60 Years or Older: Third National Health and Nutrition Examination Survey. Menopause 2013, 20, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Kobak, S.; Bes, C. An Autumn Tale: Geriatric Rheumatoid Arthritis. Ther. Adv. Musculoskelet. 2018, 10, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and Prevalence of Rheumatoid Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef] [PubMed]
- George, M.D.; Baker, J.F. The Obesity Epidemic and Consequences for Rheumatoid Arthritis Care. Curr. Rheumatol. Rep. 2016, 18, 6. [Google Scholar] [CrossRef] [PubMed]
- Alunno, A.; Carubbi, F.; Giacomelli, R.; Gerli, R. Cytokines in the Pathogenesis of Rheumatoid Arthritis: New Players and Therapeutic Targets. BMC Rheumatol. 2017, 1, 3. [Google Scholar] [CrossRef] [PubMed]
- Ortona, E.; Pierdominici, M.; Maselli, A.; Veroni, C.; Aloisi, F.; Shoenfeld, Y. Sex-based differences in autoimmune diseases. Ann. Dell’istituto Super. Di Sanita 2016, 52, 205–212. [Google Scholar]
- Carmona, L.; Aurrecoechea, E.; García De Yébenes, M.J. Tailoring Rheumatoid Arthritis Treatment through a Sex and Gender Lens. J. Clin. Med. 2023, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Syrett, C.M.; Anguera, M.C. When the Balance Is Broken: X-Linked Gene Dosage from Two X Chromosomes and Female-Biased Autoimmunity. J. Leukoc. Biol. 2019, 106, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Cooney, C.M.; Bruner, G.R.; Aberle, T.; Namjou-Khales, B.; Myers, L.K.; Feo, L.; Li, S.; D’Souza, A.; Ramirez, A.; Harley, J.B.; et al. 46,X,Del(X)(Q13) Turner’s Syndrome Women with Systemic Lupus Erythematosus in a Pedigree Multiplex for SLE. Genes. Immun. 2009, 10, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Sawalha, A.H.; Harley, J.B.; Scofield, R.H. Autoimmunity and Klinefelter’s Syndrome: When Men Have Two X Chromosomes. J. Autoimmun. 2009, 33, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Invernizzi, P.; Miozzo, M.; Oertelt-Prigione, S.; Meroni, P.L.; Persani, L.; Selmi, C.; Battezzati, P.M.; Zuin, M.; Lucchi, S.; Marasini, B.; et al. X Monosomy in Female Systemic Lupus Erythematosus. Ann. N. Y. Acad. Sci. 2007, 1110, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Scofield, R.H.; Bruner, G.R.; Namjou, B.; Kimberly, R.P.; Ramsey-Goldman, R.; Petri, M.; Reveille, J.D.; Alarcón, G.S.; Vilá, L.M.; Reid, J.; et al. Klinefelter’s Syndrome (47,XXY) in Male Systemic Lupus Erythematosus Patients: Support for the Notion of a Gene-dose Effect from the X Chromosome. Arthritis Rheum. 2008, 58, 2511–2517. [Google Scholar] [CrossRef] [PubMed]
- Harris, V.M.; Sharma, R.; Cavett, J.; Kurien, B.T.; Liu, K.; Koelsch, K.A.; Rasmussen, A.; Radfar, L.; Lewis, D.; Stone, D.U.; et al. Klinefelter’s Syndrome (47,XXY) Is in Excess among Men with Sjögren’s Syndrome. Clin. Immunol. 2016, 168, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Panimolle, F.; Tiberti, C.; Spaziani, M.; Riitano, G.; Lucania, G.; Anzuini, A.; Lenzi, A.; Gianfrilli, D.; Sorice, M.; Radicioni, A.F. Non-Organ-Specific Autoimmunity in Adult 47,XXY Klinefelter Patients and Higher-Grade X-Chromosome Aneuploidies. Clin. Exp. Immunol. 2021, 205, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Kurien, B.T.; Zimmerman, S.L.; Kaufman, K.M.; Taft, D.H.; Kottyan, L.C.; Lazaro, S.; Weaver, C.A.; Ice, J.A.; Adler, A.J.; et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjögren’s Syndrome. Arthritis Rheumatol. 2016, 68, 1290–1300. [Google Scholar] [CrossRef] [PubMed]
- Buckland, J. Autophagy: A Dual Role in the Life and Death of RASFs. Nat. Rev. Rheumatol. 2013, 9, 637. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N. Autophagy: Process and Function. Genes Dev. 2007, 21, 2861–2873. [Google Scholar] [CrossRef] [PubMed]
- Baehrecke, E.H. Autophagy: Dual Roles in Life and Death? Nat. Rev. Mol. Cell Biol. 2005, 6, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Karami, J.; Masoumi, M.; Khorramdelazad, H.; Bashiri, H.; Darvishi, P.; Sereshki, H.A.; Shekarabi, M.; Sahebkar, A. Role of Autophagy in the Pathogenesis of Rheumatoid Arthritis: Latest Evidence and Therapeutic Approaches. Life Sci. 2020, 254, 117734. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Barrera-Vargas, A.; Sandoval-Heglund, D.; Merayo-Chalico, J.; Aguirre-Aguilar, E.; Aponte, A.M.; Ruiz-Perdomo, Y.; Gucek, M.; El-Gabalawy, H.; Fox, D.A.; et al. Neutrophil-Mediated Carbamylation Promotes Articular Damage in Rheumatoid Arthritis. Sci. Adv. 2020, 6, eabd2688. [Google Scholar] [CrossRef] [PubMed]
- Manganelli, V.; Recalchi, S.; Capozzi, A.; Riitano, G.; Mattei, V.; Longo, A.; Di Franco, M.; Alessandri, C.; Bombardieri, M.; Valesini, G.; et al. Autophagy Induces Protein Carbamylation in Fibroblast-like Synoviocytes from Patients with Rheumatoid Arthritis. Rheumatology 2018, 57, 2032–2041. [Google Scholar] [CrossRef] [PubMed]
- Mydel, P.; Wang, Z.; Brisslert, M.; Hellvard, A.; Dahlberg, L.E.; Hazen, S.L.; Bokarewa, M. Carbamylation-Dependent Activation of T Cells: A Novel Mechanism in the Pathogenesis of Autoimmune Arthritis. J. Immunol. 2010, 184, 6882–6890. [Google Scholar] [CrossRef] [PubMed]
- Bánréti, Á.; Sass, M.; Graba, Y. The Emerging Role of Acetylation in the Regulation of Autophagy. Autophagy 2013, 9, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Guo, H. Acetylation Modification in the Regulation of Macroautophagy. Adv. Biotechnol. 2024, 2, 19. [Google Scholar] [CrossRef] [PubMed]
- Ireland, J.M.; Unanue, E.R. Autophagy in Antigen-Presenting Cells Results in Presentation of Citrullinated Peptides to CD4 T Cells. J. Exp. Med. 2011, 208, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yamada, R.; Yamamoto, K. Citrullination by Peptidylarginine Deiminase in Rheumatoid Arthritis. Ann. New York Acad. Sci. 2007, 1108, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Ireland, J.M.; Unanue, E.R. Processing of Proteins in Autophagy Vesicles of Antigen-Presenting Cells Generates Citrullinated Peptides Recognized by the Immune System. Autophagy 2012, 8, 429–430. [Google Scholar] [CrossRef] [PubMed]
- Matarrese, P.; Garofalo, T.; Manganelli, V.; Gambardella, L.; Marconi, M.; Grasso, M.; Tinari, A.; Misasi, R.; Malorni, W.; Sorice, M. Evidence for the Involvement of GD3 Ganglioside in Autophagosome Formation and Maturation. Autophagy 2014, 10, 750–765. [Google Scholar] [CrossRef] [PubMed]
- Sorice, M.; Iannuccelli, C.; Manganelli, V.; Capozzi, A.; Alessandri, C.; Lococo, E.; Garofalo, T.; Di Franco, M.; Bombardieri, M.; Nerviani, A.; et al. Autophagy Generates Citrullinated Peptides in Human Synoviocytes: A Possible Trigger for Anti-Citrullinated Peptide Antibodies. Rheumatology 2016, 55, 1374–1385. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.-T.; Hong, F.-F.; Yang, S.-L. Roles of Autophagy in Rheumatoid Arthritis. Clin. Exp. Rheumatol. 2022, 40, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Hao, Z.; Liu, Y. IL-38 and IL-36 Target Autophagy for Regulating Synoviocyte Proliferation, Migration, and Invasion in Rheumatoid Arthritis. Dis. Markers 2021, 2021, 7933453. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Deng, Z.; Ma, Y.; Jin, J.; Qi, F.; Li, S.; Liu, C.; Lyu, F.-J.; Zheng, Q. The Role of Autophagy and Mitophagy in Bone Metabolic Disorders. Int. J. Biol. Sci. 2020, 16, 2675–2691. [Google Scholar] [CrossRef] [PubMed]
- Sellam, J.; Proulle, V.; Jüngel, A.; Ittah, M.; Miceli Richard, C.; Gottenberg, J.-E.; Toti, F.; Benessiano, J.; Gay, S.; Freyssinet, J.-M.; et al. Increased Levels of Circulating Microparticles in Primary Sjögren’s Syndrome, Systemic Lupus Erythematosus and Rheumatoid Arthritis and Relation with Disease Activity. Arthritis Res. Ther. 2009, 11, R156. [Google Scholar] [CrossRef] [PubMed]
- Arntz, O.J.; Pieters, B.C.H.; Thurlings, R.M.; Wenink, M.H.; Van Lent, P.L.E.M.; Koenders, M.I.; Van Den Hoogen, F.H.J.; Van Der Kraan, P.M.; Van De Loo, F.A.J. Rheumatoid Arthritis Patients with Circulating Extracellular Vesicles Positive for IgM Rheumatoid Factor Have Higher Disease Activity. Front. Immunol. 2018, 9, 2388. [Google Scholar] [CrossRef] [PubMed]
- Skriner, K.; Adolph, K.; Jungblut, P.R.; Burmester, G.R. Association of Citrullinated Proteins with Synovial Exosomes. Arthritis Rheum. 2006, 54, 3809–3814. [Google Scholar] [CrossRef] [PubMed]
- Ucci, F.M.; Recalchi, S.; Barbati, C.; Manganelli, V.; Capozzi, A.; Riitano, G.; Buoncuore, G.; Garofalo, T.; Ceccarelli, F.; Spinelli, F.R.; et al. Citrullinated and Carbamylated Proteins in Extracellular Microvesicles from Plasma of Patients with Rheumatoid Arthritis. Rheumatology 2023, 62, 2312–2319. [Google Scholar] [CrossRef] [PubMed]
- Buttari, B.; Recalchi, S.; Riitano, G.; Capozzi, A.; Ucci, F.M.; Manganelli, V.; Fratini, F.; Profumo, E.; Garofalo, T.; Alessandri, C.; et al. Extracellular Microvesicles from Patients with Rheumatoid Arthritis Promote Dendritic Cell Activation in Vitro. Front. Immunol. 2025, 16, 1532114. [Google Scholar] [CrossRef] [PubMed]
- Song, J.E.; Kim, J.S.; Shin, J.H.; Moon, K.W.; Park, J.K.; Park, K.S.; Lee, E.Y. Role of Synovial Exosomes in Osteoclast Differentiation in Inflammatory Arthritis. Cells 2021, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-G.; Liu, C.; Su, K.; Yu, S.; Zhang, L.; Zhang, S.; Wang, J.; Cao, X.; Grizzle, W.; Kimberly, R.P. A Membrane Form of TNF-α Presented by Exosomes Delays T Cell Activation-Induced Cell Death. J. Immunol. 2006, 176, 7385–7393. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; Gremese, E.; McSharry, C.; Tolusso, B.; Ferraccioli, G.; McInnes, I.B.; Kurowska-Stolarska, M. MicroRNA-155—At the Critical Interface of Innate and Adaptive Immunity in Arthritis. Front. Immunol. 2018, 8, 1932. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Xue, W.; Wu, Z.; Lu, D.; Zheng, L.; Zhou, M.; Li, Y.; Wang, Y.; Liu, T. Quercetin, a Compound of the Total Flavonoids of Periploca Forrestii Schltr., Ameliorates Rheumatoid Arthritis by Targeting TNF-α. J. Inflamm. Res. 2025, 18, 2879–2898. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Wang, J.; Wang, Y.; Zhu, Z.; Zhang, S.; Wu, B.; Meng, M.; Zhao, J.; Wang, D. Sesquiterpene Lactones-Enriched Fractions from Xanthium Mongolicum Kitag Alleviate RA by Regulating M1 Macrophage Polarization via NF-κB and MAPK Signaling Pathway. Front. Pharmacol. 2023, 14, 1104153. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Feng, Q.; Cui, M.; Fan, C.; Wang, T.; Yuan, R.; Tsering, D.; Huang, S.; Li, B. Siweixizangmaoru Decoction Ameliorated Type II Collagen-Induced Arthritis in Rats via Regulating JAK2–STAT3 and NF-κB Signaling Pathway. Biol. Pharm. Bull. 2024, 47, 1511–1524. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.; Yang, Y.; Lu, H.; Shi, H.; Jiang, L.; Liao, X.; Zhao, H.; Wang, W.; Liu, J. Network Pharmacology Combines Machine Learning, Molecular Simulation Dynamics and Experimental Validation to Explore the Mechanism of Acetylbinankadsurin A in the Treatment of Liver Fibrosis. J. Ethnopharmacol. 2024, 323, 117682. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Cheng, M.-H.; Hu, L.-Q.; Shen, H.-H.; Tao, J.-H.; Li, X.-M.; Pan, H.-F.; Gao, J. Natural Products Action on Pathogenic Cues in Autoimmunity: Efficacy in Systemic Lupus Erythematosus and Rheumatoid Arthritis as Compared to Classical Treatments. Pharmacol. Res. 2020, 160, 105054. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Fan, X.; Qu, Y.; Tang, M.; Huang, Y.; Peng, Y.; Fu, Q. Magnoflorine Attenuates Inflammatory Responses in RA by Regulating the PI3K/Akt/NF-κB and Keap1-Nrf2/HO-1 Signalling Pathways in Vivo and in Vitro. Phytomedicine 2022, 104, 154339. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Liu, M.; Tang, Q.; Sun, H.; Yang, G.; Sun, J. Anti-Rheumatic Arthritis Efficacy of Pueraria Montana Extract against Type-II Collagen-Induced Rheumatoid Arthritis Rat Model an in Vitro and in Vivo Assessment. J. Ethnopharmacol. 2025, 340, 119175. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.; Breedveld, F.C.; Buch, M.; Burmester, G.; Dougados, M.; Emery, P.; Gaujoux-Viala, C.; Gossec, L.; Nam, J.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2013 Update. Ann. Rheum. Dis. 2014, 73, 492–509. [Google Scholar] [CrossRef] [PubMed]
- Rao, D.A.; Gurish, M.F.; Marshall, J.L.; Slowikowski, K.; Fonseka, C.Y.; Liu, Y.; Donlin, L.T.; Henderson, L.A.; Wei, K.; Mizoguchi, F.; et al. Pathologically Expanded Peripheral T Helper Cell Subset Drives B Cells in Rheumatoid Arthritis. Nature 2017, 542, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Wei, K.; Slowikowski, K.; Fonseka, C.Y.; Rao, D.A.; Kelly, S.; Goodman, S.M.; Tabechian, D.; Hughes, L.B.; Salomon-Escoto, K.; et al. Defining Inflammatory Cell States in Rheumatoid Arthritis Joint Synovial Tissues by Integrating Single-Cell Transcriptomics and Mass Cytometry. Nat. Immunol. 2019, 20, 928–942. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, L.J.; Kaplan, M.J. Neutrophils in Rheumatoid Arthritis: Breaking Immune Tolerance and Fueling Disease. Trends Mol. Med. 2019, 25, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Rivellese, F.; Mauro, D.; Nerviani, A.; Pagani, S.; Fossati-Jimack, L.; Messemaker, T.; Kurreeman, F.A.S.; Toes, R.E.M.; Ramming, A.; Rauber, S.; et al. Mast Cells in Early Rheumatoid Arthritis Associate with Disease Severity and Support B Cell Autoantibody Production. Ann. Rheum. Dis. 2018, 77, 1773–1781. [Google Scholar] [CrossRef] [PubMed]
- Schubert, N.; Dudeck, J.; Liu, P.; Karutz, A.; Speier, S.; Maurer, M.; Tuckermann, J.; Dudeck, A. Mast Cell Promotion of T Cell–Driven Antigen-Induced Arthritis Despite Being Dispensable for Antibody-Induced Arthritis in Which T Cells Are Bypassed. Arthritis Rheumatol. 2015, 67, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Xiong, H.; Meng, F.; Luo, M.; Chen, W.; Tian, J.; Chen, L.; Ju, Y.; Mei, Z. Anti-Inflammatory and Osteoprotective Effects of Shi-Wei-Ru-Xiang Pills on Collagen-Induced Arthritis in Rats via Inhibiting MAPK and STAT3 Pathways. J. Ethnopharmacol. 2023, 300, 115693. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Kondo, N.; Kuroda, T.; Kobayashi, D. Cytokine Networks in the Pathogenesis of Rheumatoid Arthritis. Int. J. Mol. Sci. 2021, 22, 10922. [Google Scholar] [CrossRef] [PubMed]
- Cope, A.P. T Cells in Rheumatoid Arthritis. Arthritis Res. Ther. 2008, 10, S1. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.W.; Lee, K.W.; Park, H.; Kim, H.; Lee, J.H.; Song, J.W.; Yang, J.; Kwon, Y.; Kim, T.M.; Park, J.B.; et al. Therapeutic Effects of Anti-CD154 Antibody in Cynomolgus Monkeys with Advanced Rheumatoid Arthritis. Sci. Rep. 2018, 8, 2135. [Google Scholar] [CrossRef] [PubMed]
- Cho, B.-A.; Sim, J.H.; Park, J.A.; Kim, H.W.; Yoo, W.-H.; Lee, S.-H.; Lee, D.-S.; Kang, J.S.; Hwang, Y.-I.; Lee, W.J.; et al. Characterization of Effector Memory CD8+ T Cells in the Synovial Fluid of Rheumatoid Arthritis. J. Clin. Immunol. 2012, 32, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.H.; Zhang, F.; Dunlap, G.; Gomez-Rivas, E.; Watts, G.F.M.; Faust, H.J.; Rupani, K.V.; Mears, J.R.; Meednu, N.; Wang, R.; et al. Granzyme K+ CD8 T Cells Form a Core Population in Inflamed Human Tissue. Sci. Transl. Med. 2022, 14, eabo0686. [Google Scholar] [CrossRef] [PubMed]
- Leipe, J.; Grunke, M.; Dechant, C.; Reindl, C.; Kerzendorf, U.; Schulze-Koops, H.; Skapenko, A. Role of Th17 Cells in Human Autoimmune Arthritis. Arthritis Rheum. 2010, 62, 2876–2885. [Google Scholar] [CrossRef] [PubMed]
- Van Baarsen, L.G.; Lebre, M.C.; Van Der Coelen, D.; Aarrass, S.; Tang, M.W.; Ramwadhdoebe, T.H.; Gerlag, D.M.; Tak, P.P. Heterogeneous Expression Pattern of Interleukin 17A (IL-17A), IL-17F and Their Receptors in Synovium of Rheumatoid Arthritis, Psoriatic Arthritis and Osteoarthritis: Possible Explanation for Nonresponse to Anti-IL-17 Therapy? Arthritis Res. Ther. 2014, 16, 426. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yin, H.; Zhang, K.; Wang, T.; Yang, Y.; Liu, X.; Chang, X.; Zhang, M.; Yan, X.; Ren, Y.; et al. Effector T Helper Cell Populations Are Elevated in the Bone Marrow of Rheumatoid Arthritis Patients and Correlate with Disease Severity. Sci. Rep. 2017, 7, 4776. [Google Scholar] [CrossRef] [PubMed]
- Edavalath, S.; Singh, A.; Soni, N.; Mohindra, N.; Kumar, S.; Misra, R. Peripheral Blood T Helper Type 17 Frequency Shows an Inverse Correlation with Disease Activity and Magnetic Resonance Imaging-Based Osteitis and Erosions in Disease-Modifying Anti-Rheumatic Drug- and Steroid-Naive Established Rheumatoid Arthritis. Clin. Exp. Immunol. 2016, 186, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Maston, L.D.; Jones, D.T.; Giermakowska, W.; Howard, T.A.; Cannon, J.L.; Wang, W.; Wei, Y.; Xuan, W.; Resta, T.C.; Gonzalez Bosc, L.V. Central Role of T Helper 17 Cells in Chronic Hypoxia-Induced Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2017, 312, L609–L624. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.; Tong, J.; Yang, M.; et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients with Rheumatoid Arthritis. Clin. Dev. Immunol. 2012, 2012, 827480. [Google Scholar] [CrossRef] [PubMed]
- Kondo, Y.; Yokosawa, M.; Kaneko, S.; Furuyama, K.; Segawa, S.; Tsuboi, H.; Matsumoto, I.; Sumida, T. Review: Transcriptional Regulation of CD 4+ T Cell Differentiation in Experimentally Induced Arthritis and Rheumatoid Arthritis. Arthritis Rheumatol. 2018, 70, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Yang, G.; Liu, Q.; Wang, S.; Cui, D. Function and Role of Regulatory T Cells in Rheumatoid Arthritis. Front. Immunol. 2021, 12, 626193. [Google Scholar] [CrossRef] [PubMed]
- Ambarus, C.A.; Noordenbos, T.; De Hair, M.J.; Tak, P.P.; Baeten, D.L. Intimal Lining Layer Macrophages but Not Synovial Sublining Macrophages Display an IL-10 Polarized-like Phenotype in Chronic Synovitis. Arthritis Res. Ther. 2012, 14, R74. [Google Scholar] [CrossRef] [PubMed]
- Soler Palacios, B.; Estrada-Capetillo, L.; Izquierdo, E.; Criado, G.; Nieto, C.; Municio, C.; González-Alvaro, I.; Sánchez-Mateos, P.; Pablos, J.L.; Corbí, A.L.; et al. Macrophages from the Synovium of Active Rheumatoid Arthritis Exhibit an Activin A-dependent Pro-inflammatory Profile. J. Pathol. 2015, 235, 515–526. [Google Scholar] [CrossRef] [PubMed]
- Kuo, D.; Ding, J.; Cohn, I.S.; Zhang, F.; Wei, K.; Rao, D.A.; Rozo, C.; Sokhi, U.K.; Shanaj, S.; Oliver, D.J.; et al. HBEGF+ Macrophages in Rheumatoid Arthritis Induce Fibroblast Invasiveness. Sci. Transl. Med. 2019, 11, eaau8587. [Google Scholar] [CrossRef] [PubMed]
- Alivernini, S.; MacDonald, L.; Elmesmari, A.; Finlay, S.; Tolusso, B.; Gigante, M.R.; Petricca, L.; Di Mario, C.; Bui, L.; Perniola, S.; et al. Distinct Synovial Tissue Macrophage Subsets Regulate Inflammation and Remission in Rheumatoid Arthritis. Nat. Med. 2020, 26, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Ota, Y.; Niiro, H.; Ota, S.; Ueki, N.; Tsuzuki, H.; Nakayama, T.; Mishima, K.; Higashioka, K.; Jabbarzadeh-Tabrizi, S.; Mitoma, H.; et al. Generation Mechanism of RANKL+ Effector Memory B Cells: Relevance to the Pathogenesis of Rheumatoid Arthritis. Arthritis Res. Ther. 2016, 18, 67. [Google Scholar] [CrossRef] [PubMed]
- Humby, F.; Durez, P.; Buch, M.H.; Lewis, M.J.; Rizvi, H.; Rivellese, F.; Nerviani, A.; Giorli, G.; Mahto, A.; Montecucco, C.; et al. Rituximab versus Tocilizumab in Anti-TNF Inadequate Responder Patients with Rheumatoid Arthritis (R4RA): 16-Week Outcomes of a Stratified, Biopsy-Driven, Multicentre, Open-Label, Phase 4 Randomised Controlled Trial. Lancet 2021, 397, 305–317. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.; Lom, H.; Juarez, M.; Snow, M.; Buckley, C.D.; Filer, A.; Raza, K.; Scheel-Toellner, D. Expression of FcRL4 Defines a Pro-Inflammatory, RANKL-Producing B Cell Subset in Rheumatoid Arthritis. Ann. Rheum. Dis. 2015, 74, 928–935. [Google Scholar] [CrossRef] [PubMed]
- Mizoguchi, F.; Slowikowski, K.; Wei, K.; Marshall, J.L.; Rao, D.A.; Chang, S.K.; Nguyen, H.N.; Noss, E.H.; Turner, J.D.; Earp, B.E.; et al. Functionally Distinct Disease-Associated Fibroblast Subsets in Rheumatoid Arthritis. Nat. Commun. 2018, 9, 789. [Google Scholar] [CrossRef] [PubMed]
- Yin, G.; Li, Y.; Yang, M.; Cen, X.; Xie, Q. Pim-2/mTORC1 Pathway Shapes Inflammatory Capacity in Rheumatoid Arthritis Synovial Cells Exposed to Lipid Peroxidations. BioMed Res. Int. 2015, 2015, 240210. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Zhang, Y.; Liu, X. Rheumatoid Arthritis: Pathogenesis and Therapeutic Advances. MedComm 2024, 5, e509. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.-S.; Jeong, G.H.; Yoo, S.-A. The Use of Animal Models in Rheumatoid Arthritis Research. J. Yeungnam Med. Sci. 2023, 40, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.O.; Feldmann, M.; Maini, R.N. Cartilage Destruction and Bone Erosion in Arthritis: The Role of Tumour Necrosis Factor α. Ann. Rheum. Dis. 2000, 59, i75–i80. [Google Scholar] [CrossRef] [PubMed]
- Horai, R.; Saijo, S.; Tanioka, H.; Nakae, S.; Sudo, K.; Okahara, A.; Ikuse, T.; Asano, M.; Iwakura, Y. Development of Chronic Inflammatory Arthropathy Resembling Rheumatoid Arthritis in Interleukin 1 Receptor Antagonist–Deficient Mice. J. Exp. Med. 2000, 191, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Kannan, K.; Ortmann, R.A.; Kimpel, D. Animal Models of Rheumatoid Arthritis and Their Relevance to Human Disease. Pathophysiology 2005, 12, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Razawy, W.; Asmawidjaja, P.S.; Mus, A.; Salioska, N.; Davelaar, N.; Kops, N.; Oukka, M.; Alves, C.H.; Lubberts, E. CD4+ CCR6+ T Cells, but Not Γδ T Cells, Are Important for the IL-23R-dependent Progression of Antigen-induced Inflammatory Arthritis in Mice. Eur. J. Immunol. 2020, 50, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Giant, T.T.; Mikecz, K.; Bartlett, R.R.; Deák, F.; Thonar, E.J.-M.A.; Williams, J.M.; Mattar, T.; Kuettner, K.E.; Schleyerbach, R. Immunomodulation of Proteoglycan-Induced Progressive Polyarthritis by Lefluflomide. Immunopharmacology 1992, 23, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Bäcklund, J.; Vestberg, M.; Holmdahl, R. Collagen Type II (CII)-Specific Antibodies Induce Arthritis in the Absence of T or B Cells but the Arthritis Progression Is Enhanced by CII-Reactive T Cells. Arthritis Res. Ther. 2004, 6, R544. [Google Scholar] [CrossRef] [PubMed]
- Yamada, H. Adaptive Immunity in the Joint of Rheumatoid Arthritis. Immunol. Med. 2022, 45, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Fang, Q.; Wingbro Ågren, I.; Bejmo, Z.F. Aberrant Activation of Immune and Non-Immune Cells Contributes to Joint Inflammation and Bone Degradation in Rheumatoid Arthritis. Int. J. Mol. Sci. 2023, 24, 15883. [Google Scholar] [CrossRef] [PubMed]
- Takemura, S.; Klimiuk, P.A.; Braun, A.; Goronzy, J.J.; Weyand, C.M. T Cell Activation in Rheumatoid Synovium Is B Cell Dependent. J. Immunol. 2001, 167, 4710–4718. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, P.M.; Steiert, I.; Kötter, I.; Müller, C.A. B Cells Contribute to Heterogeneity of IL-17 Producing Cells in Rheumatoid Arthritis and Healthy Controls. PLoS ONE 2013, 8, e82580. [Google Scholar] [CrossRef] [PubMed]
- Aarvak, T.; Natvig, J.B. Cell-Cell Interactions in Synovitis: Antigen Presenting Cells and T Cell Interaction in Rheumatoid Arthritis. Arthritis Res. 2001, 3, 13–17. [Google Scholar] [CrossRef] [PubMed]
- Takeshita, M.; Suzuki, K.; Kondo, Y.; Morita, R.; Okuzono, Y.; Koga, K.; Kassai, Y.; Gamo, K.; Takiguchi, M.; Kurisu, R.; et al. Multi-Dimensional Analysis Identified Rheumatoid Arthritis-Driving Pathway in Human T Cell. Ann. Rheum. Dis. 2019, 78, 1346–1356. [Google Scholar] [CrossRef] [PubMed]
- Fresneda Alarcon, M.; McLaren, Z.; Wright, H.L. Neutrophils in the Pathogenesis of Rheumatoid Arthritis and Systemic Lupus Erythematosus: Same Foe Different M.O. Front. Immunol. 2021, 12, 649693. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, Y.; Wei, W. Emerging Role of Targeting Macrophages in Rheumatoid Arthritis: Focus on Polarization, Metabolism and Apoptosis. Cell Prolif. 2020, 53, e12854. [Google Scholar] [CrossRef] [PubMed]
- Zec, K.; Schonfeldova, B.; Ai, Z.; Van Grinsven, E.; Pirgova, G.; Eames, H.L.; Berthold, D.L.; Attar, M.; Compeer, E.B.; Arnon, T.I.; et al. Macrophages in the Synovial Lining Niche Initiate Neutrophil Recruitment and Articular Inflammation. J. Exp. Med. 2023, 220, e20220595. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Smolen, J.S. Diagnosis and Management of Rheumatoid Arthritis: A Review. JAMA 2018, 320, 1360. [Google Scholar] [CrossRef] [PubMed]
- Amarasekara, D.S.; Yun, H.; Kim, S.; Lee, N.; Kim, H.; Rho, J. Regulation of Osteoclast Differentiation by Cytokine Networks. Immune Netw. 2018, 18, e8. [Google Scholar] [CrossRef] [PubMed]
- Cabral-Pacheco, G.A.; Garza-Veloz, I.; Castruita-De La Rosa, C.; Ramirez-Acuña, J.M.; Perez-Romero, B.A.; Guerrero-Rodriguez, J.F.; Martinez-Avila, N.; Martinez-Fierro, M.L. The Roles of Matrix Metalloproteinases and Their Inhibitors in Human Diseases. Int. J. Mol. Sci. 2020, 21, 9739. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Wilkinson, D.; Bou-Gharios, G. Targeting Dysregulation of Metalloproteinase Activity in Osteoarthritis. Calcif. Tissue Int. 2021, 109, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Del Buono, A.; Oliva, F.; Osti, L.; Maffulli, N. Metalloproteases and Tendinopathy. Muscle Ligaments Tendons J. 2019, 03, 51. [Google Scholar] [CrossRef]
- Van Den Steen, P.E.; Proost, P.; Brand, D.D.; Kang, A.H.; Van Damme, J.; Opdenakker, G. Generation of Glycosylated Remnant Epitopes from Human Collagen Type II by Gelatinase B. Biochemistry 2004, 43, 10809–10816. [Google Scholar] [CrossRef] [PubMed]
- Takaishi, H.; Kimura, T.; Dalal, S.; Okada, Y.; D’Armiento, J. Joint Diseases and Matrix Metalloproteinases: A Role for MMP-13. Curr. Pharm. Biotechnol. 2008, 9, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Burrage, P.S.; Mix, K.S.; Brinckerhoff, C.E. Matrix Metalloproteinases: Role in Arthritis. Front. Biosci. 2006, 11, 529. [Google Scholar] [CrossRef] [PubMed]
- Montero-Melendez, T.; Nagano, A.; Chelala, C.; Filer, A.; Buckley, C.D.; Perretti, M. Therapeutic Senescence via GPCR Activation in Synovial Fibroblasts Facilitates Resolution of Arthritis. Nat. Commun. 2020, 11, 745. [Google Scholar] [CrossRef] [PubMed]
- Zeinert, I.; Schmidt, L.; Baar, T.; Gatto, G.; De Giuseppe, A.; Korb-Pap, A.; Pap, T.; Mahabir, E.; Zaucke, F.; Brachvogel, B.; et al. Matrix-Mediated Activation of Murine Fibroblast-like Synoviocytes. Exp. Cell Res. 2025, 445, 114408. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Xiang, Z.; Wang, Y.; Ren, Q.; Chen, G.; Xiang, B.; Wang, J.; Zhang, C.; Pei, S.; Guo, S.; et al. Immunomodulatory Roles of Metalloproteinases in Rheumatoid Arthritis. Front. Pharmacol. 2023, 14, 1285455. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lü, H.; Liu, X.; Deng, Y.; Sun, T.; Li, F.; Ji, S.; Nie, X.; Yao, L. Functional Analysis of Discoidin Domain Receptor 2 in Synovial Fibroblasts in Rheumatoid Arthritis. J. Autoimmun. 2002, 19, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hao, Z.; Lu, D.; Naseem, A.; Sun, Y.; Sun, Y.; Li, J.; Kuang, H.; Liu, Y.; Yang, B. Effects of Viscum Coloratum (Kom.) Nakai on Collagen-Induced Rheumatoid Arthritis. J. Ethnopharmacol. 2024, 327, 118026. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Zhang, J.; Feng, Z.; Ji, J.; Shen, X.; Hou, X.; Mei, Z. The Antiangiogenic Effect of Total Saponins of Panax Japonicus C.A. Meyer in Rheumatoid Arthritis Is Mediated by Targeting the HIF-1α/VEGF/ANG-1 Axis. J. Ethnopharmacol. 2024, 333, 118422. [Google Scholar] [CrossRef] [PubMed]
- Duan, Z.; Jin, C.; Deng, Y.; Liu, J.; Gu, C.; Wang, J.; Cai, X.; Li, S.; Zhou, Y. Exploring the Chondroprotective Effect of Chaenomeles Speciosa on Glucose-6-Phosphate Isomerase Model Mice Using an Integrated Approach of Network Pharmacology and Experimental Validation. J. Ethnopharmacol. 2023, 314, 116553. [Google Scholar] [CrossRef] [PubMed]
- Maybee, D.V.; Ink, N.L.; Ali, M.A.M. Novel Roles of MT1-MMP and MMP-2: Beyond the Extracellular Milieu. Int. J. Mol. Sci. 2022, 23, 9513. [Google Scholar] [CrossRef] [PubMed]
- Yeon, K.Y. Role of Activating Transcription Factor 3 as a Mediator of the Protective Effects of Berberine against Lipopolysaccharide-Stimulated SW982 Cells and in Rheumatoid Arthritis Animal Models. Toxicol. Appl. Pharmacol. 2025, 497, 117279. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cheng, H.; Fan, C.; Zhou, X.; Chen, W.; Xie, C.; Hu, Y.; Chen, Y.; Wang, X.; Wu, J. LAMP3-Mediated Epithelial-Mesenchymal Transition Promotes the Invasion and Excessive Proliferation of Fibroblast-like Synoviocytes in Rheumatoid Arthritis. J. Autoimmun. 2025, 151, 103359. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Gao, C.; Sun, X.; Zhao, Y.; Zhang, H. Study on the Mechanism of Action of Wu Mei Pill in Inhibiting Rheumatoid Arthritis through TLR4-NF-κB Pathway. J. Orthop. Surg. Res. 2024, 19, 65. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Tang, Y.; Du, Y.; Zhang, J.; Hu, F.; Zou, Y.; Li, Y.; Zhu, L.; He, J.; Guo, J.; et al. Leukocyte Ig-like Receptor A3 Facilitates Inflammation, Migration and Invasion of Synovial Tissue-Derived Fibroblasts via ERK/JNK Activation. Rheumatology 2024, 63, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chang, C.; Zhou, X.; Zhang, R.; Jiang, P.; Wei, K.; Xu, L.; Shi, Y.; Yang, G.; Lv, X.; et al. LncRNA NONHSAT042241 Inhibits Rheumatoid Synovial Proliferation, Inflammation and Aggression via Inactivating WNT/β-catenin signaling pathway. Autoimmunity 2024, 57, 2387076. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.-D.; Zhou, J.-J.; Zheng, D.-H.; Chen, L.-F.; Mo, Y.-Q.; Wei, X.; Yang, L.-J.; Dai, L. Serum Matrix Metalloproteinase-3 as a Noninvasive Biomarker of Histological Synovitis for Diagnosis of Rheumatoid Arthritis. Mediat. Inflamm. 2014, 2014, 179284. [Google Scholar] [CrossRef] [PubMed]
- Leeming, D.J.; He, Y.; Veidal, S.S.; Nguyen, Q.H.T.; Larsen, D.V.; Koizumi, M.; Segovia-Silvestre, T.; Zhang, C.; Zheng, Q.; Sun, S.; et al. A Novel Marker for Assessment of Liver Matrix Remodeling: An Enzyme-Linked Immunosorbent Assay (ELISA) Detecting a MMP Generated Type I Collagen Neo-Epitope (C1M). Biomarkers 2011, 16, 616–628. [Google Scholar] [CrossRef] [PubMed]
- Veidal, S.S.; Karsdal, M.A.; Vassiliadis, E.; Nawrocki, A.; Larsen, M.R.; Nguyen, Q.H.T.; Hägglund, P.; Luo, Y.; Zheng, Q.; Vainer, B.; et al. MMP Mediated Degradation of Type VI Collagen Is Highly Associated with Liver Fibrosis—Identification and Validation of a Novel Biochemical Marker Assay. PLoS ONE 2011, 6, e24753. [Google Scholar] [CrossRef] [PubMed]
- Zewail, M.; Gaafar, P.M.E.; Abbas, H.; Elsheikh, M.A. Innovative Rheumatoid Arthritis Management Using Injection Replacement Approach via Dual Therapeutic Effects of Hyalurosomes-Encapsulated Luteolin and Dexamethasone. Colloids Surf. B Biointerfaces 2025, 249, 114497. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Shi, J.; Niu, S.; Liu, Z.; Cui, X.; Song, Y.; Tang, X.; Fan, J.; Xu, H.; Yu, W.; et al. Genistein Alleviates Rheumatoid Arthritis by Inhibiting Fibroblast-like Synovial Exosome Secretion Regulated by the Rab27/nSMase2/Mfge8 Pathway. Food Funct. 2025, 16, 1407–1422. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Liu, Q.; You, X.; Yuan, J.; Xia, J.; Tan, Y.; Hu, Y.; Wang, Q. Investigating the Molecular Mechanism of Epimedium Herb in Treating Rheumatoid Arthritis through Network Pharmacology, Molecular Docking, and Experimental Validation. Mol. Divers. 2025, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Jia, N.; Gao, Y.; Yang, L.; Xu, Y.; Zhang, Z.; Wang, J.; Zhang, L. Precise Control of the in Vivo Fate of Nanomicelles Efficiently Treats Advanced Rheumatoid Arthritis via EGFR/JNK/MMP9 Pathway. Int. J. Nanomed. 2025, 20, 5353–5375. [Google Scholar] [CrossRef] [PubMed]
- Hambardzumyan, K.; Hamsten, C.; Lourido, L.; Saevarsdottir, S.; Nilsson, P.; Van Vollenhoven, R.F.; Jakobsson, P.-J.; Idborg, H. Association of Matrix Metalloproteinase 7 and the Alpha-Chain of Fibrinogen at Baseline with Response to Methotrexate at 3 Months in Patients with Early Rheumatoid Arthritis. BMC Rheumatol. 2025, 9, 56. [Google Scholar] [CrossRef] [PubMed]
- Pulito-Cueto, V.; Atienza-Mateo, B.; Batista-Liz, J.C.; Sebastián Mora-Gil, M.; Mora-Cuesta, V.M.; Iturbe-Fernández, D.; Izquierdo Cuervo, S.; Aguirre Portilla, C.; Blanco, R.; López-Mejías, R. Matrix Metalloproteinases and Their Tissue Inhibitors as Upcoming Biomarker Signatures of Connective Tissue Diseases-Related Interstitial Lung Disease: Towards an Earlier and Accurate Diagnosis. Mol. Med. 2025, 31, 70. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.-J.; Wei, X.-L.; Liu, H.-Y.; Li, H.; Xia, Y.; Wu, D.-T.; Zhang, P.-Z.; Gandhi, G.R.; Li, H.-B.; Gan, R.-Y. State-of-the-Art Review of Dark Tea: From Chemistry to Health Benefits. Trends Food Sci. Technol. 2021, 109, 126–138. [Google Scholar] [CrossRef]
- Kany, S.; Vollrath, J.T.; Relja, B. Cytokines in Inflammatory Disease. Int. J. Mol. Sci. 2019, 20, 6008. [Google Scholar] [CrossRef] [PubMed]
- Bruno, A.; Wang, G.; Chen, Y.; Zhang, Z. Angiogenesis Is Inhibited by Arsenic Trioxide Through Downregulation of the CircHIPK3/miR-149-5p/FOXO1/VEGF Functional Module in Rheumatoid Arthritis. Front. Pharmacol. 2021, 12, 751667. [Google Scholar]
- Lesturgie-Talarek, M.; Gonzalez, V.; Combier, A.; Thomas, M.; Boisson, M.; Poiroux, L.; Wanono, S.; Hecquet, S.; Carves, S.; Cauvet, A.; et al. Inflammatory and Angiogenic Serum Profile of Refractory Rheumatoid Arthritis. Sci. Rep. 2025, 15, 7159. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Wu, Y.; Zhou, X.; Cai, Y.; Li, H.; Xia, A.; Wang, X.; Wen, J.; Duan, Q.; Xu, C.; et al. Clematichinenoside AR Alleviates Rheumatoid Arthritis by Inhibiting Synovial Angiogenesis through the HIF-1α/VEGFA/ANG2 Axis. Phytomedicine 2025, 139, 156552. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Li, Y.; Shu, Y.; Gan, P.R.; Zhu, Y.L.; Xu, J.; Jiang, X.-M.; Xia, S.-L.; Wang, Y.; Wu, H. The New Anti-Angiogenesis Perspective of Rheumatoid Arthritis with Geniposide: Reducing the Extracellular Release of HSP70 in HUVECs. Int. Immunopharmacol. 2025, 144, 113645. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fan, D.; Cao, X.; Ye, Q.; Wang, Q.; Zhang, M.; Xiao, C. The Role of Reactive Oxygen Species in the Rheumatoid Arthritis-Associated Synovial Microenvironment. Antioxidants 2022, 11, 1153. [Google Scholar] [CrossRef] [PubMed]
- Biniecka, M.; Fox, E.; Gao, W.; Ng, C.T.; Veale, D.J.; Fearon, U.; O’Sullivan, J. Hypoxia Induces Mitochondrial Mutagenesis and Dysfunction in Inflammatory Arthritis. Arthritis Rheum. 2011, 63, 2172–2182. [Google Scholar] [CrossRef] [PubMed]
- Harty, L.C.; Biniecka, M.; O’Sullivan, J.; Fox, E.; Mulhall, K.; Veale, D.J.; Fearon, U. Mitochondrial Mutagenesis Correlates with the Local Inflammatory Environment in Arthritis. Ann. Rheum. Dis. 2012, 71, 582–588. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Yu, S.; Wang, D.; Chen, S.; Chen, S.; Zheng, Y.; Wang, N.; Chen, S.; Li, J.; Shen, B. Germline and Somatic mtDNA Mutation Spectrum of Rheumatoid Arthritis Patients in the Taizhou Area, China. Rheumatology 2020, 59, 2982–2991. [Google Scholar] [CrossRef] [PubMed]
- Mateen, S.; Moin, S.; Khan, A.Q.; Zafar, A.; Fatima, N. Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis. PLoS ONE 2016, 11, e0152925. [Google Scholar] [CrossRef] [PubMed]
- Kardeş, S.; Karagülle, M.; Durak, İ.; Avcı, A.; Karagülle, M.Z. Association of Oxidative Stress with Clinical Characteristics in Patients with Rheumatoid Arthritis. Eur. J. Clin. Investig. 2018, 48, e12858. [Google Scholar] [CrossRef] [PubMed]
- Hajizadeh, S.; DeGroot, J.; TeKoppele, J.M.; Tarkowski, A.; Collins, L.V. Extracellular Mitochondrial DNA and Oxidatively Damaged DNA in Synovial Fluid of Patients with Rheumatoid Arthritis. Arthritis Res. Ther. 2003, 5, R234–R240. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Breedveld, F.C.; Burmester, G.R.; Bykerk, V.; Dougados, M.; Emery, P.; Kvien, T.K.; Navarro-Compán, M.V.; Oliver, S.; Schoels, M.; et al. Treating Rheumatoid Arthritis to Target: 2014 Update of the Recommendations of an International Task Force. Ann. Rheum. Dis. 2016, 75, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Combe, B.; Landewe, R.; Daien, C.I.; Hua, C.; Aletaha, D.; Álvaro-Gracia, J.M.; Bakkers, M.; Brodin, N.; Burmester, G.R.; Codreanu, C.; et al. 2016 Update of the EULAR Recommendations for the Management of Early Arthritis. Ann. Rheum. Dis. 2017, 76, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Hoes, J.N.; Jacobs, J.W.G.; Boers, M.; Boumpas, D.; Buttgereit, F.; Caeyers, N.; Choy, E.H.; Cutolo, M.; Da Silva, J.A.P.; Esselens, G.; et al. EULAR Evidence-Based Recommendations on the Management of Systemic Glucocorticoid Therapy in Rheumatic Diseases. Ann. Rheum. Dis. 2007, 66, 1560–1567. [Google Scholar] [CrossRef] [PubMed]
- Cronstein, B.N.; Aune, T.M. Methotrexate and Its Mechanisms of Action in Inflammatory Arthritis. Nat. Rev. Rheumatol. 2020, 16, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.M.; Pratt, A.G.; Isaacs, J.D. Mechanism of Action of Methotrexate in Rheumatoid Arthritis, and the Search for Biomarkers. Nat. Rev. Rheumatol. 2016, 12, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Solomon, D.H.; Glynn, R.J.; Karlson, E.W.; Lu, F.; Corrigan, C.; Colls, J.; Xu, C.; MacFadyen, J.; Barbhaiya, M.; Berliner, N.; et al. Adverse Effects of Low-Dose Methotrexate: A Randomized Trial. Ann. Intern. Med. 2020, 172, 369. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I.; Herrmann, M.L.; Frangou, C.G.; Wahl, G.M.; Morris, R.E.; Strand, V.; Kirschbaum, B.J. Mechanism of Action for Leflunomide in Rheumatoid Arthritis. Clin. Immunol. 1999, 93, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Leflunomide in the Treatment of Rheumatoid Arthritis. Clin. Ther. 2004, 26, 447–459. [Google Scholar] [CrossRef] [PubMed]
- Pullar, T.; Hunter, J.A.; Capell, H.A. Sulphasalazine in the Treatment of Rheumatoid Arthritis: Relationship of Dose and Serum Levels to Efficacy. Rheumatology 1985, 24, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic Mice Expressing Human Tumour Necrosis Factor: A Predictive Genetic Model of Arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Feldmann, M. Anti-TNF Biologic Agents: Still the Therapy of Choice for Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2009, 5, 578–582. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Xu, S. TNF Inhibitor Therapy for Rheumatoid Arthritis. Biomed. Rep. 2013, 1, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Mertens, M.; Singh, J.A. Anakinra for Rheumatoid Arthritis: A Systematic Review. J. Rheumatol. 2009, 36, 1118–1125. [Google Scholar] [CrossRef] [PubMed]
- Konttinen, L.; Kankaanpää, E.; Luosujärvi, R.; Blåfield, H.; Vuori, K.; Hakala, M.; Rantalaiho, V.; Savolainen, E.; Uutela, T.; Nordström, D.; et al. Effectiveness of Anakinra in Rheumatic Disease in Patients Naive to Biological Drugs or Previously on TNF Blocking Drugs: An Observational Study. Clin. Rheumatol. 2006, 25, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, Y.-C.; Fettner, S.; Rowell, L.; Gott, T.; Grimsey, P.; Unsworth, A. Pharmacokinetics and Pharmacodynamics of Tocilizumab after Subcutaneous Administration in Patients with Rheumatoid Arthritis. Int. J. Clin. Pharmacol. Ther. 2013, 51, 620–630. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.D.; Keystone, E. Rituximab for Rheumatoid Arthritis. Rheumatol. Ther. 2015, 2, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.H.; Bae, S.-C.; Song, G.G. The Efficacy and Safety of Rituximab for the Treatment of Active Rheumatoid Arthritis: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Rheumatol. Int. 2011, 31, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Kiełbowski, K.; Plewa, P.; Bratborska, A.W.; Bakinowska, E.; Pawlik, A. JAK Inhibitors in Rheumatoid Arthritis: Immunomodulatory Properties and Clinical Efficacy. Int. J. Mol. Sci. 2024, 25, 8327. [Google Scholar] [CrossRef] [PubMed]
- Harrington, R.; Al Nokhatha, S.A.; Conway, R. JAK Inhibitors in Rheumatoid Arthritis: An Evidence-Based Review on the Emerging Clinical Data. J. Inflamm. Res. 2020, 13, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewé, R.B.M.; Bergstra, S.A.; Kerschbaumer, A.; Sepriano, A.; Aletaha, D.; Caporali, R.; Edwards, C.J.; Hyrich, K.L.; Pope, J.E.; et al. EULAR Recommendations for the Management of Rheumatoid Arthritis with Synthetic and Biological Disease-Modifying Antirheumatic Drugs: 2022 Update. Ann. Rheum. Dis. 2023, 82, 3–18. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. FDA Requires Warnings About Increased Risk of Serious Heart-Related Events, Cancer, Blood Clots, and Death for JAK Inhib-Itors that Treat Certain Chronic Inflammatory Conditions|FDA. Available online: https://www.fda.gov/drugs/drug-safety-and-availability/fda-requires-warnings-about-increased-risk-serious-heart-related-events-cancer-blood-clots-and-death?utm_medium=email&utm_source=govdelivery (accessed on 26 June 2025).
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the Nomenclature for MSC: The International Society for Cellular Therapy Position Statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Krause, D.S.; Deans, R.J.; Keating, A.; Prockop, D.J.; Horwitz, E.M. Minimal Criteria for Defining Multipotent Mesenchymal Stromal Cells. The International Society for Cellular Therapy Position Statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Muguruma, Y.; Yahata, T.; Miyatake, H.; Sato, T.; Uno, T.; Itoh, J.; Kato, S.; Ito, M.; Hotta, T.; Ando, K. Reconstitution of the Functional Human Hematopoietic Microenvironment Derived from Human Mesenchymal Stem Cells in the Murine Bone Marrow Compartment. Blood 2006, 107, 1878–1887. [Google Scholar] [CrossRef] [PubMed]
- Bianco, P.; Robey, P.G.; Simmons, P.J. Mesenchymal Stem Cells: Revisiting History, Concepts, and Assays. Cell Stem Cell 2008, 2, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Nasef, A.; Chapel, A.; Mazurier, C.; Bouchet, S.; Lopez, M.; Mathieu, N.; Sensebé, L.; Zhang, Y.; Gorin, N.-C.; Thierry, D.; et al. Identification of IL-10 and TGF-β Transcripts Involved in the Inhibition of T-Lymphocyte Proliferation During Cell Contact With Human Mesenchymal Stem Cells. Gene Expr. 2006, 13, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Davies, L.C.; Heldring, N.; Kadri, N.; Le Blanc, K. Mesenchymal Stromal Cell Secretion of Programmed Death-1 Ligands Regulates T Cell Mediated Immunosuppression. Stem Cells 2017, 35, 766–776. [Google Scholar] [CrossRef] [PubMed]
- Naserian, S.; Shamdani, S.; Arouche, N.; Uzan, G. Regulatory T Cell Induction by Mesenchymal Stem Cells Depends on the Expression of TNFR2 by T Cells. Stem Cell Res. Ther. 2020, 11, 534. [Google Scholar] [CrossRef] [PubMed]
- Ansboro, S.; Roelofs, A.J.; De Bari, C. Mesenchymal Stem Cells for the Management of Rheumatoid Arthritis: Immune Modulation, Repair or Both? Curr. Opin. Rheumatol. 2017, 29, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Maumus, M.; Guérit, D.; Toupet, K.; Jorgensen, C.; Noël, D. Mesenchymal Stem Cell-Based Therapies in Regenerative Medicine: Applications in Rheumatology. Stem Cell Res. Ther. 2011, 2, 14. [Google Scholar] [CrossRef] [PubMed]
- Ullah, M.; Liu, D.D.; Thakor, A.S. Mesenchymal Stromal Cell Homing: Mechanisms and Strategies for Improvement. iScience 2019, 15, 421–438. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Nakayamada, S.; Fukuyo, S.; Kubo, S.; Yunoue, N.; Wang, S.; Yoshikawa, M.; Saito, K.; Tanaka, Y. Activation of Syk in Peripheral Blood B Cells in Patients With Rheumatoid Arthritis: A Potential Target for Abatacept Therapy. Arthritis Rheumatol. 2015, 67, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-O.; Yang, W.S.; Park, J.G.; Jeong, D.; Kim, H.G.; Yoon, K.D.; Aravinthan, A.; Kim, J.-H.; Kim, E.; Cho, J.Y. Src and Syk Contribute to the Anti-Inflammatory Activities of Achyranthes Aspera Ethanolic Extract. J. Ethnopharmacol. 2017, 206, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.K.; Prateeksha, P.; Singh, S.P.; Rao, C.V.; Singh, B.N. Nyctanthes Arbor-Tristis Bioactive Extract Ameliorates LPS-Induced Inflammation through the Inhibition of NF-κB Signalling Pathway. J. Ethnopharmacol. 2024, 320, 117382. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Teng, L.; Qu, Y.; Liu, J.; Zhu, X.; Chen, S.; Yang, L.; Huang, Y.; Song, Q.; Fu, Q. Anti-Proliferation and Anti-Inflammation Effects of Corilagin in Rheumatoid Arthritis by Downregulating NF-κB and MAPK Signaling Pathways. J. Ethnopharmacol. 2022, 284, 114791. [Google Scholar] [CrossRef] [PubMed]
- George, G.; Shyni, G.L.; Mohan, S.; Abraham, B.; Nisha, P.; Ranjith, S.; Rajankutty, K.; Raghu, K.G. In Vitro and in Vivo Anti-Inflammatory and Anti-Arthritic Effect of Tinospora Cordifolia via Modulation of JAK/STAT Pathway. Inflammopharmacol 2023, 31, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Sharif, M.; John, P.; Bhatti, A.; Paracha, R.Z.; Majeed, A. Evaluation of the Inhibitory Mechanism of Pennisetum Glaucum (Pearl Millet) Bioactive Compounds for Rheumatoid Arthritis: An in Vitro and Computational Approach. Front. Pharmacol. 2024, 15, 1488790. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Fan, D.; Li, X.; Lu, X.; Ye, Q.; Xi, X.; Wang, Q.; Zhao, H.; Xiao, C. Yi Shen Juan Bi Pill Regulates the Bone Immune Microenvironment via the JAK2/STAT3 Signaling Pathway in Vitro. Front. Pharmacol. 2021, 12, 746786. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Zhang, L.-B.; Ma, R.; Wang, M.-N.; He, J.; Wang, P.-P.; Tao, Q.-W.; Xu, Y. Jolkinolide B Ameliorates Rheumatoid Arthritis by Regulating the JAK2/STAT3 Signaling Pathway. Phytomedicine 2024, 124, 155311. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shang, W.; Zhao, Z.; Zhang, B.; Liu, C.; Cai, H. Curcumin Alleviates Rheumatoid Arthritis Progression through the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway: An in Vitro and in Vivo Study. Bioengineered 2022, 13, 12899–12911. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Tian, X.; Cao, T.; Chen, S.; Liu, C.; Zheng, L.; Zhou, M.; Peng, X.; Li, Y.; Liu, T. Emodin Mitigates Rheumatoid Arthritis through Direct Binding to TNF-α. Front. Pharmacol. 2025, 16, 1520281. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Bao, X.; Xian, H.; Wei, F.; Song, Y.; Zhao, S.; Zhang, Y.; Wang, Y.; Wang, Y. Glucocorticoid Receptors Involved in Ginsenoside Compound K Ameliorate Adjuvant Arthritis by Inhibiting the Glycolysis of Fibroblast-like Synoviocytes via the NF-κB/HIF-1α Pathway. Pharm. Biol. 2023, 61, 1162–1174. [Google Scholar] [CrossRef] [PubMed]
- Tu, Y.; Tan, L.; Lu, T.; Wang, K.; Wang, H.; Han, B.; Zhao, Y.; Chen, H.; Li, Y.; Chen, H.; et al. Glytabastan B, a Coumestan Isolated from Glycine Tabacina, Alleviated Synovial Inflammation, Osteoclastogenesis and Collagen-Induced Arthritis through Inhibiting MAPK and PI3K/AKT Pathways. Biochem. Pharmacol. 2022, 197, 114912. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Du, Q.; Sun, J.; Geng, S.; Zhang, Y. Investigation of the Mechanism of Isobavachalcone in Treating Rheumatoid Arthritis through a Combination Strategy of Network Pharmacology and Experimental Verification. J. Ethnopharmacol. 2022, 294, 115342. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lu, H.; Fan, X.; Chen, S.; Chen, X.; Gao, W. Probing the Molecular Mechanism of Kaempferol in Relieving Rheumatoid Arthritis Based on Network Pharmacology. Sci. Rep. 2025, 15, 12645. [Google Scholar] [CrossRef] [PubMed]
- Linghu, K.-G.; Zhao, G.-D.; Zhang, D.-Y.; Xiong, S.-H.; Wu, G.-P.; Shen, L.-Y.; Cui, W.-Q.; Zhang, T.; Hu, Y.-J.; Guo, B.; et al. Leocarpinolide B Attenuates Collagen Type II-Induced Arthritis by Inhibiting DNA Binding Activity of NF-κB. Molecules 2023, 28, 4241. [Google Scholar] [CrossRef] [PubMed]
- Anchi, P.; Swamy, V.; Godugu, C. Nimbolide Exerts Protective Effects in Complete Freund’s Adjuvant Induced Inflammatory Arthritis via Abrogation of STAT-3/NF-κB/Notch-1 Signaling. Life Sci. 2021, 266, 118911. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Sun, S.; Zhang, H.; Liu, S.; Xu, X.; Hu, Y.; Ma, H.; Xin, P. Sappanone A Attenuates Rheumatoid Arthritis via Inhibiting PI3K/AKT/NF-κB and JAK2/STAT3 Signaling Pathways in Vivo and in Vitro. Int. Immunopharmacol. 2024, 143, 113560. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; He, L.; Wang, J.; Wang, Q.; Sun, C.; Li, Y.; Jia, K.; Wang, J.; Xu, T.; Ming, R.; et al. Anti-Angiogenic Effect of Shikonin in Rheumatoid Arthritis by Downregulating PI3K/AKT and MAPKs Signaling Pathways. J. Ethnopharmacol. 2020, 260, 113039. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhou, W.; Huang, Y.; Tian, Y.; Wong, S.Y.; Lam, W.K.; Ying, K.Y.; Zhang, J.; Chen, H. Umbelliferone and Scopoletin Target Tyrosine Kinases on Fibroblast-like Synoviocytes to Block NF-κB Signaling to Combat Rheumatoid Arthritis. Front. Pharmacol. 2022, 13, 946210. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Li, Q.; Chen, Y.; Dong, M.; Liu, H.; Zhang, J.; Yang, L.; Yin, G.; Xie, Q. Suberosin Attenuates Rheumatoid Arthritis by Repolarizing Macrophages and Inhibiting Synovitis via the JAK/STAT Signaling Pathway. Arthritis Res. Ther. 2025, 27, 12. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Xiao, X.; Yu, J.; Yang, Y.; Yu, J.; Liu, Y.; Song, H.; Han, T.; Zhang, D.; Niu, X.; et al. Tectoridin Exhibits Anti-Rheumatoid Arthritis Activity through the Inhibition of the Inflammatory Response and the MAPK Pathway in Vivo and in Vitro. Arch. Biochem. Biophys. 2022, 727, 109328. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Peng, Y.; Li, H.; Li, C.; Wu, Y.; Wang, X.; Chang, J.; Miao, C. Wilforine Inhibits Rheumatoid Arthritis Pathology through the Wnt11/β-Catenin Signaling Pathway Axis. Arthritis Res. Ther. 2023, 25, 243. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Wang, J.; Zhang, H.; Peng, T.; Sun, Y.; Zhang, R.; Meng, X.; Lu, X.; Gao, Y.; Jin, Y.; et al. Ammopiptanthus nanus (M. Pop.) Cheng f. Stem Ethanolic Extract Ameliorates Rheumatoid Arthritis by Inhibiting PI3K/AKT/NF-κB Pathway-Mediated Macrophage Infiltration. J. Ethnopharmacol. 2025, 338, 118974. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-Y.; Kim, J.-M.; Chung, K.-S.; Jang, D.S.; Lee, J.-Y.; Kim, C.; Lee, J.Y.; Lee, J.K.; Lee, K.-T. In Vitro and in Vivo Anti-Inflammatory Effects of 5-Hydroxyconiferaldehyde via NF-κB, MAPK/AP-1, and Nrf2 Modulation. Chem. Biol. Interact. 2025, 409, 111427. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Ma, K.; Pang, X.; Liu, Y.; Song, Z.; Zhou, R.; Tang, Z. Anti-Rheumatoid Arthritis Effects of Total Saponins from Rhizoma Panacis Majoris on Adjuvant-Induced Arthritis in Rats and Rheumatoid Arthritis Fibroblast-like Synoviocytes. Phytomedicine 2023, 119, 155021. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Wang, J.; Wang, J.; Jia, X.; Xuan, Z.; Cheng, Z.; Meng, X.; Su, W. Wnt/β-Catenin Signaling Pathway Promotes Abnormal Activation of Fibroblast-like Synoviocytes and Angiogenesis in Rheumatoid Arthritis and the Intervention of Er Miao San. Phytomedicine 2023, 120, 155064. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Sun, Y.; Jiang, H.; Chen, Y.; Jiang, Y.; Han, Y.; Wang, Y. Total Saponins of Radix Clematis Regulate Fibroblast-Like Synoviocyte Proliferation in Rheumatoid Arthritis via the LncRNA OIP5-AS1/MiR-410-3p/Wnt7b Signaling Pathway. Evid. Based Complement. Altern. Med. 2022, 2022, 8393949. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Tao, Y.; Liu, J.; Sun, J.; Zeng, Y.; Meng, X.; Fan, G.; Zhang, Y. Tibetan Medicine Qi-Sai-Er-Sang-Dang-Song Decoction Inhibits TNF-α-Induced Rheumatoid Arthritis in Human Fibroblast-like Synoviocytes via Regulating NOTCH1/NF-κB/NLRP3 Pathway. J. Ethnopharmacol. 2023, 310, 116402. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.; Lu, S.; Gao, X.; Luo, Y.; Tong, B.; Wei, Z.; Lu, T.; Xia, Y.; Chou, G.; Wang, Z.; et al. Norisoboldine, an Alkaloid Compound Isolated from Radix Linderae, Inhibits Synovial Angiogenesis in Adjuvant-Induced Arthritis Rats by Moderating Notch1 Pathway-Related Endothelial Tip Cell Phenotype. Exp. Biol. Med. 2012, 237, 919–932. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R. Rheumatoid Arthritis: Pathophysiology, Current Therapeutic Strategies and Recent Advances in Targeted Drug Delivery System. Mater. Today Commun. 2023, 35, 105877. [Google Scholar] [CrossRef]
- Kim, Y.; Yang, H.-I.; Kim, K.-S. Etiology and Pathogenesis of Rheumatoid Arthritis-Interstitial Lung Disease. Int. J. Mol. Sci. 2023, 24, 14509. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. [Google Scholar] [CrossRef] [PubMed]
- Fraenkel, L.; Bathon, J.M.; England, B.R.; St. Clair, E.W.; Arayssi, T.; Carandang, K.; Deane, K.D.; Genovese, M.; Huston, K.K.; Kerr, G.; et al. 2021 American College of Rheumatology Guideline for the Treatment of Rheumatoid Arthritis. Arthritis Care Res. 2021, 73, 924–939. [Google Scholar] [CrossRef] [PubMed]
- Hein, T.R.; Peterson, L.; Bartikoski, B.J.; Portes, J.; Espírito Santo, R.C.; Xavier, R.M. The Effect of Disease-Modifying Anti-Rheumatic Drugs on Skeletal Muscle Mass in Rheumatoid Arthritis Patients: A Systematic Review with Meta-Analysis. Arthritis Res. Ther. 2022, 24, 171. [Google Scholar] [CrossRef] [PubMed]
- Gehringer, C.K.; Martin, G.P.; Hyrich, K.L.; Verstappen, S.M.M.; Sergeant, J.C. Clinical Prediction Models for Methotrexate Treatment Outcomes in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-Analysis. Semin. Arthritis Rheum. 2022, 56, 152076. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Higuchi, Y.; Kitamura, H.; Murao, N.; Saitoh, R.; Morikawa, T.; Sato, H. Novel Hyaluronic Acid–Methotrexate Conjugate Suppresses Joint Inflammation in the Rat Knee: Efficacy and Safety Evaluation in Two Rat Arthritis Models. Arthritis Res. Ther. 2016, 18, 79. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.A.; Boers, M.; Hochberg, M.; Baker, N.; Skovron, M.L.; Ray, N.; Singhal, S.; Suissa, S.; Gomez-Caminero, A. Comparative Risk of Malignancies and Infections in Patients with Rheumatoid Arthritis Initiating Abatacept versus Other Biologics: A Multi-Database Real-World Study. Arthritis Res. Ther. 2019, 21, 228. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, N.; Bhatt, L.K.; Prabhavalkar, K.S. Experimental Animal Models for Rheumatoid Arthritis. Immunopharmacol. Immunotoxicol. 2018, 40, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Bevaart, L.; Vervoordeldonk, M.J.; Tak, P.P. Evaluation of Therapeutic Targets in Animal Models of Arthritis: How Does It Relate to Rheumatoid Arthritis? Arthritis Rheum. 2010, 62, 2192–2205. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.K.; Hamilton, J.A.; Wicks, I.P. Collagen-Induced Arthritis in C57BL/6 (H-2b) Mice: New Insights into an Important Disease Model of Rheumatoid Arthritis. Eur. J. Immunol. 2000, 30, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Terato, K.; Hasty, K.A.; Reife, R.A.; Cremer, M.A.; Kang, A.H.; Stuart, J.M. Induction of Arthritis with Monoclonal Antibodies to Collagen. J. Immunol. 1992, 148, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Holmdahl, R. Efficient Promotion of Collagen Antibody Induced Arthritis (CAIA) Using Four Monoclonal Antibodies Specific for the Major Epitopes Recognized in Both Collagen Induced Arthritis and Rheumatoid Arthritis. J. Immunol. Methods 2005, 304, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Khachigian, L.M. Collagen Antibody-Induced Arthritis. Nat. Protoc. 2006, 1, 2512–2516. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, K.S.; Holmdahl, R. Collagen Antibody Induced Arthritis. In Arthritis Research; Cope, A.P., Ed.; Methods in Molecular Medicine; Humana Press: Totowa, NJ, USA, 2007; Volume 136, pp. 215–223. ISBN 978-1-58829-918-5. [Google Scholar]
- Pearson, C.M. Development of Arthritis, Periarthritis and Periostitis in Rats Given Adjuvants. Exp. Biol. Med. 1956, 91, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Negi, P.; Agarwal, S.; Garg, P.; Ali, A.; Kulshrestha, S. In Vivo Models of Understanding Inflammation (In Vivo Methods for Inflammation). In Recent Developments in Anti-Inflammatory Therapy; Elsevier: Amsterdam, The Netherlands, 2023; pp. 315–330. ISBN 978-0-323-99988-5. [Google Scholar]
- Ye, L.; Mingyue, H.; Feng, Z.; Zongshun, D.; Ying, X.; Xiong, C.; Liang, L. Systematic Review of Robust Experimental Models of Rheumatoid Arthritis for Basic Research. Digit. Chin. Med. 2021, 4, 262–272. [Google Scholar] [CrossRef]
- Hopkins, S.J.; Freemont, A.J.; Jayson, M.I.V. Pristane-Induced Arthritis in Balb/c Mice: I. Clinical and Histological Features of the Arthropathy. Rheumatol. Int. 1984, 5, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Holmdahl, R. Experimental Models for Rheumatoid Arthritis. In Kelley and Firestein’s Textbook of Rheumatology; Elsevier: Amsterdam, The Netherlands, 2017; pp. 449–460. ISBN 978-0-323-31696-5. [Google Scholar]
- van den Berg, W.B. Animal Models of Arthritis. What Have We Learned? J. Rheumatol. Suppl. 2005, 72, 7–9. [Google Scholar] [PubMed]
- Nho, J.-H.; Kim, A.-H.; Jung, H.-K.; Lee, M.-J.; Jang, J.-H.; Yang, B.-D.; Lee, H.-J.; Lee, K.-H.; Woo, K.-W.; Cho, H.-W. Water Extract of Acori graminei Rhizoma Attenuates Features of Rheumatoid Arthritis in DBA/1 Mice. Evid. Based Complement. Altern. Med. 2019, 2019, 3637453. [Google Scholar] [CrossRef] [PubMed]
- Phan, H.T.; Nguyen, T.T.; Tran, P.N.T. Evaluation of Anti-Inflammatory Effect of Fruit Peel Extracts of Annona squamosa L. on Mouse Models of Rheumatoid Arthritis. J. Microb. Biotech. Food Sci. 2021, 11, e2075. [Google Scholar] [CrossRef]
- Nho, J.-H.; Lee, H.-J.; Jung, H.-K.; Jang, J.-H.; Lee, K.-H.; Kim, A.-H.; Sung, T.-K.; Cho, H.-W. Effect of Saururus Chinensis Leaves Extract on Type II Collagen-Induced Arthritis Mouse Model. BMC Complement. Altern. Med. 2019, 19, 2. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Kim, H.; Lee, E.H.; Jo, G.; Na, C.S.; Kang, K.; Lee, T.H. Anti-Inflammatory Effect of Cudrania Tricuspidata Extract and Stewartia Koreana Extract Mixture in a Collagen-Induced Arthritis Mouse Model. Appl. Sci. 2021, 11, 6660. [Google Scholar] [CrossRef]
- Kim, S.-H.; Bang, J.; Son, C.-N.; Baek, W.-K.; Kim, J.-M. Grape Seed Proanthocyanidin Extract Ameliorates Murine Autoimmune Arthritis through Regulation of TLR4/MyD88/NF-κB Signaling Pathway. Korean J. Intern. Med. 2018, 33, 612–621. [Google Scholar] [CrossRef] [PubMed]
- Eor, J.Y.; Park, N.; Son, Y.J.; Kim, S.H. Therapeutic Effects of Gleditsia Sinensis Thorn Extract Fermented by Lactobacillus Casei 3260 in a Type II Collagen-Induced Rheumatoid Arthritis Mouse Model. Food Sci. Anim. Resour. 2021, 41, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Allam, G.; Mahdi, E.A.; Alzahrani, A.M.; Abuelsaad, A.S. Ellagic Acid Alleviates Adjuvant Induced Arthritis by Modulation of Pro- and Anti-Inflammatory Cytokines. Central Eur. J. Immunol. 2016, 4, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Ulhaq, Z.S.; Hendyatama, T.H.; Alluza, H.H.D.; Aulanni’am, A. Therapeutic Effects of Durian Wood Bark Extract on a Rat Model of Rheumatoid Arthritis. Rev. Colomb. Reumatol. 2021, 28, 118–123. [Google Scholar] [CrossRef]
- Sun, S.; Li, S.; Du, Y.; Wu, C.; Zhang, M.; Li, J.; Zhang, X. Anti-Inflammatory Effects of the Root, Stem and Leaf Extracts of Chloranthus serratus on Adjuvant-Induced Arthritis in Rats. Pharm. Biol. 2020, 58, 528–537. [Google Scholar] [CrossRef] [PubMed]
- Qu, B.; Wang, S.; Zhu, H.; Yin, T.; Zhou, R.; Hu, W.; Lu, C. Core Constituents of Caragana sinica Root for Rheumatoid Arthritis Treatment and the Potential Mechanism. ACS Omega 2023, 8, 2586–2595. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Liu, Z.; Lin, M.; Gao, N.; Wang, X. Polyphenols in Health and Food Processing: Antibacterial, Anti-Inflammatory, and Antioxidant Insights. Front. Nutr. 2024, 11, 1456730. [Google Scholar] [CrossRef] [PubMed]
- Pourhabibi-Zarandi, F.; Rafraf, M.; Zayeni, H.; Asghari-Jafarabadi, M.; Ebrahimi, A. Effects of Curcumin Supplementation on Metabolic Parameters, Inflammatory Factors and Obesity Values in Women with Rheumatoid Arthritis: A Randomized, Double-blind, Placebo-controlled Clinical Trial. Phytother. Res. 2022, 36, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Javadi, M.; Khadem Haghighian, H.; Goodarzy, S.; Abbasi, M.; Nassiri-Asl, M. Effect of Curcumin Nanomicelle on the Clinical Symptoms of Patients with Rheumatoid Arthritis: A Randomized, Double-blind, Controlled Trial. Int. J. Rheum. Dis. 2019, 22, 1857–1862. [Google Scholar] [CrossRef] [PubMed]
- Jacob, J.; Amalraj, A.; Raj, K.K.J.; Divya, C.; Kunnumakkara, A.B.; Gopi, S. A Novel Bioavailable Hydrogenated Curcuminoids Formulation (CuroWhiteTM) Improves Symptoms and Diagnostic Indicators in Rheumatoid Arthritis Patients—A Randomized, Double Blind and Placebo Controlled Study. J. Tradit. Complement. Med. 2019, 9, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Anusha, D.; Chaly, P.; Junaid, M.; Nijesh, J.; Shivashankar, K.; Sivasamy, S. Efficacy of a Mouthwash Containing Essential Oils and Curcumin as an Adjunct to Nonsurgical Periodontal Therapy among Rheumatoid Arthritis Patients with Chronic Periodontitis: A Randomized Controlled Trial. Indian. J. Dent. Res. 2019, 30, 506. [Google Scholar] [CrossRef] [PubMed]
- Aryaeian, N.; Shahram, F.; Mahmoudi, M.; Tavakoli, H.; Yousefi, B.; Arablou, T.; Jafari Karegar, S. The Effect of Ginger Supplementation on Some Immunity and Inflammation Intermediate Genes Expression in Patients with Active Rheumatoid Arthritis. Gene 2019, 698, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, Z.; Aryaeian, N.; Abolghasemi, J.; Shirani, F.; Hadidi, M.; Fallah, S.; Moradi, N. The Effect of Saffron Supplement on Clinical Outcomes and Metabolic Profiles in Patients with Active Rheumatoid Arthritis: A Randomized, Double-blind, Placebo-controlled Clinical Trial. Phytother. Res. 2020, 34, 1650–1658. [Google Scholar] [CrossRef] [PubMed]
- Aryaeian, N.; Hamidi, Z.; Shirani, F.; Hadidi, M.; Abolghasemi, J.; Moradi, N.; Fallah, S. The Effect of Saffron Supplement on Clinical Outcomes, Inflammatory and Oxidative Markers in Patients with Active Rheumatoid Arthritis. Curr. Dev. Nutr. 2021, 5, 1118. [Google Scholar] [CrossRef]
- Javadi, F.; Ahmadzadeh, A.; Eghtesadi, S.; Aryaeian, N.; Zabihiyeganeh, M.; Rahimi Foroushani, A.; Jazayeri, S. The Effect of Quercetin on Inflammatory Factors and Clinical Symptoms in Women with Rheumatoid Arthritis: A Double-Blind, Randomized Controlled Trial. J. Am. Coll. Nutr. 2017, 36, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Khojah, H.M.; Ahmed, S.; Abdel-Rahman, M.S.; Elhakeim, E.H. Resveratrol as an Effective Adjuvant Therapy in the Management of Rheumatoid Arthritis: A Clinical Study. Clin. Rheumatol. 2018, 37, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Amalraj, A.; Varma, K.; Jacob, J.; Divya, C.; Kunnumakkara, A.B.; Stohs, S.J.; Gopi, S. A Novel Highly Bioavailable Curcumin Formulation Improves Symptoms and Diagnostic Indicators in Rheumatoid Arthritis Patients: A Randomized, Double-Blind, Placebo-Controlled, Two-Dose, Three-Arm, and Parallel-Group Study. J. Med. Food 2017, 20, 1022–1030. [Google Scholar] [CrossRef] [PubMed]
- Chandran, B.; Goel, A. A Randomized, Pilot Study to Assess the Efficacy and Safety of Curcumin in Patients with Active Rheumatoid Arthritis. Phytother. Res. 2012, 26, 1719–1725. [Google Scholar] [CrossRef] [PubMed]
- Mirtaheri, E.; Khabbazi, A.; Nazemiyeh, H.; Ebrahimi, A.-A.; Hajalilou, M.; Shakibay Novin, Z.; Pirouzpanah, S. Stachys Schtschegleevii Tea, Matrix Metalloproteinase, and Disease Severity in Female Rheumatoid Arthritis Patients: A Randomized Controlled Clinical Trial. Clin. Rheumatol. 2022, 41, 1033–1044. [Google Scholar] [CrossRef] [PubMed]
- Alghadir, A.H.; Gabr, S.A.; Al-Eisa, E.S. Green Tea and Exercise Interventions as Nondrug Remedies in Geriatric Patients with Rheumatoid Arthritis. J. Phys. Ther. Sci. 2016, 28, 2820–2829. [Google Scholar] [CrossRef] [PubMed]
- Shishehbor, F.; Rezaeyan Safar, M.; Rajaei, E.; Haghighizadeh, M.H. Cinnamon Consumption Improves Clinical Symptoms and Inflammatory Markers in Women with Rheumatoid Arthritis. J. Am. Coll. Nutr. 2018, 37, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Helli, B.; Shahi, M.M.; Mowla, K.; Jalali, M.T.; Haghighian, H.K. A Randomized, Triple-blind, Placebo-controlled Clinical Trial, Evaluating the Sesamin Supplement Effects on Proteolytic Enzymes, Inflammatory Markers, and Clinical Indices in Women with Rheumatoid Arthritis. Phytother. Res. 2019, 33, 2421–2428. [Google Scholar] [CrossRef] [PubMed]
- Helli, B.; Mowla, K.; Mohammadshahi, M.; Jalali, M.T. Effect of Sesamin Supplementation on Cardiovascular Risk Factors in Women with Rheumatoid Arthritis. J. Am. Coll. Nutr. 2016, 35, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Berbert, A.A.; Kondo, C.R.M.; Almendra, C.L.; Matsuo, T.; Dichi, I. Supplementation of Fish Oil and Olive Oil in Patients with Rheumatoid Arthritis. Nutrition 2005, 21, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Nachvak, S.M.; Alipour, B.; Mahdavi, A.M.; Aghdashi, M.A.; Abdollahzad, H.; Pasdar, Y.; Samadi, M.; Mostafai, R. Effects of Coenzyme Q10 Supplementation on Matrix Metalloproteinases and DAS-28 in Patients with Rheumatoid Arthritis: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Clin. Rheumatol. 2019, 38, 3367–3374. [Google Scholar] [CrossRef] [PubMed]
- Abdollahzad, H.; Aghdashi, M.A.; Asghari Jafarabadi, M.; Alipour, B. Effects of Coenzyme Q10 Supplementation on Inflammatory Cytokines (TNF-α, IL-6) and Oxidative Stress in Rheumatoid Arthritis Patients: A Randomized Controlled Trial. Arch. Med. Res. 2015, 46, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Hang, Y.; Qin, X.; Ren, T.; Cao, J. Baicalin Reduces Blood Lipids and Inflammation in Patients with Coronary Artery Disease and Rheumatoid Arthritis: A Randomized, Double-Blind, Placebo-Controlled Trial. Lipids Health Dis. 2018, 17, 146. [Google Scholar] [CrossRef] [PubMed]
- Tao, X.; Younger, J.; Fan, F.Z.; Wang, B.; Lipsky, P.E. Benefit of an Extract of Tripterygium wilfordii Hook F in Patients with Rheumatoid Arthritis: A Double-blind, Placebo-controlled Study. Arthritis Rheum. 2002, 46, 1735–1743. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhao, L.; Chen, H.; Zhang, Y.; Wang, D.; Huang, L.; Lv, Q.; Liu, B.; Li, Z.; Wei, W.; et al. Comparison of the Impact of Tripterygium Wilfordii Hook F and Methotrexate Treatment on Radiological Progression in Active Rheumatoid Arthritis: 2-Year Follow up of a Randomized, Non-Blinded, Controlled Study. Arthritis Res. Ther. 2018, 20, 70. [Google Scholar] [CrossRef] [PubMed]
- Mur, E.; Hartig, F.; Eibl, G.; Schirmer, M. Randomized Double Blind Trial of an Extract from the Pentacyclic Alkaloid-Chemotype of Uncaria Tomentosa for the Treatment of Rheumatoid Arthritis. J. Rheumatol. 2002, 29, 678–681. [Google Scholar] [PubMed]
- Balbir-Gurman, A.; Fuhrman, B.; Braun-Moscovici, Y.; Markovits, D.; Aviram, M. Consumption of Pomegranate Decreases Serum Oxidative Stress and Reduces Disease Activity in Patients with Active Rheumatoid Arthritis: A Pilot Study. Isr. Med. Assoc. J. 2011, 13, 474–479. [Google Scholar] [PubMed]
- Ghavipour, M.; Sotoudeh, G.; Tavakoli, E.; Mowla, K.; Hasanzadeh, J.; Mazloom, Z. Pomegranate Extract Alleviates Disease Activity and Some Blood Biomarkers of Inflammation and Oxidative Stress in Rheumatoid Arthritis Patients. Eur. J. Clin. Nutr. 2017, 71, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Moosavian, S.P.; Paknahad, Z.; Habibagahi, Z. A Randomized, Double-blind, Placebo-controlled Clinical Trial, Evaluating the Garlic Supplement Effects on Some Serum Biomarkers of Oxidative Stress, and Quality of Life in Women with Rheumatoid Arthritis. Int. J. Clin. Pract. 2020, 74, e13498. [Google Scholar] [CrossRef] [PubMed]
- Sekiya, N.; Shimada, Y.; Niizawa, A.; Kogure, T.; Mantani, N.; Sakai, S.; Hikiami, H.; Terasawa, K. Suppressive Effects of Stephania tetrandra on the Neutrophil Function in Patients with Rheumatoid Arthritis. Phytother. Res. 2004, 18, 247–249. [Google Scholar] [CrossRef] [PubMed]
- Burgos, R.A.; Hancke, J.L.; Bertoglio, J.C.; Aguirre, V.; Arriagada, S.; Calvo, M.; Cáceres, D.D. Efficacy of an Andrographis Paniculata Composition for the Relief of Rheumatoid Arthritis Symptoms: A Prospective Randomized Placebo-Controlled Trial. Clin. Rheumatol. 2009, 28, 931–946. [Google Scholar] [CrossRef] [PubMed]
- López Mantecón, A.M.; Garrido, G.; Delgado-Hernández, R.; Garrido-Suárez, B.B. Combination of Mangifera indica L. Extract Supplementation Plus Methotrexate in Rheumatoid Arthritis Patients: A Pilot Study: Mangifera Indica plus Methotrexate in Rheumatoid Arthritis. Phytother. Res. 2014, 28, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Guo, M.; Luo, Y.; Yun, M.; Yan, J.; Liu, T.; Xiao, C. Effect of Artemisia Annua Extract on Treating Active Rheumatoid Arthritis: A Randomized Controlled Trial. Chin. J. Integr. Med. 2017, 23, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Aryaeian, N.; Hadidi, M.; Mahmoudi, M.; Asgari, M.; Hezaveh, Z.S.; Sadehi, S.K. The Effect of Black Barberry Hydroalcoholic Extract on Immune Mediators in Patients with Active Rheumatoid Arthritis: A Randomized, Double–Blind, Controlled Clinical Trial. Phytother. Res. 2021, 35, 1062–1068. [Google Scholar] [CrossRef] [PubMed]
- Hadi, V.; Kheirouri, S.; Alizadeh, M.; Khabbazi, A.; Hosseini, H. Effects of Nigella Sativa Oil Extract on Inflammatory Cytokine Response and Oxidative Stress Status in Patients with Rheumatoid Arthritis: A Randomized, Double-Blind, Placebo-Controlled Clinical Trial. Avicenna J. Phytomed 2016, 6, 34–43. [Google Scholar] [PubMed]
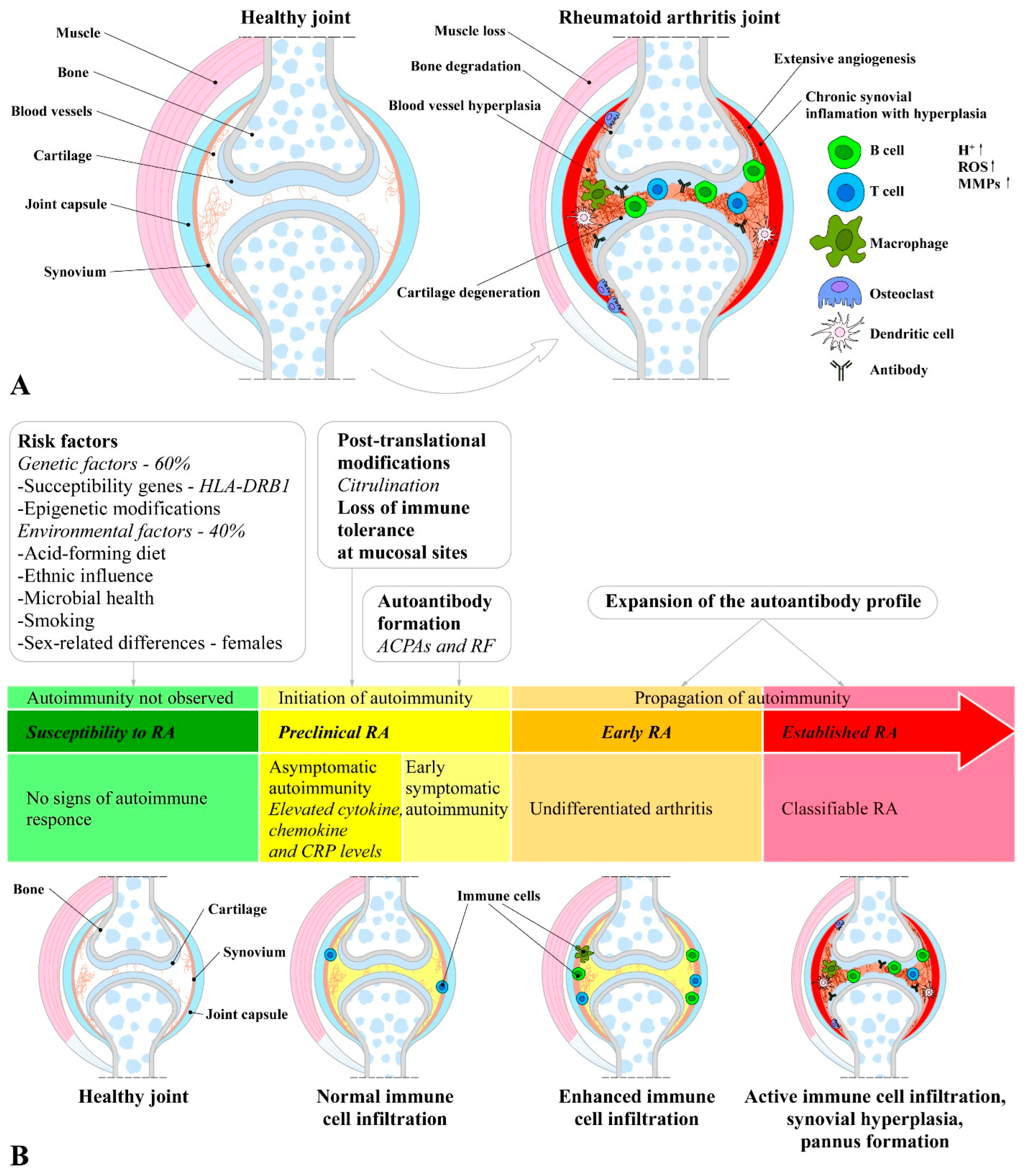
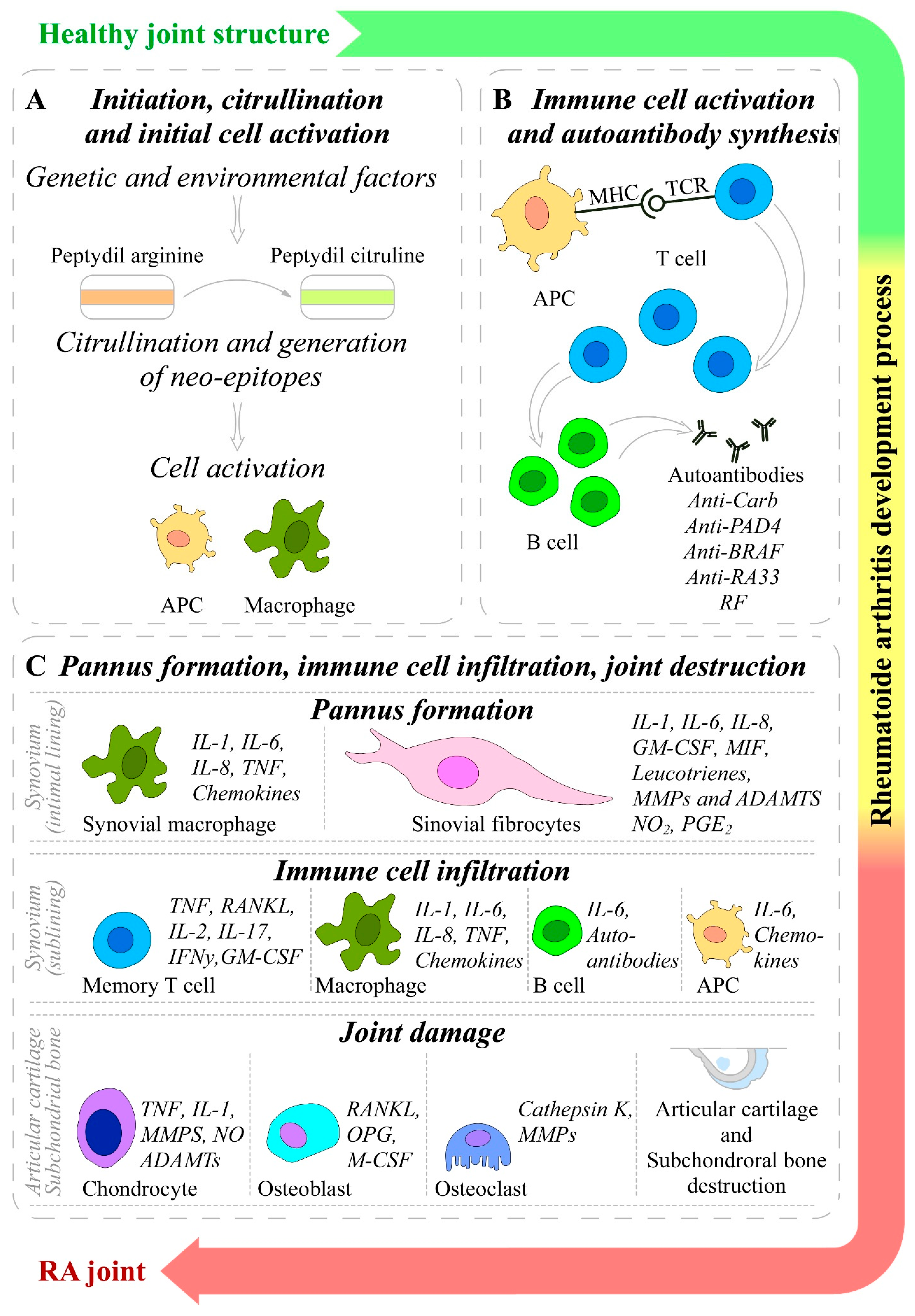
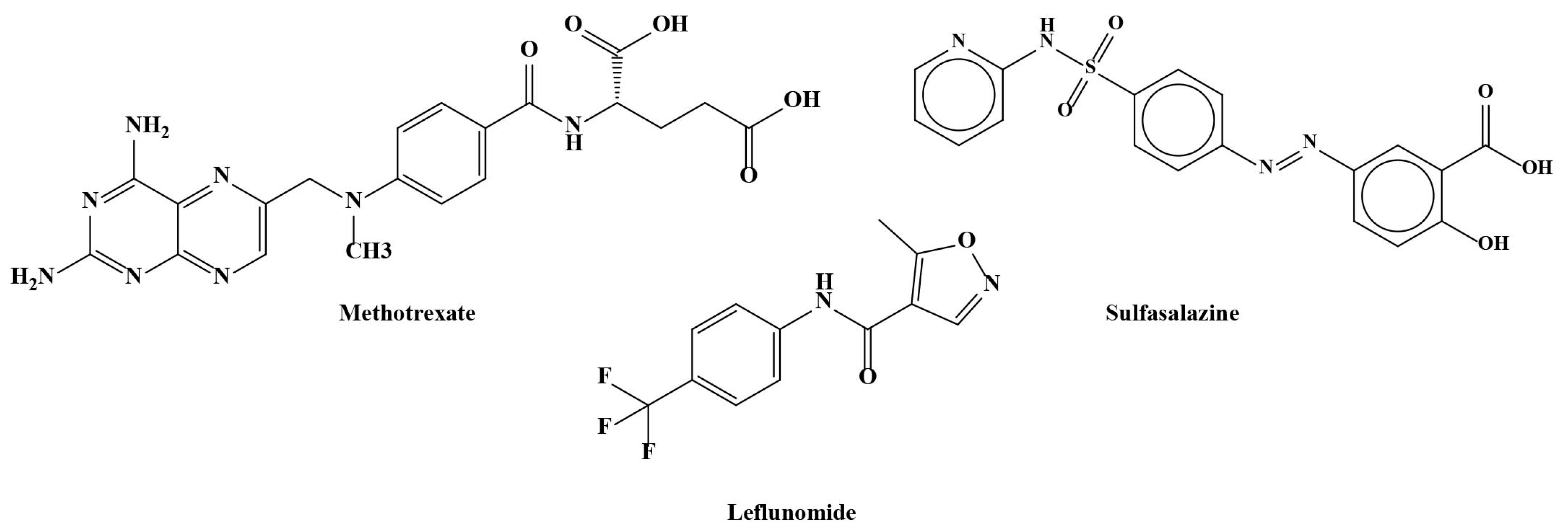
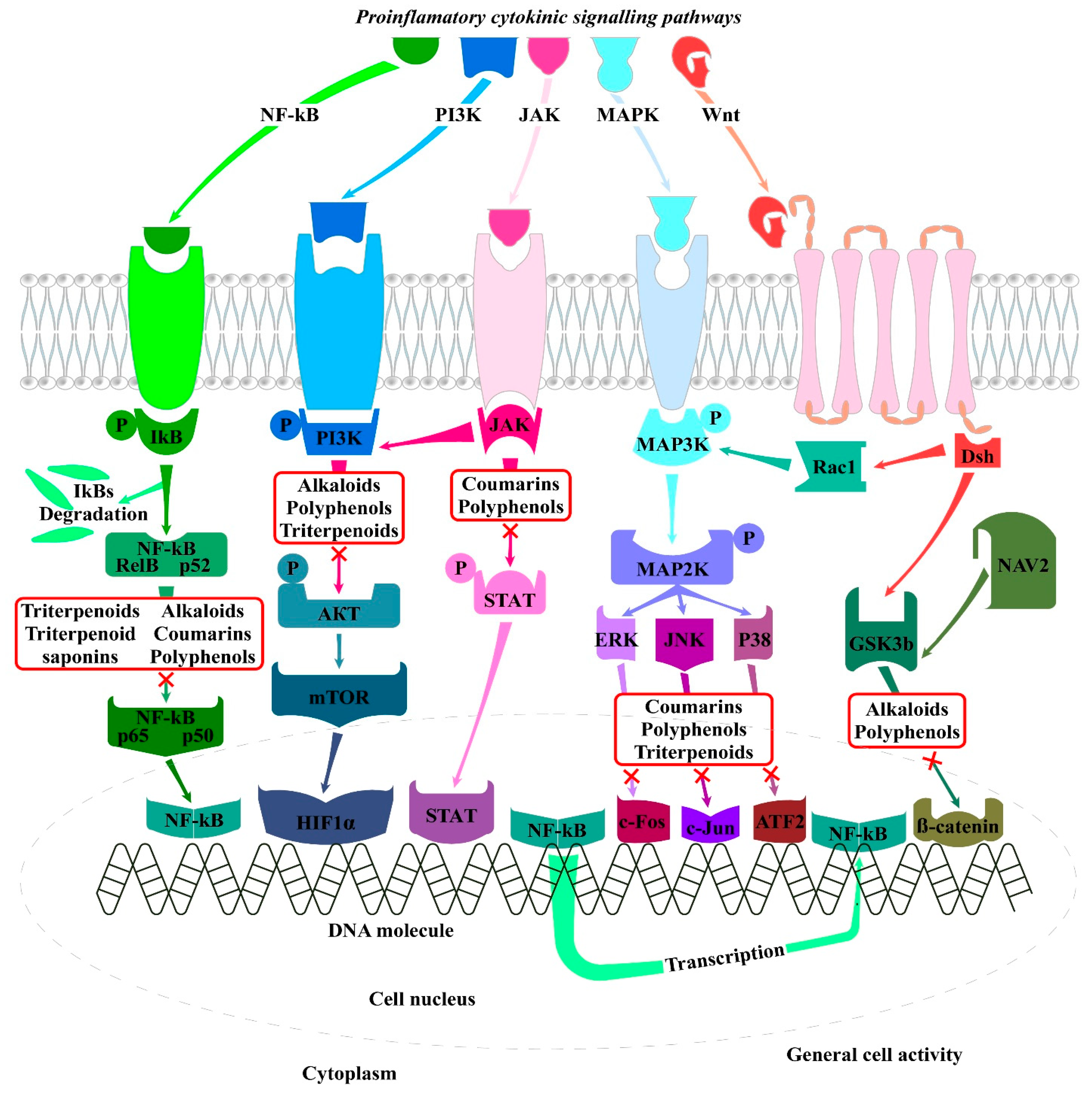
| Antibody | Molecular Structure | Administration and Dose |
|---|---|---|
| Infliximab (IFX, Remicade) | Chimeric IgG1 monoclonal antibody | Intravenous injections 3–10 mg/kg every 4–8 weeks |
| Etanercept (ETN, Enbrel) | Recombinant human fusion protein (TNF-α receptor bound to Fc fragment) | Subcutaneous injections 50 mg/week or 25 mg/twice a week |
| Adalimumab (ADA, Humira) | Recombinant human IgG monoclonal antibody | Subcutaneous injections 25 mg/twice a week |
| Golimumab (GOL, Simponi) | Human IgG monoclonal antibody | Subcutaneous injections 50 mg/month |
| Certolizumab Pegol (CZP, Cimzia) | Recombinant humanized Fab fragment | Subcutaneous injections 400 mg at weeks 0, 2, and 4 followed by 200 mg/every 2 weeks |
| JAK Inhibitor | Target | Dose |
|---|---|---|
| Tofacitinib (Xeljanz) | JAK1 and JAK3 | 5 mg twice a day or 11 mg once a day |
| Baricitinib (Olumiant) | JAK1 and JAK2 | 2–4 mg once a day |
| Upadacitinib (Rinvoq) | JAK1 | once a day |
| Host | Animal Model | Biological Agent | Mode of Action | Reference |
|---|---|---|---|---|
| Mouse | Male DBA/1 mice (6 weeks old); CIA (bovine type II collagen + CFA/IFA) | Acori graminei | Reduction in inflammation indicators including IL-6 and TNF-α | [338] |
| Mouse | Male Swiss Albino mice (6 weeks old); AIA (complete Freund’s adjuvant (CFA)) | Fruit peel extracts of Annona squamosa L. | Decrease in leukocytes in serum | [339] |
| Mouse | Male DBA/1 mice (6 weeks of age); CIA (bovine type II collagen + CFA/IFA) | Saururus chinensis | Reduction in inflammatory cytokines | [340] |
| Mouse | Male DBA/1J mice (7 weeks of age); CIA (bovine type II collagen + CFA/IFA) | Cudrania tricuspidata and Stewartia koreana | Decrease in inflammatory cytokine levels, NOS inhibitors | [341] |
| Mouse | Male DBA/1J mice (6 to 8 weeks of age); CIA (bovine type II collagen + CFA/IFA) | Grape seed proanthocyanidin extract | Inhibition of TLR4/ MyD88/NF-κB signaling pathway | [342] |
| Mouse | Male DBA/1 mice (6 weeks old); CIA (bovine type II collagen + CFA/IFA) | Gleditsia sinensis thorn extract fermented by Lactobacillus | Reduction in inflammatory cytokine levels | [343] |
| Mouse | Male MF1 mice (8 weeks old); AIA (complete Freund’s adjuvant (CFA)) | Ellagic acid | Downregulation of pro-inflammatory cytokines and upregulation of anti-inflammatory cytokines | [344] |
| Rat | Female Wistar rat (3–4 months of age); AIA (complete Freund’s adjuvant (CFA)) | Duran wood bark extract | iNOS suppression/NOS inhibitor | [345] |
| Rat | Male Sprague Dawley (SD) rats (6 weeks of age); AIA (complete Freund′s adjuvant (CFA)) | Chloranthus serratus | Inhibition of release of inflammatory cytokines and amelioration of antioxidant capacity | [346] |
| Rat | Male Sprague–Dawley (SD) rats; AIA (complete Freund’s adjuvant (CFA)) | Caragana sinica | Negative regulation of NF-κB pathway | [347] |
| Phytochemical | Duration | Dosage | Outcome Measures | Results | Reference |
|---|---|---|---|---|---|
| Curcumin | 90 days | 250 or 500 mg twice a day | ACR response; VAS; CRP; DAS28; ESR; RF | Significant improvement in ESR, CRP, VAS, RF, DAS28, and ACR responses compared to placebo | [358] |
| 8 weeks | 500 mg curcumin or 50 mg diclofenac sodium or their combination twice a day | ACR; DAS28 | Curcumin group showed highest percentage of improvement in overall DAS and ACR scores (ACR 20, 50 and 70), and these scores were significantly better than patients in diclofenac sodium group | [359] | |
| 8 weeks | 500 mg curcumin daily | Fasting blood samples; anthropometric measurements; dietary intakes; physical activity levels | Significantly decreased HOMA-IR, ESR, CRP, triglycerides, weight, and body mass index compared with placebo | [349] | |
| 12 weeks | 40 mg curcumin nanomicelle and placebo capsules 3 times a day | DAS-28; ESR | Within-group values of DAS-28, TJC, and SJC in curcumin nanomicelle and placebo groups reduced significantly compared to baseline | [350] | |
| 3 months | 250 or 500 mg hydrogenated curcuminoid formulation or placebo daily | ACR; DAS 28; ESR; CRP; RF | Significant changes in DAS28 (50–64%), VAS (63–72%), ESR (88–89%), CRP (31–45%), and RF (80–84%) values and ACR response for curcumin-treated groups in comparison with placebo | [351] | |
| 6 weeks | 0.2% chlorhexidine with scaling and root planing mouthwash; curcumin with scaling and root planing; scaling and root planing alone | CRP; ESR; RF; anti-citrullinated protein antibody; plaque index; pocket depth | Significant reduction in periodontal and RA disease activity parameters was observed from baseline; highest percentage of mean reduction in plaque index and RA parameters from baseline was observed in group treated with curcumin | [352] | |
| Ginger | 12 weeks | 1500 mg ginger powder or placebo daily | DAS28; gene expression of NF-κB, PPAR-γ, FoxP3, T-bet, GATA-3, RORγt | Statistically significant reduction in DAS28 within ginger group and between two groups; significant increase in FoxP3 gene expression within ginger group and between two groups; T-bet and RORγt gene expression decreased significantly between two groups; in ginger group, PPAR-γ gene expression increased significantly, but difference between two groups was not statistically significant | [353] |
| 12 weeks | 1.5 g ginger per day; placebo | Serum hs-CRP and mRNA levels of IL-1β, IL-2, TNF-α | Ginger powder supplementation caused significant decline in CRP and IL-1β mRNA levels; TNFα mRNA levels reduced in ginger group compared to placebo group but was statistically insignificant; no effects on IL2 gene expression | [353] | |
| Saffron | 12 weeks | 100 mg/day saffron supplement or placebo | DAS28; ESR; hs-CRP; TNF-α; IFN-γ | Saffron supplementation significantly decreased number of tender and swollen joints; DAS28, hs-CRP, TNF-α, IFN-γ, and malondialdehyde were decreased and total antioxidant capacity was increased | [354,355] |
| Quercetin | 8 weeks | 500 mg/day quercetin or placebo | hs-TNFα; ESR; EMS; TSC; SJC; DAS-28; PGA | Significantly reduced EMS, DAS-28, and plasma hs-TNFα levels in quercetin group; no significant differences in TJC and SJC between groups | [356] |
| Resveratrol | 3 months | 1 g resveratrol with conventional treatment and control group with regular treatment | Clinical and biochemical markers | DAS-28 was significantly lowered in resveratrol-treated group; ESR, CRP, MMP3, TNF-alpha, and IL6 were also decreased | [357] |
| Tea polyphenols | 8 weeks | 2.4 g/day Stachys schtschegleevii + 2.4 g/day black tea; placebo—2.4 g/day black tea; | s-CRP; IL-1β; MMPs; DAS28 | Stachys schtschegleevii intervention caused significant reductions in number and percent changes in tender joints and DAS28 and caused significant MMP-3 reductions | [360] |
| 6 months | i.v. infusion of Infliximab at dose of 3 mg/kg at baseline, at 2 and 6 weeks later, then every 8 weeks; green tea was supplemented at dose of 4–6 cups/day (60 to 125 mg catechins) | CRP; ESR; DAS28-ESR | Significant decrease (p < 0.01) in disease activity parameters (CRP, ESR, and DAS28-ESR) was observed in patients treated with green tea compared with those treated with infliximab or exercise program; TJC and SJC were significantly decreased after 6 months of therapy | [361] | |
| Cinnamon | 8 weeks | 2000 mg cinnamon powder or placebo daily | Fasting blood sugar (FBS); lipid profile; liver enzymes; CRP; TNF-α; ESR; blood pressure; clinical symptoms | Significant decrease in serum levels of CRP and TNF-α in cinnamon group compared to placebo group; cinnamon intake significantly reduced DAS-28, VAS, TJC, and SJC; no significant changes observed for FBS, lipid profile, liver enzymes, or ESR | [362] |
| Sesamin | 6 weeks | 200 mg/day sesamin supplement or placebo | Serum levels of hyaluronidase, aggrecanase, MMP-3; hs-CRP, IL-1β, IL-6, TNF-α, cyclooxygenase-2 | Serum levels of hyaluronidase and MMP-3, hs-CRP, TNF-α, and cyclooxygenase-2 decreased significantly in sesamin group compared with placebo group; sesamin supplementation also caused significant reduction in number of tender joints and severity of pain in these patients | [363] |
| 6 weeks | 200 mg/day sesamin supplement or placebo | Serum concentrations of lipid profile; malondialdehyde (MDA); total antioxidant capacity (TAC) | Sesamin supplementation significantly decreased serum levels of MDA and increased TAC and HDL-C levels in patients with RA; means of weight, body mass index, waist-to-hip ratio, body fat, systolic blood pressure, and concentration of other lipid profiles (triglycerides, total cholesterol, and low-density lipoprotein cholesterol [LDL-C]) were also significantly decreased at end of study compared to baseline values | [364] | |
| Olive oil | 12 and 24 weeks | 3 g/d fish oil omega-3 fatty acids, 3 g/d fish oil omega-3 fatty acids, and 9.6 mL of olive oil; placebo soy oil supplementation | Clinical and laboratory indicators | Significant improvement in joint pain intensity, right and left handgrip strength after 12 and 24 wk, and Ritchie’s articular index for pain joints after 24 wk | [365] |
| Coenzyme Q10 | 2 months | 100 mg/day CoQ10 or placebo | Serum MMP-1 and MMP-3; DAS-28 | Significant reduction was observed in both CoQ10 and placebo groups in medians of serum MMP-1 and swollen joint count and means of DAS-28; significant reductions were only observed in ESR, pain score, and tender joint count in CoQ10 group compared with baseline | [366] |
| 2 months | 100 mg/day capsules of CoQ10 and placebo | MDA; total antioxidant capacity (TAC); IL-6; TNF-α | Serum MDA significantly decreased in supplemented group; suppressed overexpression of TNF-α; no significant difference in TAC and IL-6 levels between groups | [367] | |
| Baicalin | 12 weeks | 500 mg baicalin or placebo/daily | Lipid profile; cardiotrophin-1 (CT-1); high-sensitivity C-reactive protein (hs-CRP) | Levels of triglycerides, total cholesterol, LDL-cholesterol, and apolipoproteins, as well as CT-1 and hs-CRP, were all significantly improved in baicalin group compared to placebo group | [368] |
| Extract of Tripterygium wilfordii Hook F | 20 weeks | 180 mg/day or 360 mg/day extract or placebo | Disease activity and treatment response were evaluated according to ACR criteria; morning stiffness and serum titers of RF | 80% of high-dose group and 40% in low-dose group experienced disease improvement, fulfilling ACR 20% improvement criteria; no patients in placebo group attained these criteria Significant decreases were also found in number of tender joints, number of swollen joints, and physician’s global assessment in low-dose group | [369] |
| 24 weeks with 18 additional months for monitoring | 60 mg TwHF/day; 7.5 mg MTX/weekly; 60 mg TwHF plus 7.5 mg MTX | ACR criteria, HAQ, ESR or serum CRP level, EULAR; cDAI DAS28 | Disease activity in patients from combination and MTX monotherapy groups; significant differences in ACR20, EULAR good response, and DAS remission rate at year 2; all treatment groups had decreases in DAS28 and HAQ scores and increases in SF36 scores | [370] | |
| Uncaria tomentosa | 52 weeks; 2-phase study | First phase (24 weeks), patients were treated with Uncaria tomentosa extract or placebo; second phase (28 weeks), all patients received plant extract | Number of swollen and painful joints and Ritchie Index; VAS; HAQ; ESR, CRP, RF, antinuclear antibodies, complete blood count, hepatic and renal variables | Uncaria tomentosa extract resulted in reduction in number of painful joints compared to placebo; there was no change in HAQ | [371] |
| Punica granatum | 12 weeks | 10 mL/day pomegranate extract | Joint status and serum oxidative status (lipid peroxidation, total thiols group, paraoxonase 1 activity) | Pomegranate extract consumption significantly reduced DAS28; reduction in serum oxidative status; increase in serum high-density lipoprotein-associated paraoxonase 1 (PON1) activity | [372] |
| 8 weeks | 500 mg pomegranate extract per day and 500 mg cellulose for control group | HAQ; DAS- 28; CRP; MMP3; MDA; glutathione peroxidase (GPx); erythrocyte sedimentation rate (ESR) | Pomegranate extract supplementation significantly reduced score of DAS28 and tender joint count, pain intensity, and ESR levels compared to placebo; HAQ score and morning stiffness were also decreased; no differences in MMP3, CRP, and MDA levels between two groups | [373] | |
| Garlic | 8 weeks | 1000 mg garlic or placebo daily | Serum levels of total antioxidant capacity (TAC) and malondialdehyde (MDA); HAQ | Significant increase in serum levels of TAC in garlic group compared with placebo group; MDA levels were significantly decreased in intervention group compared with control group; HAQ scores decreased in garlic group compared with placebo | [374] |
| Stephania tetrandra | 12 weeks | 10 g Stephania tetrandra | Tender joint count, swollen joint count, patient’s assessment of pain, patient’s global assessment of disease activity, physician’s global assessment of disease activity, HAQ, CRP, ACR score IgM rheumatoid factor (IgMRF) concentration | Proportion of granulocytes and granulocyte count in peripheral blood decreased significantly; lipid peroxide and human granulocyte elastase levels of stored plasma declined significantly | [375] |
| Andrographis paniculata | 14 weeks | 30 mg of andrographolides three times a day; placebo group | Clinical signs and symptoms of pain and swelling evaluated by VAPS, ACR, EULAR, SF36 | Decreased intensity of joint pain in active group vs. placebo group; reduction in rheumatoid factor, IgA, and C4 | [376] |
| Mangifera indica L. Extract | 180 days | 900 mg/day Mangifera indica extract combined with 12.5 mg/week methotrexate and 5–10 mg/day NSAIDs and/or prednisone; control group on usual treatment | Tender and swollen joint counts, ESR, DAS 28, VAS, HAQ; treatment’s efficacy demonstrated with ACR criteria | Only patients in MTX Mangifera indica extract group revealed statistically significant improvement in DAS 28 parameters with respect to baseline data but no differences were observed between groups; ACR improvements amounted to 80% only in MTX Mangifera indica extract group at 90 days and decreased NSAID administration | [377] |
| Artemisia annua extract | 12, 24 and 48 weeks | 30 g/day extract of Artemisia annua L.; control group treated with leflflunomide and methotrexate | Pain score, tenderness score, number of painful joints, number of swollen joints, HAQ score for quality of life, RF levels, CCP-Ab, ESR, CRP, VAS score | At 24 and 48 weeks, overall efficacy of extract of Artemisia annua L-treated group was significantly higher than control group; tenderness score, number of painful joints, number of swollen joints, HAQ, CRP, RF, and CCP-Ab were significantly improved compared with control group after 48 weeks of treatment | [378] |
| Black barberry hydroalcoholic extract | 12 weeks | 2000 mg black barberry extract or maltodextrin as placebo/daily | Cytokines IL-2, IL-4, IL-10, and IL-17 in blood sample; physical activity; dietary intake; disease activity | Black barberry supplementation reduced severity of RA; IL-17 levels decreased significantly after intervention within black barberry group, while IL-10 had significant increase in this group; no significant effect on IL-2 and IL-4 cytokines | [379] |
| Nigella sativa oil extract | 8 weeks | 1000 mg Nigella sativa oil/daily or placebo | Serum TNF-α, IL-10, and whole blood levels of oxidative stress parameters | Serum level of IL-10 was increased in Nigella sativa group; significant reduction in serum MDA and NO compared with baseline; no significant differences in TNF-α, SOD, catalase, and TAS values between or within groups | [380] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nikolova-Ganeva, K.A.; Mihaylova, N.M.; Kechidzhieva, L.A.; Ivanova, K.I.; Zarkov, A.S.; Parzhanov, D.L.; Ivanov, M.M.; Marchev, A.S. The Therapeutic Potential of Phytochemicals Unlocks New Avenues in the Management of Rheumatoid Arthritis. Int. J. Mol. Sci. 2025, 26, 6813. https://doi.org/10.3390/ijms26146813
Nikolova-Ganeva KA, Mihaylova NM, Kechidzhieva LA, Ivanova KI, Zarkov AS, Parzhanov DL, Ivanov MM, Marchev AS. The Therapeutic Potential of Phytochemicals Unlocks New Avenues in the Management of Rheumatoid Arthritis. International Journal of Molecular Sciences. 2025; 26(14):6813. https://doi.org/10.3390/ijms26146813
Chicago/Turabian StyleNikolova-Ganeva, Kalina A., Nikolina M. Mihaylova, Lidiya A. Kechidzhieva, Kristina I. Ivanova, Alexander S. Zarkov, Daniel L. Parzhanov, Momchil M. Ivanov, and Andrey S. Marchev. 2025. "The Therapeutic Potential of Phytochemicals Unlocks New Avenues in the Management of Rheumatoid Arthritis" International Journal of Molecular Sciences 26, no. 14: 6813. https://doi.org/10.3390/ijms26146813
APA StyleNikolova-Ganeva, K. A., Mihaylova, N. M., Kechidzhieva, L. A., Ivanova, K. I., Zarkov, A. S., Parzhanov, D. L., Ivanov, M. M., & Marchev, A. S. (2025). The Therapeutic Potential of Phytochemicals Unlocks New Avenues in the Management of Rheumatoid Arthritis. International Journal of Molecular Sciences, 26(14), 6813. https://doi.org/10.3390/ijms26146813