
Sarcopenia: Current Insights into Molecular Mechanisms, Diagnostics, and Emerging Interventional Approaches


 , ,
, ,  and
and 
Abstract
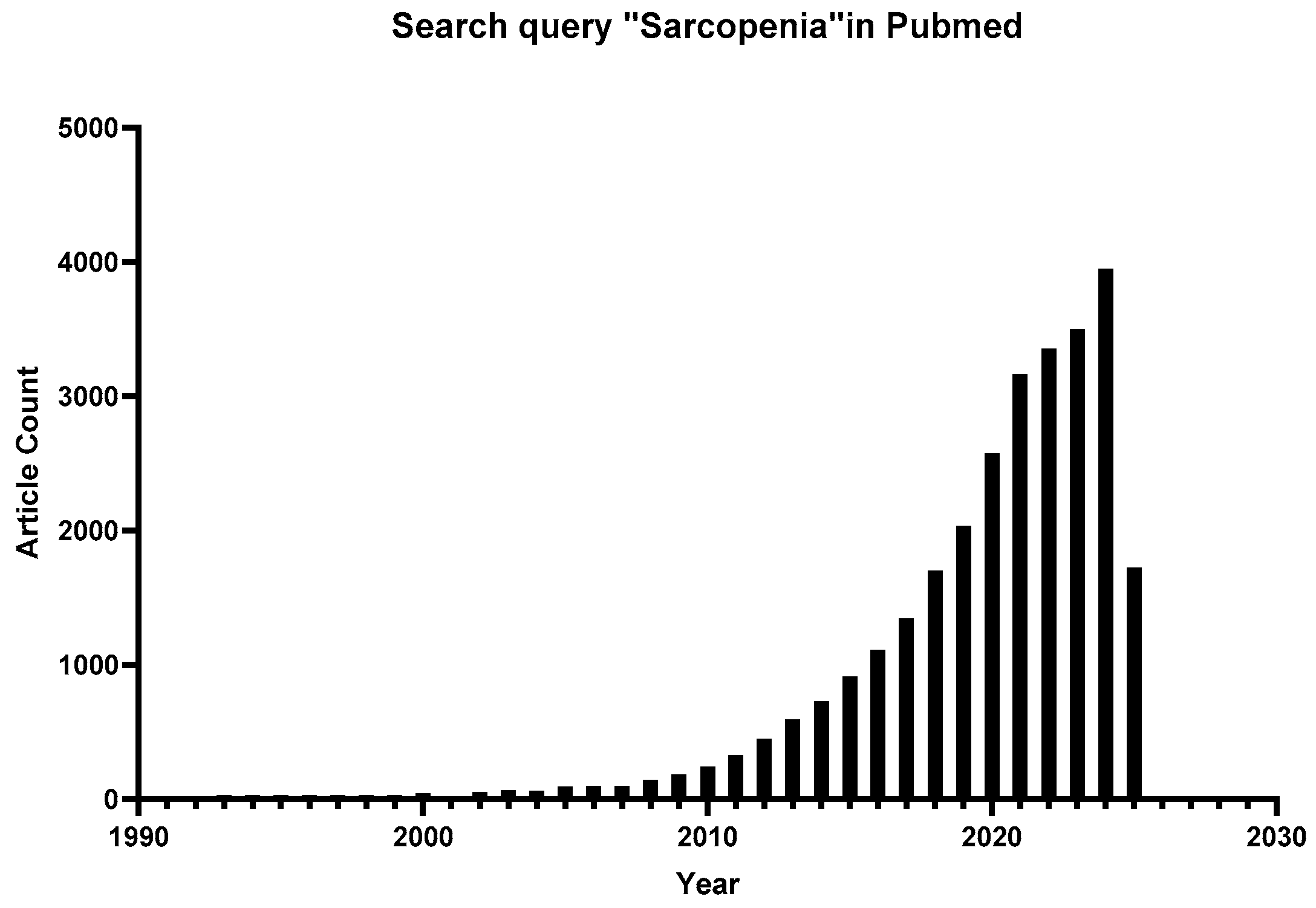
1. Introduction
2. Molecular Mechanisms: Decoding the Multisystem Interplay
2.1. Upstream Triggers
2.1.1. Mitochondrial Disorders and Oxidative Injury
2.1.2. Chronic Low-Grade Inflammation
2.1.3. Hormone and Growth Factor Imbalances
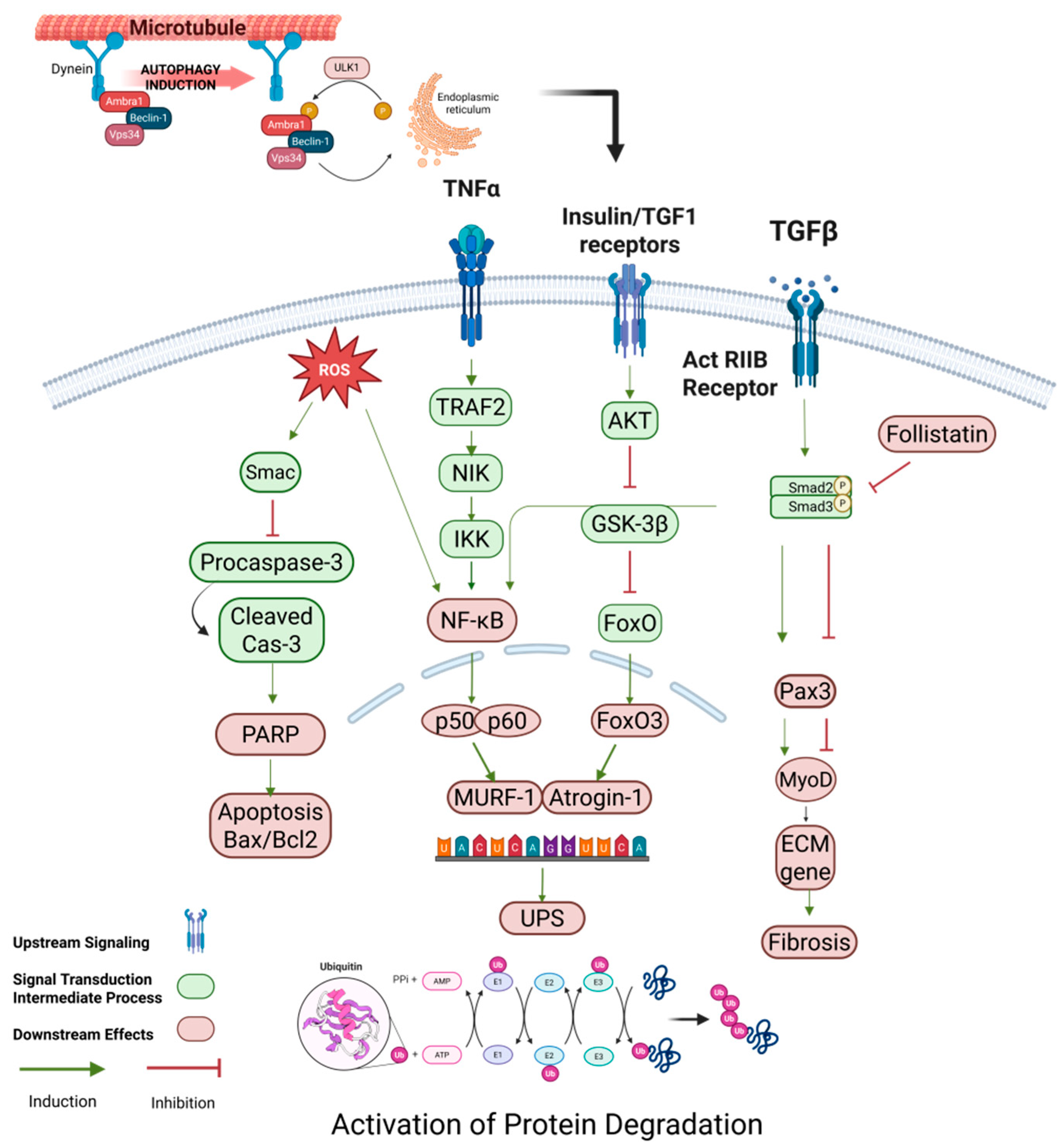
2.2. Activation of Protein Degradation Pathways
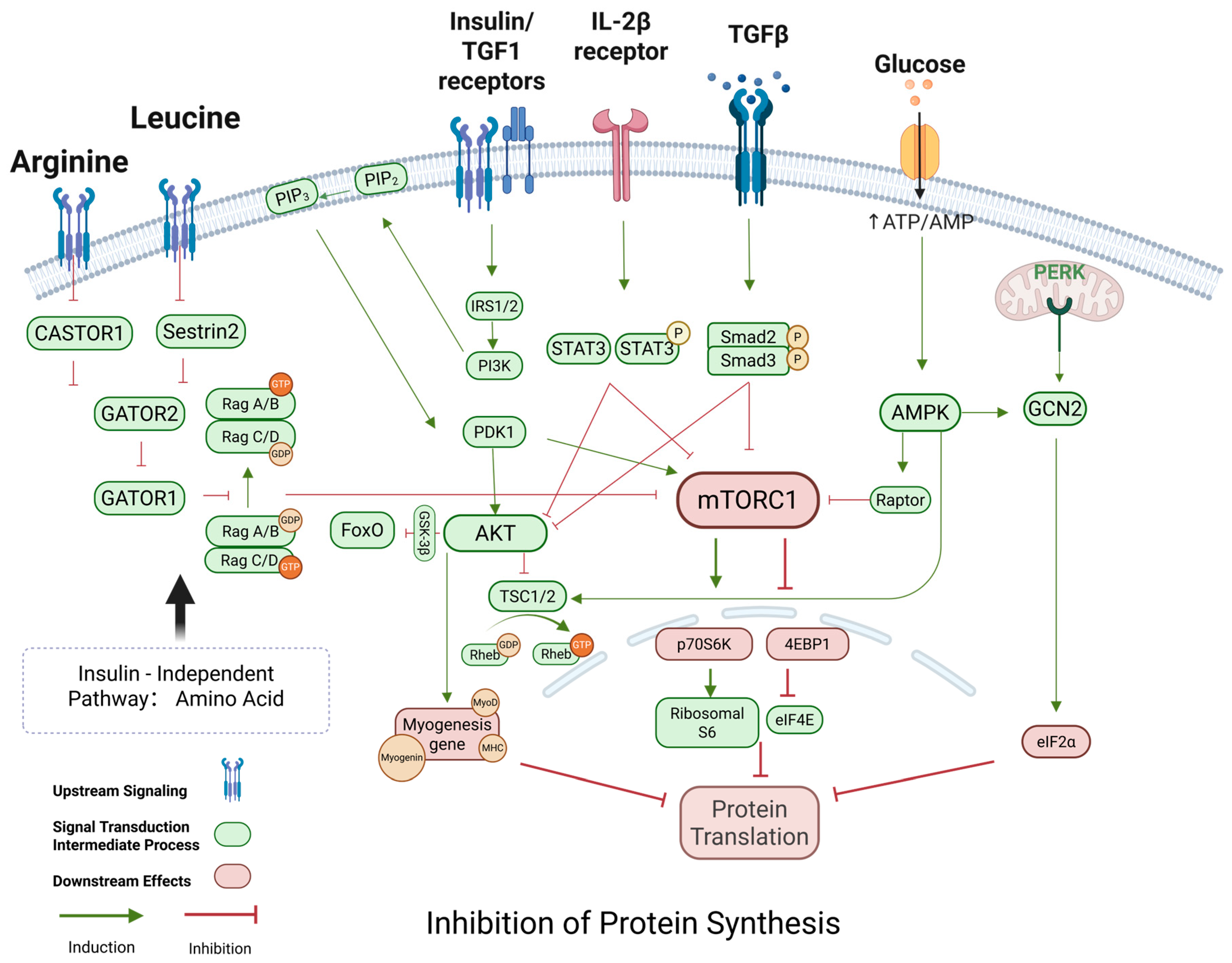
2.3. Protein Synthesis Inhibition
2.4. Stem Cells and Regenerative Disorders
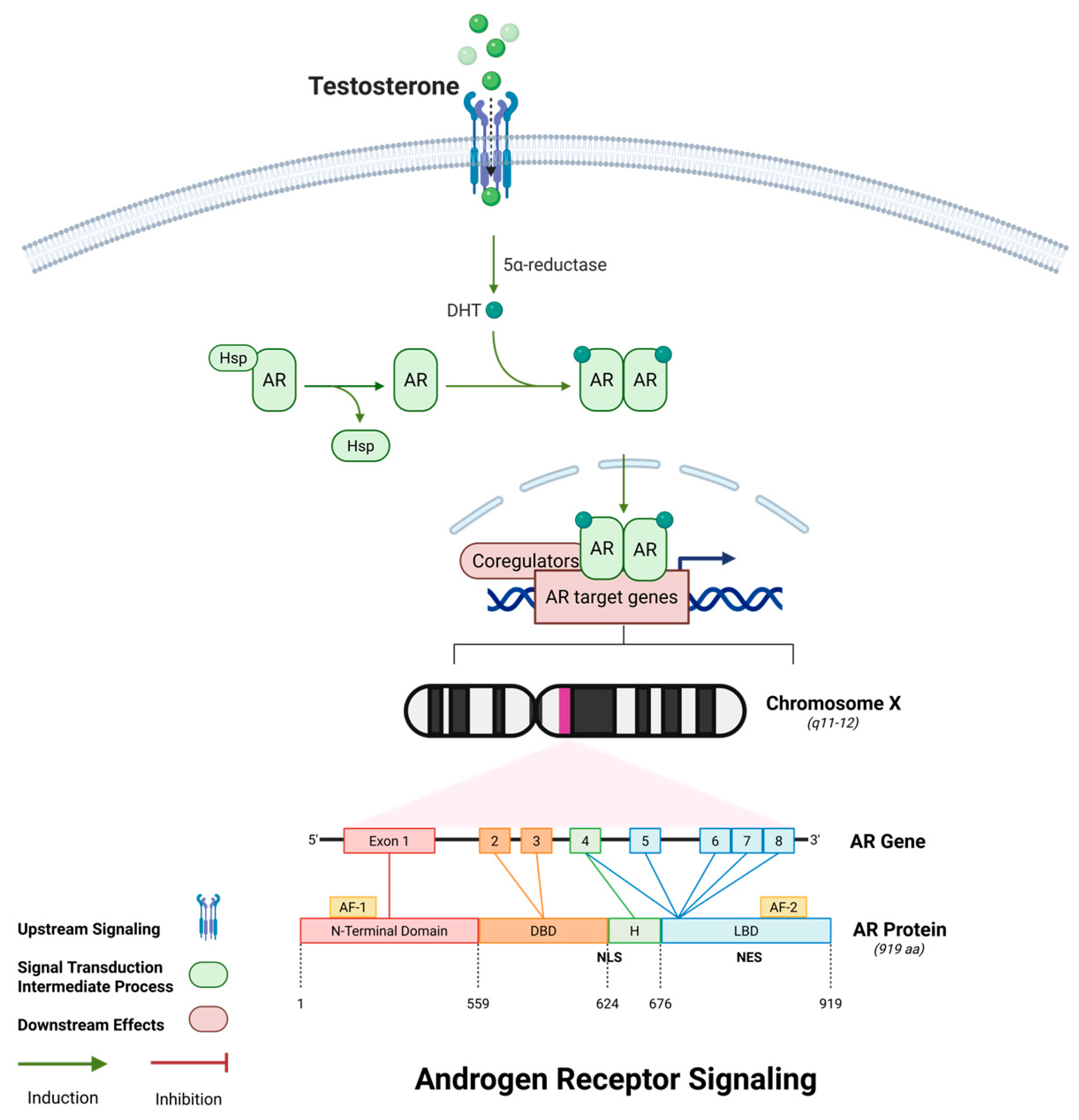
2.5. Androgen Receptor Signaling and Cytoskeletal Stability
3. Diagnostic Advancements: From Traditional to Precision Tools
3.1. Traditional Tools
3.1.1. Screening Questionnaires
3.1.2. Recommendations for Diagnosing
3.2. Precision Tool
3.2.1. Dual-Energy X-Ray Absorptiometry (DXA) in Diagnosis of Sarcopenia
3.2.2. Bioelectrical Impedance (BIA) in Diagnosis of Sarcopenia
3.2.3. Computed Tomography (CT)/Magnetic Resonance Imaging (MRI) as the Gold Standards
3.2.4. Ultrasound Imaging
3.3. Current Landscape of Sarcopenia Biomarkers
4. Interventions and Strategies for Skeletal Muscle Atrophy
4.1. Nutritional and Exercise Interventions
4.2. Pharmacological and Molecular Interventions
5. Challenges and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Argilés, J.M.; Campos, N.; Lopez-Pedrosa, J.M.; Rueda, R.; Rodriguez-Mañas, L. Skeletal Muscle Regulates Metabolism via Interorgan Crosstalk: Roles in Health and Disease. J. Am. Med. Dir. Assoc. 2016, 17, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.-P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Jentoft, A.J.; Landi, F.; Schneider, S.M.; Zuniga, C.; Arai, H.; Boirie, Y.; Chen, L.-K.; Fielding, R.A.; Martin, F.C.; Michel, J.-P.; et al. Prevalence of and interventions for sarcopenia in ageing adults: A systematic review. Report of the International Sarcopenia Initiative (EWGSOP and IWGS). Age Ageing 2014, 43, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Petermann-Rocha, F.; Balntzi, V.; Gray, S.R.; Lara, J.; Ho, F.K.; Pell, J.P.; Celis-Morales, C. Global prevalence of sarcopenia and severe sarcopenia: A systematic review and meta-analysis. J. Cachexia Sarcopenia Muscle 2022, 13, 86–99. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyere, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Beaudart, C.; Alcazar, J.; Aprahamian, I.; Batsis, J.A.; Yamada, Y.; Prado, C.M.; Reginster, J.Y.; Sanchez-Rodriguez, D.; Lim, W.S.; Sim, M.; et al. Health outcomes of sarcopenia: A consensus report by the outcome working group of the Global Leadership Initiative in Sarcopenia (GLIS). Aging Clin. Exp. Res. 2025, 37, 100. [Google Scholar] [CrossRef]
- Kirk, B.; Cawthon, P.M.; Arai, H.; Ávila-Funes, J.A.; Barazzoni, R.; Bhasin, S.; Binder, E.F.; Bruyere, O.; Cederholm, T.; Chen, L.K.; et al. The Conceptual Definition of Sarcopenia: Delphi Consensus from the Global Leadership Initiative in Sarcopenia (GLIS). Age Ageing 2024, 53, afae052. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Bauer, J.; Biolo, G.; Cederholm, T.; Cesari, M.; Cruz-Jentoft, A.J.; Morley, J.E.; Phillips, S.; Sieber, C.; Stehle, P.; Teta, D.; et al. Evidence-based recommendations for optimal dietary protein intake in older people: A position paper from the PROT-AGE Study Group. J. Am. Med. Dir. Assoc. 2013, 14, 542–559. [Google Scholar] [CrossRef]
- Rizzoli, R.; Biver, E.; Brennan-Speranza, T.C. Nutritional intake and bone health. Lancet Diabetes Endocrinol. 2021, 9, 606–621. [Google Scholar] [CrossRef] [PubMed]
- Rooks, D.; Petricoul, O.; Praestgaard, J.; Bartlett, M.; Laurent, D.; Roubenoff, R. Safety and pharmacokinetics of bimagrumab in healthy older and obese adults with body composition changes in the older cohort. J. Cachexia Sarcopenia Muscle 2020, 11, 1525–1534. [Google Scholar] [CrossRef] [PubMed]
- Jean, W.H.; Hsieh, Y.W.; Lai, L.F.; Dewi, L.; Liao, Y.C.; Ye, M.; Yu, S.H.; Kao, C.L.; Huang, C.Y.; Kuo, C.H. Senolytic effect of high intensity interval exercise on human skeletal muscle. Aging (Albany NY) 2023, 15, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Ocampo, A.; Reddy, P.; Martinez-Redondo, P.; Platero-Luengo, A.; Hatanaka, F.; Hishida, T.; Li, M.; Lam, D.; Kurita, M.; Beyret, E.; et al. In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell 2016, 167, 1719–1733. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. Hallmarks of aging: An expanding universe. Cell 2023, 186, 243–278. [Google Scholar] [CrossRef]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Chen, T.; Dai, Z.; Mo, P.; Li, X.; Ma, Z.; Song, S.; Chen, X.; Luo, M.; Liang, K.; Gao, S.; et al. Clinical Characteristics and Outcomes of Older Patients with Coronavirus Disease 2019 (COVID-19) in Wuhan, China: A Single-Centered, Retrospective Study. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1788–1795. [Google Scholar] [CrossRef]
- Cho, M.R.; Lee, S.; Song, S.K. A Review of Sarcopenia Pathophysiology, Diagnosis, Treatment and Future Direction. J. Korean Med. Sci. 2022, 37, e146. [Google Scholar] [CrossRef]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musaro, A. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef]
- Agostini, D.; Gervasi, M.; Ferrini, F.; Bartolacci, A.; Stranieri, A.; Piccoli, G.; Barbieri, E.; Sestili, P.; Patti, A.; Stocchi, V.; et al. An Integrated Approach to Skeletal Muscle Health in Aging. Nutrients 2023, 15, 1802. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.; Garcia, J.M. Sarcopenia, Cachexia and Aging: Diagnosis, Mechanisms and Therapeutic Options—A Mini-Review. Gerontology 2014, 60, 294–305. [Google Scholar] [CrossRef] [PubMed]
- Arnold, W.D.; Clark, B.C. Neuromuscular junction transmission failure in aging and sarcopenia: The nexus of the neurological and muscular systems. Ageing Res. Rev. 2023, 89, 101966. [Google Scholar] [CrossRef] [PubMed]
- Bernet, J.D.; Doles, J.D.; Hall, J.K.; Tanaka, K.K.; Carter, T.A.; Olwin, B.B. p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat. Med. 2014, 20, 265–271. [Google Scholar] [CrossRef]
- Zong, Y.; Li, H.; Liao, P.; Chen, L.; Pan, Y.; Zheng, Y.; Zhang, C.; Liu, D.; Zheng, M.; Gao, J. Mitochondrial dysfunction: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2024, 9, 124. [Google Scholar] [CrossRef]
- Chen, X.; Ji, Y.; Liu, R.; Zhu, X.; Wang, K.; Yang, X.; Liu, B.; Gao, Z.; Huang, Y.; Shen, Y.; et al. Mitochondrial dysfunction: Roles in skeletal muscle atrophy. J. Transl. Med. 2023, 21, 503. [Google Scholar] [CrossRef]
- Kandarian, S.C.; Jackman, R.W. Intracellular signaling during skeletal muscle atrophy. Muscle Nerve 2006, 33, 155–165. [Google Scholar] [CrossRef]
- Liguori, I.; Russo, G.; Curcio, F.; Bulli, G.; Aran, L.; Della-Morte, D.; Gargiulo, G.; Testa, G.; Cacciatore, F.; Bonaduce, D.; et al. Oxidative stress, aging, and diseases. Clin. Interv. Aging 2018, 13, 757–772. [Google Scholar] [CrossRef]
- Casanova, A.; Wevers, A.; Navarro-Ledesma, S.; Pruimboom, L. Mitochondria: It is all about energy. Front. Physiol. 2023, 14, 1114231. [Google Scholar] [CrossRef]
- Marzetti, E.; Calvani, R.; Cesari, M.; Buford, T.W.; Lorenzi, M.; Behnke, B.J.; Leeuwenburgh, C. Mitochondrial dysfunction and sarcopenia of aging: From signaling pathways to clinical trials. Int. J. Biochem. Cell Biol. 2013, 45, 2288–2301. [Google Scholar] [CrossRef]
- Guo, Y.; Guan, T.; Shafiq, K.; Yu, Q.; Jiao, X.; Na, D.; Li, M.; Zhang, G.; Kong, J. Mitochondrial dysfunction in aging. Ageing Res. Rev. 2023, 88, 101955. [Google Scholar] [CrossRef] [PubMed]
- Priego, T.; Martin, A.I.; Gonzalez-Hedstrom, D.; Granado, M.; Lopez-Calderon, A. Role of hormones in sarcopenia. Vitam. Horm. 2021, 115, 535–570. [Google Scholar] [CrossRef] [PubMed]
- Tournadre, A.; Vial, G.; Capel, F.; Soubrier, M.; Boirie, Y. Sarcopenia. Jt. Bone Spine 2019, 86, 309–314. [Google Scholar] [CrossRef]
- Ryall, J.G.; Schertzer, J.D.; Lynch, G.S. Cellular and molecular mechanisms underlying age-related skeletal muscle wasting and weakness. Biogerontology 2008, 9, 213–228. [Google Scholar] [CrossRef]
- Siebel, C.; Lendahl, U. Notch Signaling in Development, Tissue Homeostasis, and Disease. Physiol. Rev. 2017, 97, 1235–1294. [Google Scholar] [CrossRef]
- Zhou, J.; Liu, B.; Liang, C.; Li, Y.; Song, Y.H. Cytokine Signaling in Skeletal Muscle Wasting. Trends Endocrinol. Metab. 2016, 27, 335–347. [Google Scholar] [CrossRef]
- Anand, A.; Nambirajan, A.; Kumar, V.; Agarwal, S.; Sharma, S.; Mohta, S.; Gopi, S.; Kaushal, K.; Gunjan, D.; Singh, N.; et al. Alterations in Autophagy and Mammalian Target of Rapamycin (mTOR) Pathways Mediate Sarcopenia in Patients with Cirrhosis. J. Clin. Exp. Hepatol. 2022, 12, 510–518. [Google Scholar] [CrossRef]
- Savova, M.S.; Mihaylova, L.V.; Tews, D.; Wabitsch, M.; Georgiev, M.I. Targeting PI3K/AKT signaling pathway in obesity. Biomed. Pharmacother. 2023, 159, 114244. [Google Scholar] [CrossRef]
- Zanou, N.; Gailly, P. Skeletal muscle hypertrophy and regeneration: Interplay between the myogenic regulatory factors (MRFs) and insulin-like growth factors (IGFs) pathways. Cell Mol. Life Sci. 2013, 70, 4117–4130. [Google Scholar] [CrossRef]
- Lee, S.J.; McPherron, A.C. Regulation of myostatin activity and muscle growth. Proc. Natl. Acad. Sci. USA 2001, 98, 9306–9311. [Google Scholar] [CrossRef]
- Sakuma, K.; Aoi, W.; Yamaguchi, A. Molecular mechanism of sarcopenia and cachexia: Recent research advances. Pflug. Arch. 2017, 469, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Kalinkovich, A.; Livshits, G. Sarcopenia—The search for emerging biomarkers. Ageing Res. Rev. 2015, 22, 58–71. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Zhao, H.; Li, Y. Mitochondrial dynamics in health and disease: Mechanisms and potential targets. Signal Transduct. Target. Ther. 2023, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, J.M. Androgens, bone and muscles. Osteoporos. Int. 2020, 31, S78. [Google Scholar] [CrossRef]
- Rom, O.; Reznick, A.Z. The role of E3 ubiquitin-ligases MuRF-1 and MAFbx in loss of skeletal muscle mass. Free. Radic. Biol. Med. 2016, 98, 218–230. [Google Scholar] [CrossRef]
- Wiedmer, P.; Jung, T.; Castro, J.P.; Pomatto, L.C.D.; Sun, P.Y.; Davies, K.J.A.; Grune, T. Sarcopenia - Molecu-lar mechanisms and open questions. Ageing Res. Rev. 2021, 65, 101200. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-κB pathway for the therapy of diseases: Mechanism and clinical study. Signal Transduct. Target. Ther. 2020, 5, 209. [Google Scholar] [CrossRef]
- Wang, J.; Qin, X.; Huang, Y.; Zhang, Q.; Pei, J.; Wang, Y.; Goren, I.; Ma, S.; Song, Z.; Liu, Y.; et al. TRIM7/RNF90 promotes autophagy via regulation of ATG7 ubiquitination during L. monocytogenes infec-tion. Autophagy 2023, 19, 1844–1862. [Google Scholar] [CrossRef]
- Magnone, M.; Emionite, L.; Guida, L.; Vigliarolo, T.; Sturla, L.; Spinelli, S.; Buschiazzo, A.; Marini, C.; Sam-buceti, G.; De Flora, A.; et al. Insulin-independent stimulation of skeletal muscle glucose uptake by low-dose abscisic acid via AMPK activation. Sci. Rep. 2020, 10, 1454. [Google Scholar] [CrossRef]
- Salucci, S.; Falcieri, E. Polyphenols and their potential role in preventing skeletal muscle atrophy. Nutr. Res. 2020, 74, 10–22. [Google Scholar] [CrossRef]
- Thalacker-Mercer, A.; Riddle, E.; Barre, L. Protein and amino acids for skeletal muscle health in aging. Adv. Food Nutr. Res. 2020, 91, 29–64. [Google Scholar] [CrossRef] [PubMed]
- Therdyothin, A.; Phiphopthatsanee, N.; Isanejad, M. The Effect of Omega-3 Fatty Acids on Sarcopenia: Mechanism of Action and Potential Efficacy. Mar. Drugs 2023, 21, 399. [Google Scholar] [CrossRef] [PubMed]
- Pullen, N.; Thomas, G. The modular phosphorylation and activation of p70s6k. FEBS Lett. 1997, 410, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Cleasby, M.E.; Jamieson, P.M.; Atherton, P.J. Insulin resistance and sarcopenia: Mechanistic links between common co-morbidities. J. Endocrinol. 2016, 229, R67–R81. [Google Scholar] [CrossRef]
- Cohen, S.; Nathan, J.A.; Goldberg, A.L. Muscle wasting in disease: Molecular mechanisms and promising therapies. Nat. Rev. Drug Discov. 2015, 14, 58–74. [Google Scholar] [CrossRef]
- Zhang, N.; Zhang, Y.; Xu, J.; Wang, P.; Wu, B.; Lu, S.; Lu, X.; You, S.; Huang, X.; Li, M.; et al. α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart fail-ure. Cell Res. 2023, 33, 679–698. [Google Scholar] [CrossRef]
- Silva, K.A.; Dong, J.; Dong, Y.; Dong, Y.; Schor, N.; Tweardy, D.J.; Zhang, L.; Mitch, W.E. Inhibition of Stat3 activation suppresses caspase-3 and the ubiquitin-proteasome system, leading to preservation of muscle mass in cancer cachexia. J. Biol. Chem. 2015, 290, 11177–11187. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, Z.; Shen, W.; Huang, G.; Sedivy, J.M.; Wang, H.; Ju, Z. Inflammation, epigenetics, and metabo-lism converge to cell senescence and ageing: The regulation and intervention. Signal Transduct. Tar-Geted Ther. 2021, 6, 245. [Google Scholar] [CrossRef]
- Buccitelli, C.; Selbach, M. mRNAs, proteins and the emerging principles of gene expression control. Nat. Rev. Genet. 2020, 21, 630–644. [Google Scholar] [CrossRef]
- Fujimaki, S.; Hidaka, R.; Asashima, M.; Takemasa, T.; Kuwabara, T. Wnt protein-mediated satellite cell conversion in adult and aged mice following voluntary wheel running. J. Biol. Chem. 2014, 289, 7399–7412. [Google Scholar] [CrossRef]
- Liu, F.; Liang, Z.; Xu, J.; Li, W.; Zhao, D.; Zhao, Y.; Yan, C. Activation of the wnt/β-Catenin Signaling Path-way in Polymyositis, Dermatomyositis and Duchenne Muscular Dystrophy. J. Clin. Neurol. 2016, 12, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Acar, A.; Hidalgo-Sastre, A.; Leverentz, M.K.; Mills, C.G.; Woodcock, S.; Baron, M.; Collu, G.M.; Brennan, K. Inhibition of Wnt signalling by Notch via two distinct mechanisms. Sci. Rep. 2021, 11, 9096. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Tian, X.; He, L.; Liu, D.; Li, J.; Mei, Z.; Zhou, T.; Liu, C.; He, J.; Jia, X.; et al. CREG1 deficiency im-paired myoblast differentiation and skeletal muscle regeneration. J. Cachexia Sarcopenia Muscle 2024, 15, 587–602. [Google Scholar] [CrossRef]
- Ma, J.; Li, Y.; Yang, X.; Liu, K.; Zhang, X.; Zuo, X.; Ye, R.; Wang, Z.; Shi, R.; Meng, Q.; et al. Signaling pathways in vascular function and hypertension: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 168. [Google Scholar] [CrossRef]
- Pang, X.; He, X.; Qiu, Z.; Zhang, H.; Xie, R.; Liu, Z.; Gu, Y.; Zhao, N.; Xiang, Q.; Cui, Y. Targeting integrin pathways: Mechanisms and advances in therapy. Signal Transduct. Target. Ther. 2023, 8, 1. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, Q.L.; Sun, W.; Chandrasekharan, P.; Cheng, H.S.; Ying, Z.; Lakshmanan, M.; Raju, A.; Tenen, D.G.; Cheng, S.Y.; et al. Non-canonical NF-κB signalling and ETS1/2 cooperatively drive C250T mutant TERT promoter activation. Nat. Cell Biol. 2015, 17, 1327–1338. [Google Scholar] [CrossRef]
- Li, C.W.; Yu, K.; Shyh-Chang, N.; Li, G.X.; Jiang, L.J.; Yu, S.L.; Xu, L.Y.; Liu, R.J.; Guo, Z.J.; Xie, H.Y.; et al. Circulating factors associated with sarcopenia during ageing and after intensive lifestyle intervention. J. Ca-Chexia Sarcopenia Muscle 2019, 10, 586–600. [Google Scholar] [CrossRef]
- Andersen, P.; Uosaki, H.; Shenje, L.T.; Kwon, C. Non-canonical Notch signaling: Emerging role and mechanism. Trends Cell Biol. 2012, 22, 257–265. [Google Scholar] [CrossRef]
- Yeh, C.-J.; Sattler, K.M.; Lepper, C. Molecular regulation of satellite cells via intercellular signaling. Gene 2023, 858, 147172. [Google Scholar] [CrossRef]
- Lennicke, C.; Cochemé, H.M. Redox metabolism: ROS as specific molecular regulators of cell signaling and function. Mol. Cell 2021, 81, 3691–3707. [Google Scholar] [CrossRef]
- Tajrishi, M.M.; Zheng, T.S.; Burkly, L.C.; Kumar, A. The TWEAK-Fn14 pathway: A potent regulator of skeletal muscle biology in health and disease. Cytokine Growth Factor Rev. 2014, 25, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Rozpędek-Kamińska, W.; Siwecka, N.; Wawrzynkiewicz, A.; Wojtczak, R.; Pytel, D.; Diehl, J.A.; Majsterek, I. The PERK-Dependent Molecular Mechanisms as a Novel Therapeutic Target for Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 2108. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, P.M.; Corbel, S.; Doyonnas, R.; Havenstrite, K.; Magnusson, K.E.; Blau, H.M. A single cell bioengineering approach to elucidate mechanisms of adult stem cell self-renewal. Integr. Biol. 2012, 4, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Xu, Y.Q.; Yuan, Y.X.; Xu, P.W.; Zhang, C.; Li, F.; Wang, L.N.; Yin, C.; Zhang, L.; Cai, X.C.; et al. Succinate induces skeletal muscle fiber remodeling via SUNCR1 signaling. EMBO Rep. 2019, 20, e47892. [Google Scholar] [CrossRef]
- Huang, T.; Zhou, M.Y.; Zou, G.L.; Hu, R.H.; Han, L.; Zhang, Q.X.; Zhao, X.K. Focal adhesion kinase promotes aerobic glycolysis in hepatic stellate cells via the cyclin D1/c-Myc/MCT-1 pathway to induce liver fibrosis. Sci. Rep. 2025, 15, 4552. [Google Scholar] [CrossRef]
- Zhao, L.; Liu, X.; Zhang, J.; Dong, G.; Xiao, W.; Xu, X. Hydrogen Sulfide Alleviates Skeletal Muscle Fibrosis via Attenuating Inflammation and Oxidative Stress. Front. Physiol. 2020, 11, 533690. [Google Scholar] [CrossRef]
- Moiseeva, V.; Cisneros, A.; Sica, V.; Deryagin, O.; Lai, Y.; Jung, S.; Andrés, E.; An, J.; Segalés, J.; Ortet, L.; et al. Senescence atlas reveals an aged-like inflamed niche that blunts muscle regeneration. Nature 2023, 613, 169–178. [Google Scholar] [CrossRef]
- Chellini, F.; Tani, A.; Zecchi-Orlandini, S.; Sassoli, C. Influence of Platelet-Rich and Platelet-Poor Plasma on Endogenous Mechanisms of Skeletal Muscle Repair/Regeneration. Int. J. Mol. Sci. 2019, 20, 683. [Google Scholar] [CrossRef]
- Dudley, A.C.; Griffioen, A.W. Nectins and Nectin-like molecules drive vascular development and barrier function. Angiogenesis 2023, 26, 313–347. [Google Scholar] [CrossRef]
- Cosgrove, B.D.; Sacco, A.; Gilbert, P.M.; Blau, H.M. A home away from home: Challenges and opportunities in engineering in vitro muscle satellite cell niches. Differentiation 2009, 78, 185–194. [Google Scholar] [CrossRef]
- Carlson, M.E.; Hsu, M.; Conboy, I.M. Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 2008, 454, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Rodgers, K.D.; San Antonio, J.D.; Jacenko, O. Heparan sulfate proteoglycans: A GAGgle of skeletal-hematopoietic regulators. Dev. Dyn. 2008, 237, 2622–2642. [Google Scholar] [CrossRef] [PubMed]
- Sansone, A.; Romanelli, F. Hormones in aging. In Human Aging; Elsevier: Amsterdam, The Netherlands, 2021; pp. 207–217. [Google Scholar]
- Di, X.; Gao, X.; Peng, L.; Ai, J.; Jin, X.; Qi, S.; Li, H.; Wang, K.; Luo, D. Cellular mechanotransduction in health and diseases: From molecular mechanism to therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Cai, H.L.; Bao, J.P.; Wu, L.D. Dehydroepiandrosterone and age-related musculoskeletal diseases: Connections and therapeutic implications. Ageing Res. Rev. 2020, 62, 101132. [Google Scholar] [CrossRef]
- La Colla, A.; Pronsato, L.; Milanesi, L.; Vasconsuelo, A. 17beta-Estradiol and testosterone in sarcopenia: Role of satellite cells. Ageing Res. Rev. 2015, 24, 166–177. [Google Scholar] [CrossRef]
- Liu, L.; Charville, G.W.; Cheung, T.H.; Yoo, B.; Santos, P.J.; Schroeder, M.; Rando, T.A. Impaired Notch Signaling Leads to a Decrease in p53 Activity and Mitotic Catastrophe in Aged Muscle Stem Cells. Cell Stem Cell 2018, 23, 544–556. [Google Scholar] [CrossRef]
- Girardi, F.; Le Grand, F. Chapter Five—Wnt Signaling in Skeletal Muscle Development and Regeneration. In Progress in Molecular Biology and Translational Science; Larraín, J., Olivares, G., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 153, pp. 157–179. [Google Scholar]
- Domingues-Faria, C.; Vasson, M.P.; Goncalves-Mendes, N.; Boirie, Y.; Walrand, S. Skeletal muscle regeneration and impact of aging and nutrition. Ageing Res. Rev. 2016, 26, 22–36. [Google Scholar] [CrossRef]
- Tan, M.H.E.; Li, J.; Xu, H.E.; Melcher, K.; Yong, E.-l. Androgen receptor: Structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 2015, 36, 3–23. [Google Scholar] [CrossRef]
- Sakai, H.; Uno, H.; Yamakawa, H.; Tanaka, K.; Ikedo, A.; Uezumi, A.; Ohkawa, Y.; Imai, Y. The androgen receptor in mesenchymal progenitors regulates skeletal muscle mass via Igf1 expression in male mice. Proc. Natl. Acad. Sci. USA 2024, 121, e2407768121. [Google Scholar] [CrossRef]
- Bhasin, S.; Krishnan, V.; Storer, T.W.; Steiner, M.; Dobs, A.S. Androgens and Selective Androgen Receptor Modulators to Treat Functional Limitations Associated with Aging and Chronic Disease. J. Gerontol. Ser. A-Biol. Sci. Med. Sci. 2023, 78, 25–31. [Google Scholar] [CrossRef]
- Dillon, E.L.; Durham, W.J.; Urban, R.J.; Sheffield-Moore, M. Hormone treatment and muscle anabolism during aging: Androgens. Clin. Nutr. 2010, 29, 697–700. [Google Scholar] [CrossRef] [PubMed]
- Ghanim, H.; Dhindsa, S.; Batra, M.; Green, K.; Abuaysheh, S.; Kuhadiya, N.D.; Makdissi, A.; Chaudhuri, A.; Dandona, P. Effect of testosterone on FGF2, MRF4, and myostatin in hypogonadotropic hypogonadism: Relevance to muscle growth. J. Clin. Endocrinol. Metab. 2019, 104, 2094–2102. [Google Scholar] [CrossRef] [PubMed]
- Gentile, G.; De Stefano, F.; Sorrentino, C.; D’Angiolo, R.; Lauretta, C.; Giovannelli, P.; Migliaccio, A.; Castoria, G.; Di Donato, M. Androgens as the “old age stick” in skeletal muscle. Cell Commun. Signal 2025, 23, 167. [Google Scholar] [CrossRef] [PubMed]
- Barsky, S.T.; Monks, D.A. Androgen action on myogenesis throughout the lifespan; comparison with neurogenesis. Front. Neuroendocrinol. 2023, 71, 101101. [Google Scholar] [CrossRef]
- Di Donato, M.; Moretti, A.; Sorrentino, C.; Toro, G.; Gentile, G.; Iolascon, G.; Castoria, G.; Migliaccio, A. Filamin A cooperates with the androgen receptor in preventing skeletal muscle senescence. Cell Death Discov. 2023, 9, 437. [Google Scholar] [CrossRef]
- Gromova, A.; Cha, B.; Nguyen, N.; Garg, D.; Coscolluela, C.; Strickland, L.M.; Luong, D.; Longo, F.; Sopher, B.L.; ElMallah, M.K.; et al. Neuromuscular junction transcriptome analysis of spinal and bulbar muscular atrophy mice implicates sarcomere gene expression and calcium flux dysregulation in disease pathogenesis. Hum. Mol. Genet. 2025, 34, 1238–1251. [Google Scholar] [CrossRef]
- Lee, T.W.; Kao, P.Y.; Chen, Y.C.; Wang, S.T. Effects of Testosterone Replacement Therapy on Muscle Strength in Older Men with Low to Low-Normal Testosterone Levels: A Systematic Review and Meta-Analysis. Gerontology 2023, 69, 1157–1166. [Google Scholar] [CrossRef]
- Chen, L.-K.; Liu, L.-K.; Woo, J.; Assantachai, P.; Auyeung, T.-W.; Bahyah, K.S.; Chou, M.-Y.; Chen, L.-Y.; Hsu, P.-S.; Krairit, O.; et al. Sarcopenia in Asia: Consensus Report of the Asian Working Group for Sarcopenia. J. Am. Med. Dir. Assoc. 2014, 15, 95–101. [Google Scholar] [CrossRef]
- Chen, L.-K.; Woo, J.; Assantachai, P.; Auyeung, T.-W.; Chou, M.-Y.; Iijima, K.; Jang, H.C.; Kang, L.; Kim, M.; Kim, S.; et al. Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment. J. Am. Med. Dir. Assoc. 2020, 21, 300–307. [Google Scholar] [CrossRef]
- Fielding, R.A.; Vellas, B.; Evans, W.J.; Bhasin, S.; Morley, J.E.; Newman, A.B.; van Kan, G.A.; Andrieu, S.; Bauer, J.; Breuille, D.; et al. Sarcopenia: An Undiagnosed Condition in Older Adults. Current Consensus Definition: Prevalence, Etiology, and Consequences. International Working Group on Sarcopenia. J. Am. Med. Dir. Assoc. 2011, 12, 249–256. [Google Scholar] [CrossRef]
- Studenski, S.A.; Peters, K.W.; Alley, D.E.; Cawthon, P.M.; McLean, R.R.; Harris, T.B.; Ferrucci, L.; Guralnik, J.M.; Fragala, M.S.; Kenny, A.M.; et al. The FNIH Sarcopenia Project: Rationale, Study Description, Conference Recommendations, and Final Estimates. J. Gerontol. Ser. A-Biol. Sci. Med. Sci. 2014, 69, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Bauer, J.; Morley, J.E.; Schols, A.; Ferrucci, L.; Cruz-Jentoft, A.J.; Dent, E.; Baracos, V.E.; Crawford, J.A.; Doehner, W.; Heymsfield, S.B.; et al. Sarcopenia: A Time for Action. An SCWD Position Paper. J. Cachexia Sarcopenia Muscle 2019, 10, 956–961. [Google Scholar] [CrossRef] [PubMed]
- Bhasin, S.; Travison, T.G.; Manini, T.M.; Patel, S.; Pencina, K.M.; Fielding, R.A.; Magaziner, J.M.; Newman, A.B.; Kiel, D.P.; Cooper, C.; et al. Sarcopenia Definition: The Position Statements of the Sarcopenia Definition and Outcomes Consortium. J. Am. Geriatr. Soc. 2020, 68, 1410–1418. [Google Scholar] [CrossRef]
- Malmstrom, T.K.; Morley, J.E. SARC-F: A simple questionnaire to rapidly diagnose sarcopenia. J. Am. Med. Dir. Assoc. 2013, 14, 531–532. [Google Scholar] [CrossRef]
- Woo, J.; Leung, J.; Morley, J.E. Validating the SARC-F: A suitable community screening tool for sarcopenia? J. Am. Med. Dir. Assoc. 2014, 15, 630–634. [Google Scholar] [CrossRef]
- Bahat, G.; Oren, M.M.; Yilmaz, O.; Kılıç, C.; Aydin, K.; Karan, M.A. Comparing SARC-F with SARC-CalF to Screen Sarcopenia in Community Living Older Adults. J. Nutr. Health Aging 2018, 22, 1034–1038. [Google Scholar] [CrossRef]
- Krzymińska-Siemaszko, R.; Deskur-Śmielecka, E.; Kaluźniak-Szymanowska, A.; Murawiak, M.; Wieczorowska-Tobis, K. Comparison of Diagnostic Value of the SARC-F and Its Four Modified Versions in Polish Community-Dwelling Older Adults. Clin. Interv. Aging 2023, 18, 783–797. [Google Scholar] [CrossRef]
- Krzymińska-Siemaszko, R.; Deskur-Śmielecka, E.; Styszyński, A.; Wieczorowska-Tobis, K. Polish Translation and Validation of the Mini Sarcopenia Risk Assessment (MSRA) Questionnaire to Assess Nutritional and Non-Nutritional Risk Factors of Sarcopenia in Older Adults. Nutrients 2021, 13, 1061. [Google Scholar] [CrossRef]
- Rossi, A.P.; Micciolo, R.; Rubele, S.; Fantin, F.; Caliari, C.; Zoico, E.; Mazzali, G.; Ferrari, E.; Volpato, S.; Zamboni, M. Assessing the Risk of Sarcopenia in the Elderly: The Mini Sarcopenia Risk Assessment (MSRA) Questionnaire. J. Nutr. Health Aging 2017, 21, 743–749. [Google Scholar] [CrossRef]
- Yang, M.; Hu, X.; Xie, L.; Zhang, L.; Zhou, J.; Lin, J.; Wang, Y.; Li, Y.; Han, Z.; Zhang, D.; et al. Comparing Mini Sarcopenia Risk Assessment with SARC-F for Screening Sarcopenia in Community-Dwelling Older Adults. J. Am. Med. Dir. Assoc. 2019, 20, 53–57. [Google Scholar] [CrossRef]
- Jones, C.J.; Rikli, R.E.; Beam, W.C. A 30-s chair-stand test as a measure of lower body strength in community-residing older adults. Res. Q. Exerc. Sport 1999, 70, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Guralnik, J.M.; Simonsick, E.M.; Ferrucci, L.; Glynn, R.J.; Berkman, L.F.; Blazer, D.G.; Scherr, P.A.; Wallace, R.B. A short physical performance battery assessing lower extremity function: Association with self-reported disability and prediction of mortality and nursing home admission. J. Gerontol. 1994, 49, M85–M94. [Google Scholar] [CrossRef] [PubMed]
- Podsiadlo, D.; Richardson, S. The timed “Up & Go”: A test of basic functional mobility for frail elderly persons. J. Am. Geriatr. Soc. 1991, 39, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Albano, D.; Messina, C.; Vitale, J.; Sconfienza, L.M. Imaging of sarcopenia: Old evidence and new insights. Eur. Radiol. 2020, 30, 2199–2208. [Google Scholar] [CrossRef]
- Yuan, S.; Larsson, S.C. Epidemiology of sarcopenia: Prevalence, risk factors, and consequences. Metabolism 2023, 144, 155533. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Xu, M.; Zhang, Z.; He, L.; Li, Y. Sarcopenia prevalence and associated factors among older Chinese population: Findings from the China Health and Retirement Longitudinal Study. PLoS ONE 2021, 16, e0247617. [Google Scholar] [CrossRef]
- Sergi, G.; De Rui, M.; Veronese, N.; Bolzetta, F.; Berton, L.; Carraro, S.; Bano, G.; Coin, A.; Manzato, E.; Perissinotto, E. Assessing appendicular skeletal muscle mass with bioelectrical impedance analysis in free-living Caucasian older adults. Clin. Nutr. 2015, 34, 667–673. [Google Scholar] [CrossRef]
- Dent, E.; Morley, J.E.; Cruz-Jentoft, A.J.; Arai, H.; Kritchevsky, S.B.; Guralnik, J.; Bauer, J.M.; Pahor, M.; Clark, B.C.; Cesari, M.; et al. International Clinical Practice Guidelines for Sarcopenia (ICFSR): Screening, Diagnosis and Management. J. Nutr. Health Aging 2018, 22, 1148–1161. [Google Scholar] [CrossRef]
- González-Correa, C.H. Body Composition by Bioelectrical Impedance Analysis. In Bioimpedance in Biomedical Applications and Research; Simini, F., Bertemes-Filho, P., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 219–241. [Google Scholar]
- Ticinesi, A.; Meschi, T.; Narici, M.V.; Lauretani, F.; Maggio, M. Muscle Ultrasound and Sarcopenia in Older Individuals: A Clinical Perspective. J. Am. Med. Dir. Assoc. 2017, 18, 290–300. [Google Scholar] [CrossRef]
- Tosato, M.; Marzetti, E.; Cesari, M.; Savera, G.; Miller, R.R.; Bernabei, R.; Landi, F.; Calvani, R. Measurement of muscle mass in sarcopenia: From imaging to biochemical markers. Aging Clin. Exp. Res. 2017, 29, 19–27. [Google Scholar] [CrossRef]
- Yang, M.; Liu, Y.; Zuo, Y.; Tang, H. Sarcopenia for predicting falls and hospitalization in community-dwelling older adults: EWGSOP versus EWGSOP2. Sci. Rep. 2019, 9, 17636. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Shin, Y.; Huh, J.; Sung, Y.S.; Lee, I.-S.; Yoon, K.-H.; Kim, K.W. Recent Issues on Body Composition Imaging for Sarcopenia Evaluation. Korean J. Radiol. 2019, 20, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Engelke, K.; Museyko, O.; Wang, L.; Laredo, J.D. Quantitative analysis of skeletal muscle by computed tomography imaging-State of the art. J. Orthop. Transl. 2018, 15, 91–103. [Google Scholar] [CrossRef]
- Yang, F.; Zhu, L.; Cao, B.; Zeng, L.; Yuan, Z.; Tian, Y.; Li, Y.; Chen, X. Accuracy of Ultrasound Measurements of Muscle Thickness in Identifying Older Patients with Sarcopenia and Its Impact on Frailty: A Systematic Review and Meta-Analysis. J. Am. Med. Dir. Assoc. 2025, 26, 105419. [Google Scholar] [CrossRef]
- Nijholt, W.; Scafoglieri, A.; Jager-Wittenaar, H.; Hobbelen, J.S.M.; van der Schans, C.P. The reliability and validity of ultrasound to quantify muscles in older adults: A systematic review. J. Cachexia Sarcopenia Muscle 2017, 8, 702–712. [Google Scholar] [CrossRef]
- Rojas-Martínez, M.; Serna, L.Y.; Jordanic, M.; Marateb, H.R.; Merletti, R.; Mañanas, M.Á. High-density surface electromyography signals during isometric contractions of elbow muscles of healthy humans. Sci. Data 2020, 7, 397. [Google Scholar] [CrossRef]
- Kobylarz, M.D.; Valera-Calero, J.A.; Sánchez-Jorge, S.; Buffet-García, J.; Díaz-Arribas, M.J.; Ortega-Santiago, R.; Klich, S. Shear Wave Elastography for Measuring the Stiffness of Latent Trigger Points and Surrounding Areas in the Infraspinatus Muscle: Intra- and Interexaminer Reliability Analysis. Arch. Phys. Med. Rehabil. 2025, S0003-9993, 00616-1. [Google Scholar] [CrossRef]
- Jones, S.; Chiesa, S.T.; Chaturvedi, N.; Hughes, A.D. Recent developments in near-infrared spectroscopy (NIRS) for the assessment of local skeletal muscle microvascular function and capacity to utilise oxygen. Artery Res. 2016, 16, 25–33. [Google Scholar] [CrossRef]
- Schaap, L.A.; Pluijm, S.M.F.; Deeg, D.J.H.; Visser, M. Inflammatory Markers and Loss of Muscle Mass (Sarcopenia) and Strength. Am. J. Med. 2006, 119, 526.E9–526.E17. [Google Scholar] [CrossRef]
- Muzembo, B.A.; Nagano, Y.; Eitoku, M.; Ngatu, N.R.; Matsui, T.; Bhatti, S.A.; Hirota, R.; Ishida, K.; Suganuma, N. A cross-sectional assessment of oxidative DNA damage and muscle strength among elderly people living in the community. Environ. Health Prev. Med. 2014, 19, 21–29. [Google Scholar] [CrossRef]
- Rizk, J.; Sahu, R.; Duteil, D. An overview on androgen-mediated actions in skeletal muscle and adipose tissue. Steroids 2023, 199, 109306. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, S.; Burd, N.A.; van Loon, L.J. The Skeletal Muscle Anabolic Response to Plant- versus Animal-Based Protein Consumption. J. Nutr. 2015, 145, 1981–1991. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, T.; Carstens, M.; Millen, A.M. Whey or Casein Hydrolysate with Carbohydrate for Metabolism and Performance in Cycling. Int. J. Sports Med. 2015, 36, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.; Granic, A.; Cruz-Jentoft, A.J.; Sayer, A.A. The role of nutrition in the prevention of sarcopenia. Am. J. Clin. Nutr. 2023, 118, 852–864. [Google Scholar] [CrossRef]
- Visser, M.; Deeg, D.J.H.; Lips, P. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (Sarcopenia): The Longitudinal Aging Study Amsterdam. J. Clin. Endocrinol. Metab. 2003, 88, 5766–5772. [Google Scholar] [CrossRef]
- Zhang, F.; Li, W. Vitamin D and Sarcopenia in the Senior People: A Review of Mechanisms and Comprehensive Prevention and Treatment Strategies. Ther. Clin. Risk Manag. 2024, 20, 577–595. [Google Scholar] [CrossRef]
- Feng, L.; Li, B.; Yong, S.S.; Wu, X.; Tian, Z. Exercise and nutrition benefit skeletal muscle: From influence factor and intervention strategy to molecular mechanism. Sports Med. Health Sci. 2024, 6, 302–314. [Google Scholar] [CrossRef]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef]
- Deutz, N.E.P.; Bauer, J.M.; Barazzoni, R.; Biolo, G.; Boirie, Y.; Bosy-Westphal, A.; Cederholm, T.; Cruz-Jentoft, A.; Krznaric, Z.; Nair, K.S.; et al. Protein intake and exercise for optimal muscle function with aging: Recommendations from the ESPEN Expert Group. Clin. Nutr. 2014, 33, 929–936. [Google Scholar] [CrossRef]
- Mao, D.; Chen, F.; Wang, R.; Bai, P.; Zhang, Y.; Zhao, W.; Chen, J.; Yang, L.; Yang, X.; Li, M. Protein Requirements of Elderly Chinese Adults Are Higher than Current Recommendations. J. Nutr. 2020, 150, 1208–1213. [Google Scholar] [CrossRef]
- Huang, G.; Pencina, K.; Li, Z.; Apovian, C.M.; Travison, T.G.; Storer, T.W.; Gagliano-Jucá, T.; Basaria, S.; Bhasin, S. Effect of Protein Intake on Visceral Abdominal Fat and Metabolic Biomarkers in Older Men with Functional Limitations: Results From a Randomized Clinical Trial. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 1084–1089. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Yarasheski, K.E.; Campbell, J.A.; Smith, K.; Rennie, M.J.; Holloszy, J.O.; Bier, D.M. Effect of growth hormone and resistance exercise on muscle growth in young men. Am. J. Physiol. 1992, 262, E261–E267. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Alberts, I.; Li, X. Dysregulation of the IGF-I/PI3K/AKT/mTOR signaling pathway in autism spectrum disorders. Int. J. Dev. Neurosci. 2014, 35, 35–41. [Google Scholar] [CrossRef]
- Yoshida, T.; Delafontaine, P. Mechanisms of IGF-1-Mediated Regulation of Skeletal Muscle Hypertrophy and Atrophy. Cells 2020, 9, 1970. [Google Scholar] [CrossRef]
- Brown, D.M.; Goljanek-Whysall, K. microRNAs: Modulators of the underlying pathophysiology of sarcopenia? Ageing Res. Rev. 2015, 24, 263–273. [Google Scholar] [CrossRef]
- Analay, P.; Kara, M.; Özçakar, L. ISarcoPRM algorithm for global operationalization of sarcopenia diagnosis. Aging (Albany NY) 2024, 16, 13434–13435. [Google Scholar] [CrossRef]
- Liu, D.; Wang, S.; Liu, S.; Wang, Q.; Che, X.; Wu, G. Frontiers in sarcopenia: Advancements in diagnostics, molecular mechanisms, and therapeutic strategies. Mol. Asp. Med. 2024, 97, 101270. [Google Scholar] [CrossRef]
- Shin, Y.; Yang, J.; Lee, Y.H.; Kim, S. Artificial intelligence in musculoskeletal ultrasound imaging. Ultrasonography 2021, 40, 30–44. [Google Scholar] [CrossRef]
- Guo, H.; Cao, J.; He, S.; Wei, M.; Meng, D.; Yu, I.; Wang, Z.; Chang, X.; Yang, G.; Wang, Z. Quantifying the Enhancement of Sarcopenic Skeletal Muscle Preservation Through a Hybrid Exercise Program: Randomized Controlled Trial. JMIR Aging 2024, 7, e58175. [Google Scholar] [CrossRef]
- Takegaki, J.; Sase, K.; Yasuda, J.; Shindo, D.; Kato, H.; Toyoda, S.; Yamada, T.; Shinohara, Y.; Fujita, S. The Effect of Leucine-Enriched Essential Amino Acid Supplementation on Anabolic and Catabolic Signaling in Human Skeletal Muscle after Acute Resistance Exercise: A Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Comparison Trial. Nutrients 2020, 12, 2421. [Google Scholar] [CrossRef] [PubMed]
- Cereda, E.; Pisati, R.; Rondanelli, M.; Caccialanza, R. Whey Protein, Leucine- and Vitamin-D-Enriched Oral Nutritional Supplementation for the Treatment of Sarcopenia. Nutrients 2022, 14, 1524. [Google Scholar] [CrossRef] [PubMed]
- Bruno, B.J.; Weavil, J.C.; Ogle, J.; Chidambaram, N.; Carey, E.J.; Danford, C.J.; Fricker, Z.P.; Galati, J.S.; Lee, W.M.; Mantry, P.S.; et al. Oral LPCN 1148 improves sarcopenia and hepatic encephalopathy in male patients with cirrhosis: A randomized, placebo-controlled phase 2 trial. Hepatology 2025, 81, 1764–1775. [Google Scholar] [CrossRef] [PubMed]
- Di Dalmazi, G.; Chalan, P.; Caturegli, P. MYMD-1, a Novel Immunometabolic Regulator, Ameliorates Autoimmune Thyroiditis via Suppression of Th1 Responses and TNF-α Release. J. Immunol. 2019, 202, 1350–1362. [Google Scholar] [CrossRef]
- Cacciatore, S.; Calvani, R.; Esposito, I.; Massaro, C.; Gava, G.; Picca, A.; Tosato, M.; Marzetti, E.; Landi, F. Emerging Targets and Treatments for Sarcopenia: A Narrative Review. Nutrients 2024, 16, 3271. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and associated dietary extremes impact on gut microbial diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Zhang, Z.; Guo, Q.; Yang, Z.; Sun, Y.; Jiang, S.; He, Y.; Li, J.; Zhang, J. Bifidobacterium adolescentis-derived nicotinic acid improves host skeletal muscle mitochondrial function to ameliorate sarcopenia. Cell Rep. 2025, 44, 115265. [Google Scholar] [CrossRef]
- Jiang, Y.; Wang, Y.; Chen, G.; Sun, F.; Wu, Q.; Huang, Q.; Zeng, D.; Qiu, W.; Wang, J.; Yao, Z.; et al. Nicotinamide metabolism face-off between macrophages and fibroblasts manipulates the microenvironment in gastric cancer. Cell Metab. 2024, 36, 1806–1822.e1811. [Google Scholar] [CrossRef]
- Parker, B.A.; Walton, C.M.; Carr, S.T.; Andrus, J.L.; Cheung, E.C.K.; Duplisea, M.J.; Wilson, E.K.; Draney, C.; Lathen, D.R.; Kenner, K.B.; et al. β-Hydroxybutyrate Elicits Favorable Mitochondrial Changes in Skeletal Muscle. Int. J. Mol. Sci. 2018, 19, 2247. [Google Scholar] [CrossRef]
- Genserová, L.; Duška, F.; Krajčová, A. β-hydroxybutyrate exposure restores mitochondrial function in skeletal muscle satellite cells of critically ill patients. Clin. Nutr. 2024, 43, 1250–1260. [Google Scholar] [CrossRef]
- Sincennes, M.-C.; Brun, C.E.; Lin, A.Y.T.; Rosembert, T.; Datzkiw, D.; Saber, J.; Ming, H.; Kawabe, Y.-i.; Rudnicki, M.A. Acetylation of PAX7 controls muscle stem cell self-renewal and differentiation potential in mice. Nat. Commun. 2021, 12, 3253. [Google Scholar] [CrossRef] [PubMed]
- Coogan, M.; Alston, V.; Su, B.; Khalil, K.; Elaswad, A.; Khan, M.; Simora, R.M.C.; Johnson, A.; Xing, D.; Li, S.; et al. CRISPR/Cas-9 induced knockout of myostatin gene improves growth and disease resistance in channel catfish (Ictalurus punctatus). Aquaculture 2022, 557, 738290. [Google Scholar] [CrossRef]
- Mukae, H.; Yotsuyanagi, H.; Ohmagari, N.; Doi, Y.; Sakaguchi, H.; Sonoyama, T.; Ichihashi, G.; Sanaki, T.; Baba, K.; Tsuge, Y.; et al. Efficacy and Safety of Ensitrelvir in Patients with Mild-to-Moderate Coronavirus Disease 2019: The Phase 2b Part of a Randomized, Placebo-Controlled, Phase 2/3 Study. Clin. Infect. Dis. 2023, 76, 1403–1411. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| International Working Group | Year | Recommendation for Diagnosing Sarcopenia | Notes |
|---|---|---|---|
| EWGSOP [2] | 2010 | Diagnosis requires both low muscle mass and either low muscle strength or low physical performance. Defines stages: pre-sarcopenia, sarcopenia, severe sarcopenia. | Introduced muscle function into definition. Muscle mass cut-offs: 2 SD below healthy young adults. |
| EWGSOP2 (Revision) [6] | 2018 | Low muscle strength is the primary screening criterion; confirmed by low muscle mass/quality. Poor physical performance defines severity. | Provided clear thresholds: Grip strength < 27 kg (men), <16 kg (women); gait speed ≤0.8 m/s. Prioritized strength as key prognostic factor. |
| AWGS [100] | 2014 | Diagnosis requires low muscle mass plus low muscle strength and/or low physical performance. | Regional thresholds for Asians: ASM (DXA) < 7.0 kg/m2 (men), <5.4 kg/m2 (women); grip strength < 26 kg (men), <18 kg (women); gait speed < 0.8 m/s. |
| AWGS2 (Revision) [101] | 2019 | Introduced “possible sarcopenia” (low strength or performance only). Full diagnosis: low muscle mass + low strength or performance. | Gait speed threshold raised to <1.0 m/s; emphasized early detection and community screening. |
| IWGS [102] | 2011 | Diagnosis: low muscle mass + poor physical performance (gait speed < 1.0 m/s). Muscle strength not required. | Recommended DXA to assess lean mass. Focused on older adults with mobility limitations. |
| FNIH Sarcopenia Project [103] | 2014 | Low grip strength (<26 kg men, <16 kg women) and low ALM/BMI (<0.789 men, <0.512 women) define sarcopenia. | Introduced ALM/BMI as a muscle mass index. Emphasized function-based approach. |
| SDOC [105] | 2020 | Sarcopenia defined as low muscle strength (grip: <35.5 kg men, <20 kg women) and slow gait speed (<0.8 m/s). Muscle mass not included. | Prioritized functional measures to predict outcomes like falls and disability. |
| SCWD [104] | 2019 | Classification into pre-sarcopenia (only low mass), sarcopenia (mass + strength/performance loss), and severe sarcopenia (all three impaired). | Proposed clinical staging framework. No specific numeric thresholds. |
| GLIS [8] | 2024 | Conceptual definition: includes muscle mass, muscle strength, and muscle-specific force; physical performance viewed as an outcome. | First global conceptual consensus; sets foundation for future harmonized operational criteria. |
| Tool | Items | Sensitivity | Specificity | Notes |
|---|---|---|---|---|
| SARC-F [106] | 5 self-reported domains: strength, walking assistance, chair rise, stairs, falls | 4–35% | 80–98% | High specificity but poor sensitivity |
| SARC-Calf [109] | SARC-F + calf circumference | 66.1% | 73.8% | Adds calf circumference to boost both sensitivity and specificity |
| MSRA-5 [112] | Age, hospitalization, activity, meals, unintentional weight loss | 59.7% | 46.4% | More sensitive than SARC-F but lower specificity |
| MSRA-7 [112] | MSRA-5 items + dairy intake, protein intake | 14.5% | 88.7% | Highest specificity of MSRA versions, lowest sensitivity |
| Modality | Principle | Diagnostic Performance | Advantages | Limitations | Notes | Cut-Off Points |
|---|---|---|---|---|---|---|
| BIA | Measures body impedance to estimate lean and fat mass based on conductor properties of tissues. | Sensitivity ~60%, specificity ≥ 85% (vs DXA) | Portable, rapid, low-cost, non-invasive; suitable for bedridden/mobile settings | Affected by hydration, electrode placement, equations; cannot measure intramuscular fat | Recognized by EWGSOP2 and AWGS as valid screening in resource-limited contexts | SMI < 7.0 kg/m2 (men), <5.7 kg/m2 (women) (EWGSOP2) |
| DXA | Low-dose X-rays differentiate bone, fat, and lean tissue to quantify appendicular lean mass. | Correlates strongly with CT/MRI; used as reference standard | Fast (<20 min), reproducible (same machine), regional analysis, low radiation | Cannot detect intramuscular adipose tissue; less accessible in low-resource settings; hydration effects | Endorsed by EWGSOP, AWGS, FNIH and IWGS; part of ICD-10 codes M62.84 | ALM/height2 < 7.0 kg/m2 (men), <5.5 kg/m2 (women) (EWGSOP2); ALM/BMI < 0.789/0.512 (FNIH) |
| CT | X-ray attenuation differentiates tissues by Hounsfield units, quantifying muscle cross-sectional area and fat infiltration. | High accuracy; excellent agreement with MRI | Precise quantification of muscle and fat; gold standard for muscle-quality assessment | High radiation dose; costly; limited portability; no standardized global cut-offs | Used mainly in research and oncology follow-up | SMI < 55 cm2/m2 (men), <39 cm2/m2 (women) |
| MRI | Magnetic fields and radiofrequency pulses produce high-contrast images of muscle volume and fat infiltration. | Comparable to CT; no ionizing radiation | Non-ionizing, multiplanar, excellent soft-tissue contrast; assesses composition and architecture | High cost; time-consuming; limited access; space and technologist requirements | Interchangeable with CT for muscle mass assessment | SMI < 55 cm2/m2 (men), <39 cm2/m2 (women) |
| Ultrasound Imaging | High-frequency sound waves measure muscle thickness, cross-sectional area, and echogenicity. | ICC 0.92–0.99 for thickness measures; correlates with DXA/CT | Portable; radiation-free; low-cost; real-time; assesses muscle quality via echogenicity | Operator-dependent; lack of standardized protocols and cut-offs; limited penetration depth | Growing interest for bedside and community use; needs further validation | Rectus femoris thickness < 1.0–1.5 cm (varies; no universal cut-off) |
| Category | Biomarker | Function/Mechanism |
|---|---|---|
| Inflammatory | IL-6, TNF-α, CRP, IL-1β, IL-18, suPAR | Promote catabolism via STAT3, NF-κB; inhibit regeneration |
| Oxidative Stress/Mitochondria | SOD, GPx, Catalase, 8-OHdG, Nrf2, PGC-1α, Humanin | Indicate mitochondrial dysfunction and redox imbalance |
| NMJ and Motor Units | CAF, NFL, BDNF, Neuregulin-1 | Reflect NMJ degradation and axonal injury |
| Hormonal | IGF-1, Testosterone, GH, Estrogen, DHEA-S, FGF21, Melatonin | Regulate protein synthesis and satellite cells via PI3K/Akt/mTOR |
| Muscle Growth Regulators | Myostatin, Activin A/B, GDF-15, follistatin, BMP-7, BDNF, Irisin, Myonectin | Balance anabolic and catabolic signals |
| Muscle Damage and Turnover | CK, LDH, Atrogin-1, MuRF1, MyoD, Myogenin, FGF2 methylation, Periostin | Reflect muscle atrophy and impaired regeneration |
| Intervention | Mechanism of Action | Key Outcomes |
|---|---|---|
| Whey Protein | Activates mTORC1 via leucine-rich amino acid profile | Increase muscle protein synthesis (~30–40% vs. soy) will lead an increase of strength |
| Plant Protein | Provides essential AAs but lower leucine bioavailability | Requires higher intake (~60 g) to match whey effect |
| n-3 PUFAs (EPA/DHA) | Decrease TNF-α signaling, MuRF1 inhibition | Increase Myotube survival (25%) will decrease muscle catabolism |
| Polyphenols | Inhibit NF-κB activation | Decrease Inflammation will decrease creatine kinase and protection against muscle damage |
| Vitamin D | Modulates Notch signaling, enhances regeneration | Increase Post-injury recovery (19%) lead to regenerative decline, but risk of hypercalcemia at high doses |
| Triple Supplement | Combined anabolic, anti-inflammatory, and regenerative effects | Increase muscle strength by 28% (Phase III trial) |
| Resistance Training | Activates IGF-1/Akt/mTOR, stimulates satellite cells | Increase muscle mass and strength will decrease catabolism and increase Type II fiber activation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tu, S.; Hao, X.; Xu, S.; Jin, X.; Liao, W.; Xia, H.; Wang, S.; Sun, G. Sarcopenia: Current Insights into Molecular Mechanisms, Diagnostics, and Emerging Interventional Approaches. Int. J. Mol. Sci. 2025, 26, 6740. https://doi.org/10.3390/ijms26146740
Tu S, Hao X, Xu S, Jin X, Liao W, Xia H, Wang S, Sun G. Sarcopenia: Current Insights into Molecular Mechanisms, Diagnostics, and Emerging Interventional Approaches. International Journal of Molecular Sciences. 2025; 26(14):6740. https://doi.org/10.3390/ijms26146740
Chicago/Turabian StyleTu, Siying, Xiaoyu Hao, Shan Xu, Xingyi Jin, Wang Liao, Hui Xia, Shaokang Wang, and Guiju Sun. 2025. "Sarcopenia: Current Insights into Molecular Mechanisms, Diagnostics, and Emerging Interventional Approaches" International Journal of Molecular Sciences 26, no. 14: 6740. https://doi.org/10.3390/ijms26146740
APA StyleTu, S., Hao, X., Xu, S., Jin, X., Liao, W., Xia, H., Wang, S., & Sun, G. (2025). Sarcopenia: Current Insights into Molecular Mechanisms, Diagnostics, and Emerging Interventional Approaches. International Journal of Molecular Sciences, 26(14), 6740. https://doi.org/10.3390/ijms26146740