
Oxidative Stress in HIV-Associated Neurodegeneration: Mechanisms of Pathogenesis and Therapeutic Targets


 , ,
, ,  ,
,  and
and 
Abstract
1. Introduction
2. Neurocognitive Disorders Caused by HIV Infection
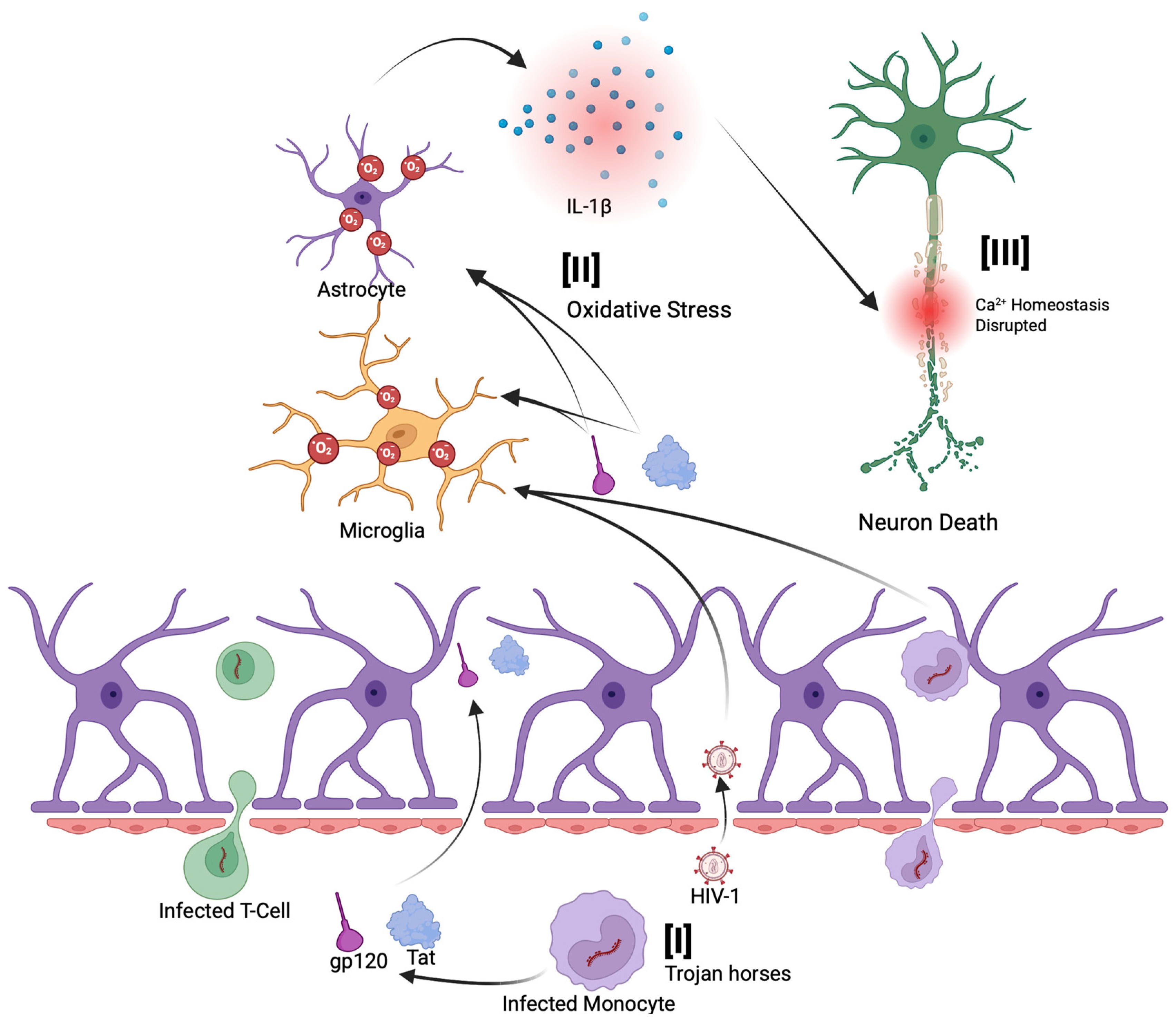
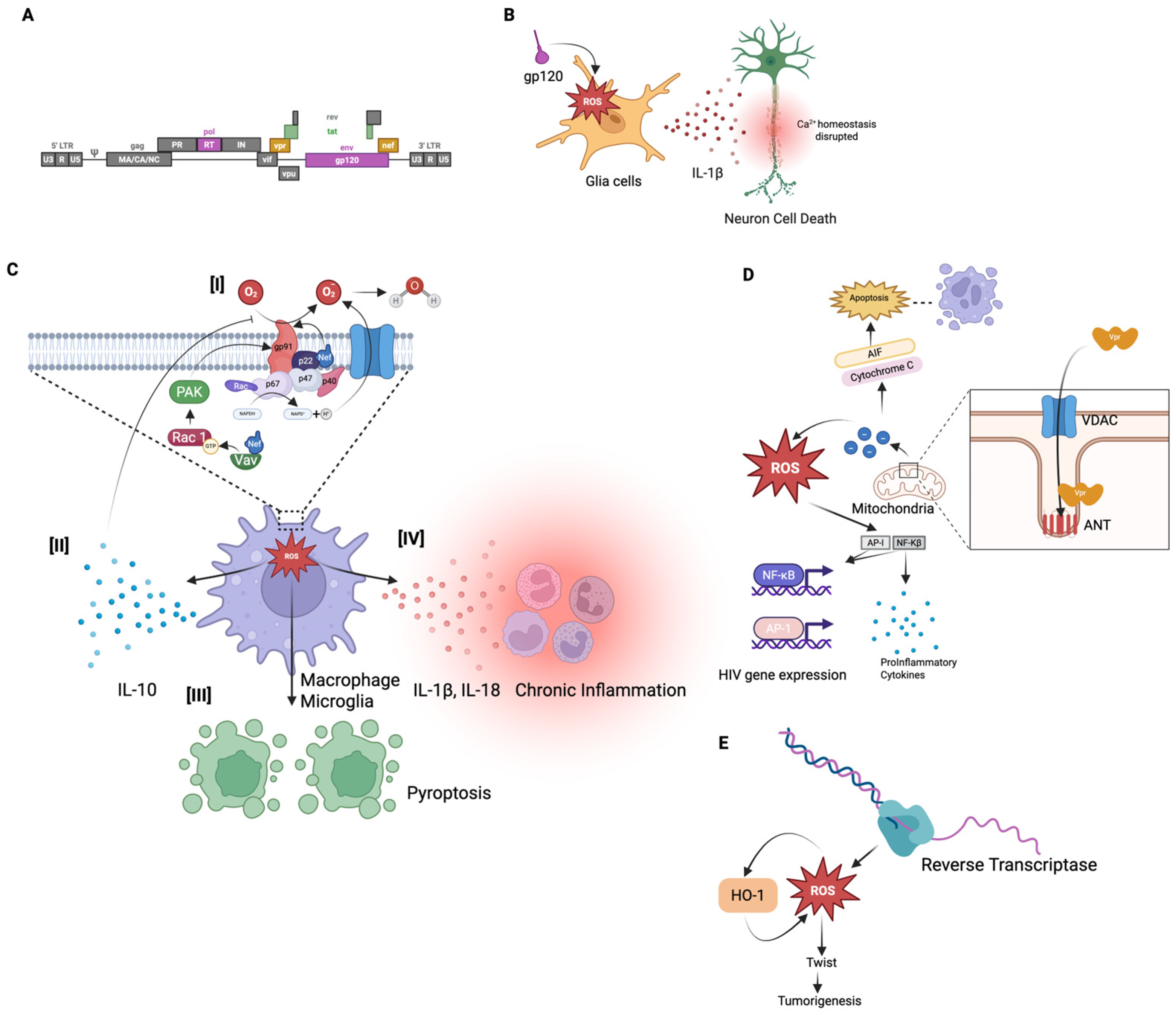
3. Mechanisms of Reactive Oxygen Species (ROS) Production by Human Immunodeficiency Virus
3.1. HIV Pathogenesis and ROS Production
3.2. Envelope Glycoprotein (gp120) and Tat Mediated ROS Production
3.3. Nef-Mediated ROS Production
3.4. Vpr-Mediated ROS Production
3.5. Reverse Transcriptase (RT) Mediated ROS Production
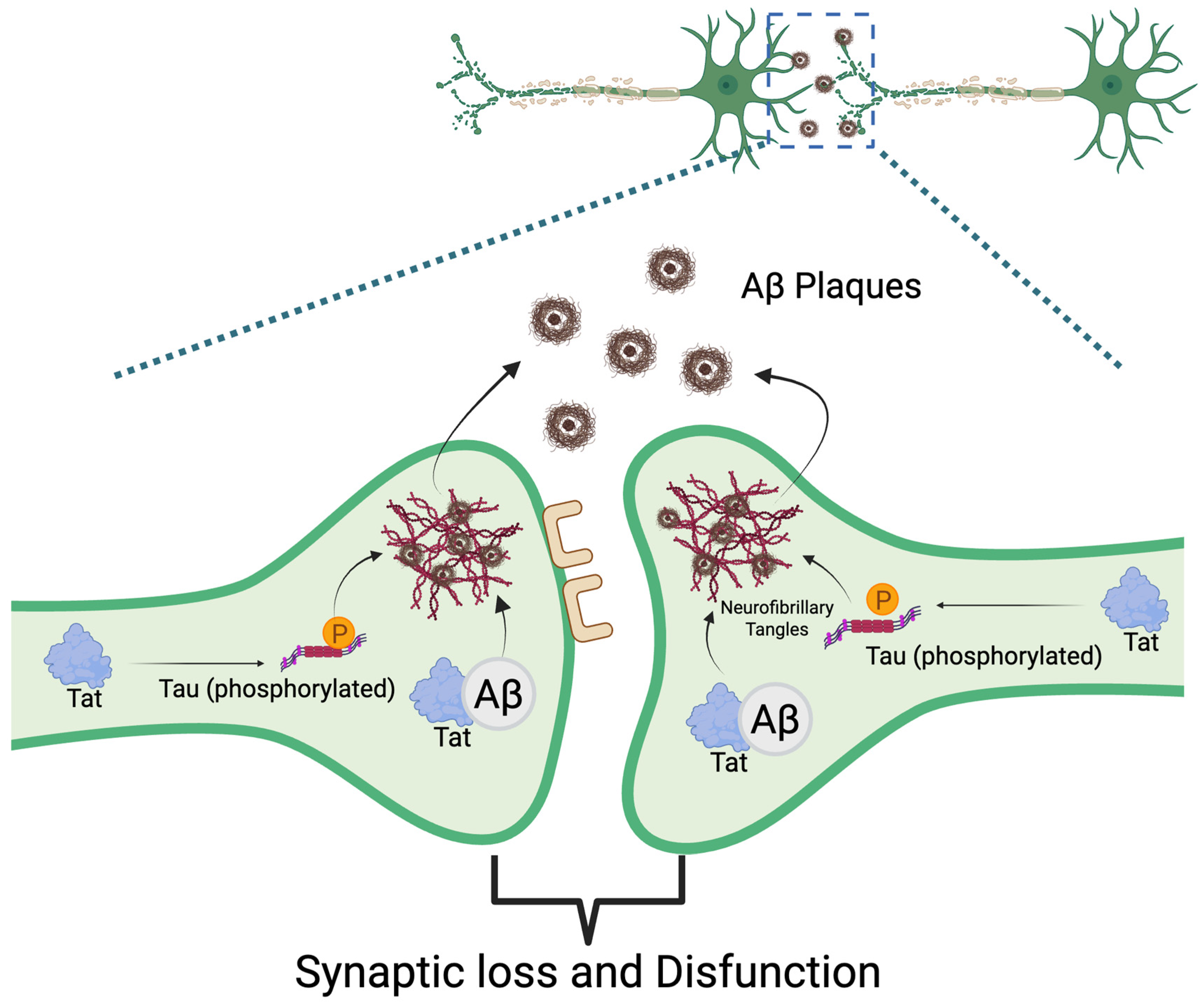
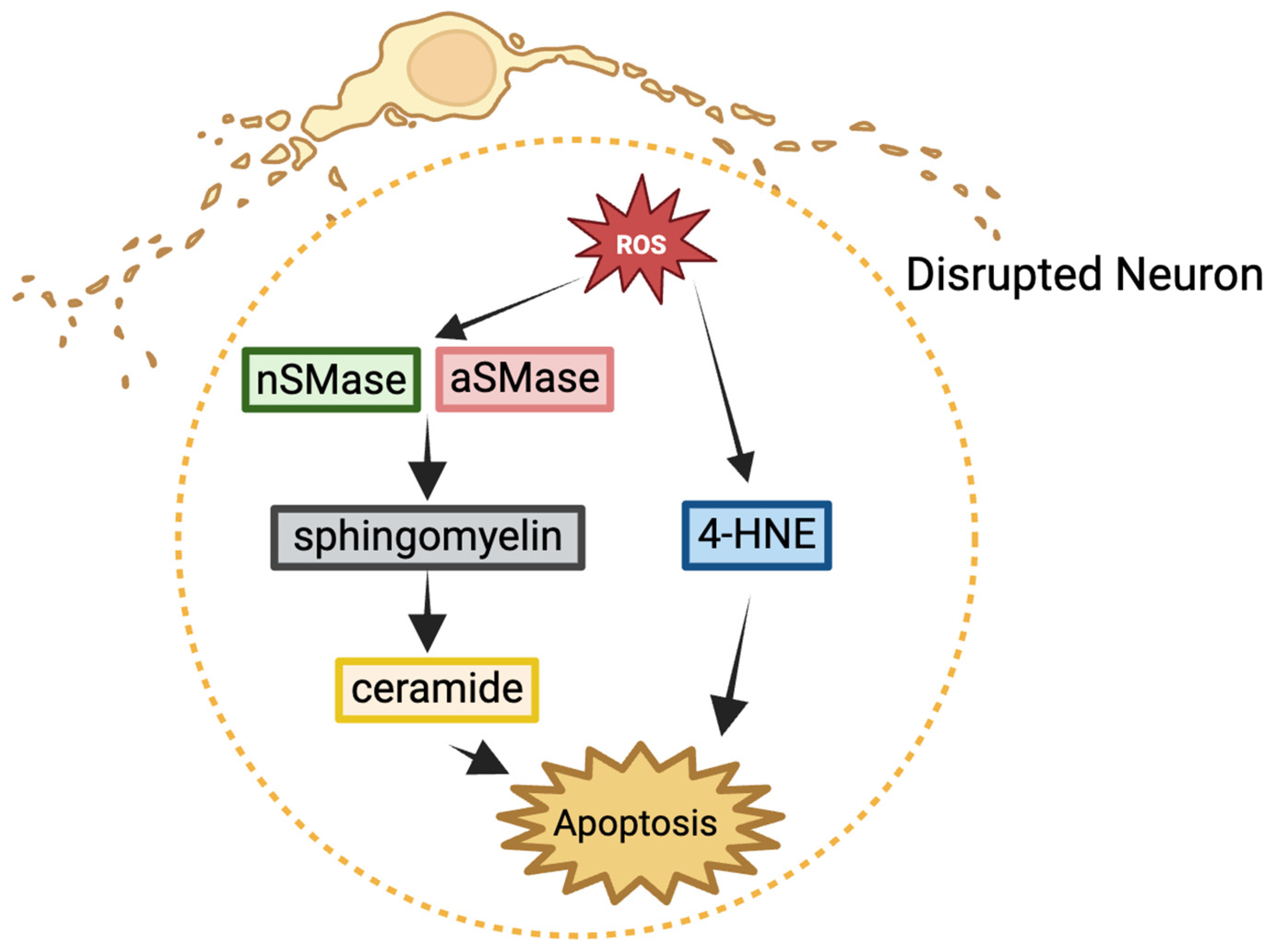
4. Reactive Oxygen Species (ROS) and Chronic HIV-Associated Neurodegeneration
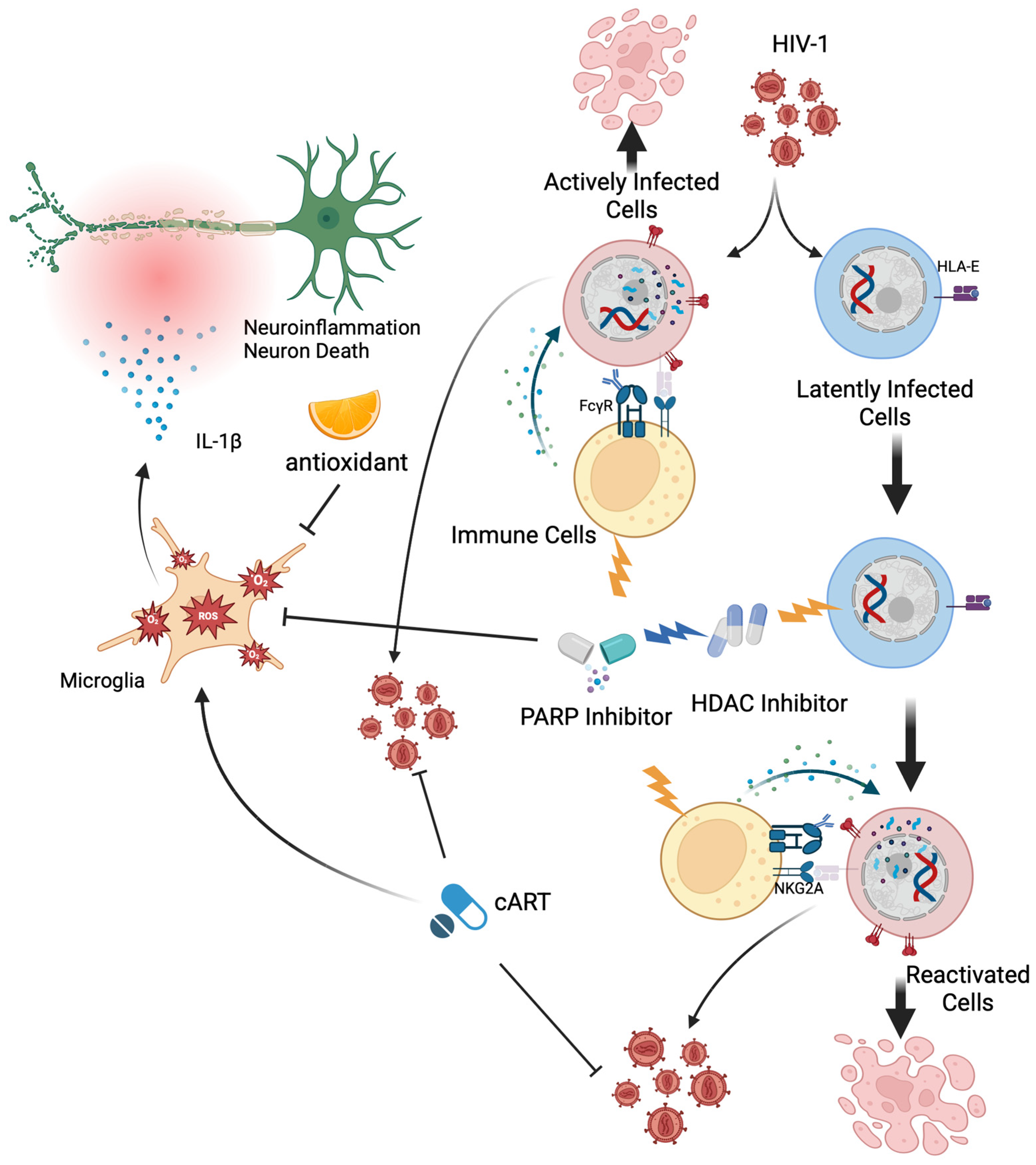
5. Therapeutic Interventions for HIV-Associated Neurocognitive Disorder
5.1. Antioxidant
5.2. Poly (ADP-Ribose) Polymerase Inhibitors
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Giovanetti, M.; Ciccozzi, M.; Parolin, C.; Borsetti, A. Molecular Epidemiology of HIV-1 in African Countries: A Comprehensive Overview. Pathogens 2020, 9, 1072. [Google Scholar] [CrossRef]
- Chen, Y.; Li, A.D.; Yang, Y.; Lu, J.; Xu, Y.; Ji, X.; Wu, L.; Han, L.; Zhu, B.; Xu, M. Global, regional and national burden of HIV/AIDS among individuals aged 15–79 from 1990 to 2021. AIDS Res. Ther. 2025, 22, 51. [Google Scholar] [CrossRef]
- Ayieko, J.; Thorp, M.; Ghebremichael, M. Renewing Our Focus on Vulnerable Populations Among People Living with HIV. Trop. Med. Infect. Dis. 2024, 9, 278. [Google Scholar] [CrossRef]
- Sudharshan, S.; Biswas, J. Introduction and immunopathogenesis of acquired immune deficiency syndrome. Indian J. Ophthalmol. 2008, 56, 357–362. [Google Scholar] [CrossRef]
- Balasubramaniam, M.; Pandhare, J.; Dash, C. Immune Control of HIV. J. Life Sci. 2019, 1, 4–37. [Google Scholar] [CrossRef]
- Weichseldorfer, M.; Reitz, M.; Latinovic, O.S. Past HIV-1 Medications and the Current Status of Combined Antiretroviral Therapy Options for HIV-1 Patients. Pharmaceutics 2021, 13, 1798. [Google Scholar] [CrossRef]
- McGraw, A.; Hillmer, G.; Medehincu, S.M.; Hikichi, Y.; Gagliardi, S.; Narayan, K.; Tibebe, H.; Marquez, D.; Mei Bose, L.; Keating, A.; et al. Exploring HIV-1 Maturation: A New Frontier in Antiviral Development. Viruses 2024, 16, 1423. [Google Scholar] [CrossRef]
- Nachega, J.B.; Scarsi, K.K.; Gandhi, M.; Scott, R.K.; Mofenson, L.M.; Archary, M.; Nachman, S.; Decloedt, E.; Geng, E.H.; Wilson, L.; et al. Long-acting antiretrovirals and HIV treatment adherence. Lancet HIV 2023, 10, e332–e342. [Google Scholar] [CrossRef]
- Smith, L.K.; Kuhn, T.B.; Chen, J.; Bamburg, J.R. HIV Associated Neurodegenerative Disorders: A New Perspective on the Role of Lipid Rafts in Gp120-Mediated Neurotoxicity. Curr. HIV Res. 2018, 16, 258–269. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Simioni, S.; Cavassini, M.; Annoni, J.M.; Rimbault Abraham, A.; Bourquin, I.; Schiffer, V.; Calmy, A.; Chave, J.P.; Giacobini, E.; Hirschel, B.; et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010, 24, 1243–1250. [Google Scholar] [CrossRef]
- Chemparthy, D.T.; Kannan, M.; Gordon, L.; Buch, S.; Sil, S. Alzheimer’s-Like Pathology at the Crossroads of HIV-Associated Neurological Disorders. Vaccines 2021, 9, 930. [Google Scholar] [CrossRef]
- Breijyeh, Z.; Karaman, R. Comprehensive Review on Alzheimer’s Disease: Causes and Treatment. Molecules 2020, 25, 5789. [Google Scholar] [CrossRef]
- Stephenson, J.; Nutma, E.; van der Valk, P.; Amor, S. Inflammation in CNS neurodegenerative diseases. Immunology 2018, 154, 204–219. [Google Scholar] [CrossRef]
- Richards, R.I.; Robertson, S.A.; Kastner, D.L. Neurodegenerative diseases have genetic hallmarks of autoinflammatory disease. Hum. Mol. Genet. 2018, 27, R108–R118. [Google Scholar] [CrossRef]
- Madore, C.; Yin, Z.; Leibowitz, J.; Butovsky, O. Microglia, Lifestyle Stress, and Neurodegeneration. Immunity 2020, 52, 222–240. [Google Scholar] [CrossRef]
- Campbell, A. Inflammation, neurodegenerative diseases, and environmental exposures. Ann. N.Y. Acad. Sci. 2004, 1035, 117–132. [Google Scholar] [CrossRef]
- Soraci, L.; Corsonello, A.; Paparazzo, E.; Montesanto, A.; Piacenza, F.; Olivieri, F.; Gambuzza, M.E.; Savedra, E.V.; Marino, S.; Lattanzio, F.; et al. Neuroinflammaging: A Tight Line Between Normal Aging and Age-Related Neurodegenerative Disorders. Aging Dis. 2024, 15, 1726–1747. [Google Scholar] [CrossRef]
- Stoccoro, A.; Coppede, F. Exposure to Metals, Pesticides, and Air Pollutants: Focus on Resulting DNA Methylation Changes in Neurodegenerative Diseases. Biomolecules 2024, 14, 1366. [Google Scholar] [CrossRef]
- Xu, L.; He, D.; Bai, Y. Microglia-Mediated Inflammation and Neurodegenerative Disease. Mol. Neurobiol. 2016, 53, 6709–6715. [Google Scholar] [CrossRef]
- Subhramanyam, C.S.; Wang, C.; Hu, Q.; Dheen, S.T. Microglia-mediated neuroinflammation in neurodegenerative diseases. Semin. Cell Dev. Biol. 2019, 94, 112–120. [Google Scholar] [CrossRef]
- Bachiller, S.; Jimenez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Dheen, S.T.; Kaur, C.; Ling, E.A. Microglial activation and its implications in the brain diseases. Curr. Med. Chem. 2007, 14, 1189–1197. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef]
- Rodriguez-Gomez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia development and function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef]
- Oddo, S.; Caccamo, A.; Shepherd, J.D.; Murphy, M.P.; Golde, T.E.; Kayed, R.; Metherate, R.; Mattson, M.P.; Akbari, Y.; LaFerla, F.M. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: Intracellular Abeta and synaptic dysfunction. Neuron 2003, 39, 409–421. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial Cells: The Main HIV-1 Reservoir in the Brain. Front. Cell. Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef]
- Tang, Y.; Chaillon, A.; Gianella, S.; Wong, L.M.; Li, D.; Simermeyer, T.L.; Porrachia, M.; Ignacio, C.; Woodworth, B.; Zhong, D.; et al. Brain microglia serve as a persistent HIV reservoir despite durable antiretroviral therapy. J. Clin. Investig. 2023, 133, e167417. [Google Scholar] [CrossRef]
- Gumbs, S.B.H.; Berdenis van Berlekom, A.; Kubler, R.; Schipper, P.J.; Gharu, L.; Boks, M.P.; Ormel, P.R.; Wensing, A.M.J.; de Witte, L.D.; Nijhuis, M. Characterization of HIV-1 Infection in Microglia-Containing Human Cerebral Organoids. Viruses 2022, 14, 829. [Google Scholar] [CrossRef] [PubMed]
- Schlachetzki, J.C.M.; Zhou, Y.; Glass, C.K. Human microglia phenotypes in the brain associated with HIV infection. Curr. Opin. Neurobiol. 2022, 77, 102637. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Jennings, A.; Akbarian, S. Genomic Exploration of the Brain in People Infected with HIV-Recent Progress and the Road Ahead. Curr. HIV/AIDS Rep. 2023, 20, 357–367. [Google Scholar] [CrossRef]
- Strazza, M.; Pirrone, V.; Wigdahl, B.; Nonnemacher, M.R. Breaking down the barrier: The effects of HIV-1 on the blood-brain barrier. Brain Res. 2011, 1399, 96–115. [Google Scholar] [CrossRef]
- Woodburn, B.M.; Kanchi, K.; Zhou, S.; Colaianni, N.; Joseph, S.B.; Swanstrom, R. Characterization of Macrophage-Tropic HIV-1 Infection of Central Nervous System Cells and the Influence of Inflammation. J. Virol. 2022, 96, e0095722. [Google Scholar] [CrossRef]
- Gisslen, M.; Keating, S.M.; Spudich, S.; Arechiga, V.; Stephenson, S.; Zetterberg, H.; Di Germanio, C.; Blennow, K.; Fuchs, D.; Hagberg, L.; et al. Compartmentalization of cerebrospinal fluid inflammation across the spectrum of untreated HIV-1 infection, central nervous system injury and viral suppression. PLoS ONE 2021, 16, e0250987. [Google Scholar] [CrossRef] [PubMed]
- Uwishema, O.; Ayoub, G.; Badri, R.; Onyeaka, H.; Berjaoui, C.; Karabulut, E.; Anis, H.; Sammour, C.; Mohammed Yagoub, F.E.A.; Chalhoub, E. Neurological disorders in HIV: Hope despite challenges. Immun. Inflamm. Dis. 2022, 10, e591. [Google Scholar] [CrossRef]
- McRae, M. HIV and viral protein effects on the blood brain barrier. Tissue Barriers 2016, 4, e1143543. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Ouyang, Y.B.; Liu, L.G.; Chen, D.X. Blood-brain barrier and neuro-AIDS. Eur. Rev. Med. Pharmacol. Sci. 2015, 19, 4927–4939. [Google Scholar]
- Atluri, V.S.; Hidalgo, M.; Samikkannu, T.; Kurapati, K.R.; Jayant, R.D.; Sagar, V.; Nair, M.P. Effect of human immunodeficiency virus on blood-brain barrier integrity and function: An update. Front. Cell. Neurosci. 2015, 9, 212. [Google Scholar] [CrossRef]
- Al-Obaidi, M.M.J.; Bahadoran, A.; Wang, S.M.; Manikam, R.; Raju, C.S.; Sekaran, S.D. Disruption of the blood brain barrier is vital property of neurotropic viral infection of the central nervous system. Acta Virol. 2018, 62, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 2019, 142, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Cai, M.; Liang, Y.; Zhang, Y. Disruption of blood-brain barrier: Effects of HIV Tat on brain microvascular endothelial cells and tight junction proteins. J. Neurovirol. 2023, 29, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Lorin, V.; Danckaert, A.; Porrot, F.; Schwartz, O.; Afonso, P.V.; Mouquet, H. Antibody Neutralization of HIV-1 Crossing the Blood-Brain Barrier. mBio 2020, 11, e02424-20. [Google Scholar] [CrossRef]
- Barker, C.T.; Vaidya, N.K. Modeling HIV-1 infection in the brain. PLoS Comput. Biol. 2020, 16, e1008305. [Google Scholar] [CrossRef]
- Couret, J.; Chang, T.L. Reactive Oxygen Species in HIV Infection. EC Microbiol. 2016, 3, 597–604. [Google Scholar]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef]
- Didonna, A. Tau at the interface between neurodegeneration and neuroinflammation. Genes Immun. 2020, 21, 288–300. [Google Scholar] [CrossRef]
- Cotto, B.; Natarajanseenivasan, K.; Langford, D. HIV-1 infection alters energy metabolism in the brain: Contributions to HIV-associated neurocognitive disorders. Prog. Neurobiol. 2019, 181, 101616. [Google Scholar] [CrossRef]
- Jha, N.K.; Sharma, A.; Jha, S.K.; Ojha, S.; Chellappan, D.K.; Gupta, G.; Kesari, K.K.; Bhardwaj, S.; Shukla, S.D.; Tambuwala, M.M.; et al. Alzheimer’s disease-like perturbations in HIV-mediated neuronal dysfunctions: Understanding mechanisms and developing therapeutic strategies. Open Biol. 2020, 10, 200286. [Google Scholar] [CrossRef]
- Williams, M.E.; Naude, P.J.W. The relationship between HIV-1 neuroinflammation, neurocognitive impairment and encephalitis pathology: A systematic review of studies investigating post-mortem brain tissue. Rev. Med. Virol. 2024, 34, e2519. [Google Scholar] [CrossRef] [PubMed]
- Kodidela, S.; Gerth, K.; Haque, S.; Gong, Y.; Ismael, S.; Singh, A.; Tauheed, I.; Kumar, S. Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells 2019, 8, 968. [Google Scholar] [CrossRef]
- Gonzalez, J.; Wilson, A.; Byrd, D.; Cortes, E.P.; Crary, J.F.; Morgello, S. Neuronal accumulation of hyperphosphorylated tau protein predicts stable memory impairment in people living with HIV. AIDS 2023, 37, 1247–1256. [Google Scholar] [CrossRef]
- Fields, J.A.; Swinton, M.K.; Soontornniyomkij, B.; Carson, A.; Achim, C.L. Beta amyloid levels in cerebrospinal fluid of HIV-infected people vary by exposure to antiretroviral therapy. AIDS 2020, 34, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Ozturk, T.; Kollhoff, A.; Anderson, A.M.; Christina Howell, J.; Loring, D.W.; Waldrop-Valverde, D.; Franklin, D.; Letendre, S.; Tyor, W.R.; Hu, W.T. Linked CSF reduction of phosphorylated tau and IL-8 in HIV associated neurocognitive disorder. Sci. Rep. 2019, 9, 8733. [Google Scholar] [CrossRef]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef]
- Pulliam, L.; Calosing, C.; Sun, B.; Grunfeld, C.; Rempel, H. Monocyte activation from interferon-alpha in HIV infection increases acetylated LDL uptake and ROS production. J. Interferon Cytokine Res. 2014, 34, 822–828. [Google Scholar] [CrossRef] [PubMed]
- Harshithkumar, R.; Shah, P.; Jadaun, P.; Mukherjee, A. ROS Chronicles in HIV Infection: Genesis of Oxidative Stress, Associated Pathologies, and Therapeutic Strategies. Curr. Issues Mol. Biol. 2024, 46, 8852–8873. [Google Scholar] [CrossRef]
- Cevallos, C.; Ojeda, D.S.; Sanchez, L.; Urquiza, J.; Delpino, M.V.; Quarleri, J. HIV-induced bystander cell death in astrocytes requires cell-to-cell viral transmission. J. Neurochem. 2022, 163, 338–356. [Google Scholar] [CrossRef]
- Viviani, B.; Corsini, E.; Binaglia, M.; Galli, C.L.; Marinovich, M. Reactive oxygen species generated by glia are responsible for neuron death induced by human immunodeficiency virus-glycoprotein 120 in vitro. Neuroscience 2001, 107, 51–58. [Google Scholar] [CrossRef]
- Ronaldson, P.T.; Bendayan, R. HIV-1 viral envelope glycoprotein gp120 produces oxidative stress and regulates the functional expression of multidrug resistance protein-1 (Mrp1) in glial cells. J. Neurochem. 2008, 106, 1298–1313. [Google Scholar] [CrossRef]
- Zakirova, N.F.; Kondrashova, A.S.; Golikov, M.V.; Ivanova, O.N.; Ivanov, A.V.; Isaguliants, M.G.; Bayurova, E.O. Expression of HIV-1 Reverse Transcriptase in Murine Cancer Cells Increases Mitochondrial Respiration. Mol. Biol. 2022, 56, 795–807. [Google Scholar] [CrossRef]
- El-Amine, R.; Germini, D.; Zakharova, V.V.; Tsfasman, T.; Sheval, E.V.; Louzada, R.A.N.; Dupuy, C.; Bilhou-Nabera, C.; Hamade, A.; Najjar, F.; et al. HIV-1 Tat protein induces DNA damage in human peripheral blood B-lymphocytes via mitochondrial ROS production. Redox Biol. 2018, 15, 97–108. [Google Scholar] [CrossRef]
- Shah, A.; Kumar, S.; Simon, S.D.; Singh, D.P.; Kumar, A. HIV gp120- and methamphetamine-mediated oxidative stress induces astrocyte apoptosis via cytochrome P450 2E1. Cell Death Dis. 2013, 4, e850. [Google Scholar] [CrossRef]
- Butler, S.L.; Johnson, E.P.; Bushman, F.D. Human immunodeficiency virus cDNA metabolism: Notable stability of two-long terminal repeat circles. J. Virol. 2002, 76, 3739–3747. [Google Scholar] [CrossRef]
- Zhou, R.; Xie, X.; Li, X.; Qin, Z.; Wei, C.; Liu, J.; Luo, Y. The triggers of the cGAS-STING pathway and the connection with inflammatory and autoimmune diseases. Infect. Genet. Evol. 2020, 77, 104094. [Google Scholar] [CrossRef]
- Zhang, Z.; Yang, J.; Zhou, Q.; Zhong, S.; Liu, J.; Zhang, X.; Chang, X.; Wang, H. The cGAS-STING-mediated ROS and ferroptosis are involved in manganese neurotoxicity. J. Environ. Sci. 2025, 152, 71–86. [Google Scholar] [CrossRef]
- Sui, H.; Sun, Z.; Liu, C.; Xi, H. Ferritinophagy promotes microglia ferroptosis to aggravate neuroinflammation induced by cerebral ischemia-reperfusion injury via activation of the cGAS-STING signaling pathway. Neurochem. Int. 2025, 183, 105920. [Google Scholar] [CrossRef]
- Ding, L.; Zhang, R.; Du, W.; Wang, Q.; Pei, D. The role of cGAS-STING signaling pathway in ferroptosis. J. Adv. Res. 2024. [Google Scholar] [CrossRef]
- Akang, E.N. Combination antiretroviral therapy (cART)-induced hippocampal disorders: Highlights on therapeutic potential of Naringenin and Quercetin. IBRO Rep. 2019, 6, 137–146. [Google Scholar] [CrossRef]
- Akay, C.; Cooper, M.; Odeleye, A.; Jensen, B.K.; White, M.G.; Vassoler, F.; Gannon, P.J.; Mankowski, J.; Dorsey, J.L.; Buch, A.M.; et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J. Neurovirol. 2014, 20, 39–53. [Google Scholar] [CrossRef]
- Haughey, N.J.; Mattson, M.P. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J. Acquir. Immune. Defic. Syndr. 2002, 31 (Suppl. 2), S55–S61. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Reyes, B.A.; Agrawal, L.; Van Bockstaele, E.J.; Strayer, D.S. HIV-1 gp120-induced neuroinflammation: Relationship to neuron loss and protection by rSV40-delivered antioxidant enzymes. Exp. Neurol. 2010, 221, 231–245. [Google Scholar] [CrossRef]
- Viviani, B.; Gardoni, F.; Bartesaghi, S.; Corsini, E.; Facchi, A.; Galli, C.L.; Di Luca, M.; Marinovich, M. Interleukin-1 beta released by gp120 drives neural death through tyrosine phosphorylation and trafficking of NMDA receptors. J. Biol. Chem. 2006, 281, 30212–30222. [Google Scholar] [CrossRef]
- Steiner, J.; Haughey, N.; Li, W.; Venkatesan, A.; Anderson, C.; Reid, R.; Malpica, T.; Pocernich, C.; Butterfield, D.A.; Nath, A. Oxidative stress and therapeutic approaches in HIV dementia. Antioxid. Redox Signal. 2006, 8, 2089–2100. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.P.; Marusich, E.; Reyes, B.A.; Van Bockstaele, E.J.; Strayer, D.S. Dopaminergic neurotoxicity of HIV-1 gp120: Reactive oxygen species as signaling intermediates. Brain Res. 2010, 1306, 116–130. [Google Scholar] [CrossRef]
- Denton, A.R.; Samaranayake, S.A.; Kirchner, K.N.; Roscoe, R.F., Jr.; Berger, S.N.; Harrod, S.B.; Mactutus, C.F.; Hashemi, P.; Booze, R.M. Selective monoaminergic and histaminergic circuit dysregulation following long-term HIV-1 protein exposure. J. Neurovirol. 2019, 25, 540–550. [Google Scholar] [CrossRef]
- Cross, S.A.; Cook, D.R.; Chi, A.W.; Vance, P.J.; Kolson, L.L.; Wong, B.J.; Jordan-Sciutto, K.L.; Kolson, D.L. Dimethyl fumarate, an immune modulator and inducer of the antioxidant response, suppresses HIV replication and macrophage-mediated neurotoxicity: A novel candidate for HIV neuroprotection. J. Immunol. 2011, 187, 5015–5025. [Google Scholar] [CrossRef]
- Bachis, A.; Aden, S.A.; Nosheny, R.L.; Andrews, P.M.; Mocchetti, I. Axonal transport of human immunodeficiency virus type 1 envelope protein glycoprotein 120 is found in association with neuronal apoptosis. J. Neurosci. 2006, 26, 6771–6780. [Google Scholar] [CrossRef]
- Guengerich, F.P. Cytochrome P450 2E1 and its roles in disease. Chem. Biol. Interact. 2020, 322, 109056. [Google Scholar] [CrossRef]
- Biedrzycki, G.; Wolszczak-Biedrzycka, B.; Dorf, J.; Maciejczyk, M. The antioxidant barrier, oxidative/nitrosative stress, and protein glycation in allergy: From basic research to clinical practice. Front. Immunol. 2024, 15, 1440313. [Google Scholar] [CrossRef]
- Skonieczna, M.; Hejmo, T.; Poterala-Hejmo, A.; Cieslar-Pobuda, A.; Buldak, R.J. NADPH Oxidases: Insights into Selected Functions and Mechanisms of Action in Cancer and Stem Cells. Oxid. Med. Cell. Longev. 2017, 2017, 9420539. [Google Scholar] [CrossRef]
- Pandhare, J.; Dash, S.; Jones, B.; Villalta, F.; Dash, C. A Novel Role of Proline Oxidase in HIV-1 Envelope Glycoprotein-induced Neuronal Autophagy. J. Biol. Chem. 2015, 290, 25439–25451. [Google Scholar] [CrossRef] [PubMed]
- Hategan, A.; Masliah, E.; Nath, A. HIV and Alzheimer’s disease: Complex interactions of HIV-Tat with amyloid beta peptide and Tau protein. J. Neurovirol. 2019, 25, 648–660. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Niu, F.; Zheng, Y.; Xu, Y. CART mitigates oxidative stress and DNA damage in memory deficits of APP/PS1 mice via upregulating beta-amyloid metabolism-associated enzymes. Mol. Med. Rep. 2021, 23, 280. [Google Scholar] [CrossRef]
- Olivetta, E.; Pietraforte, D.; Schiavoni, I.; Minetti, M.; Federico, M.; Sanchez, M. HIV-1 Nef regulates the release of superoxide anions from human macrophages. Biochem. J. 2005, 390 Pt 2, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Green, L.A.; Gupta, S.K.; Kim, C.; Wang, L.; Almodovar, S.; Flores, S.C.; Prudovsky, I.A.; Jolicoeur, P.; Liu, Z.; et al. Transfer of intracellular HIV Nef to endothelium causes endothelial dysfunction. PLoS ONE 2014, 9, e91063. [Google Scholar] [CrossRef]
- Jacotot, E.; Ferri, K.F.; El Hamel, C.; Brenner, C.; Druillennec, S.; Hoebeke, J.; Rustin, P.; Metivier, D.; Lenoir, C.; Geuskens, M.; et al. Control of mitochondrial membrane permeabilization by adenine nucleotide translocator interacting with HIV-1 viral protein rR and Bcl-2. J. Exp. Med. 2001, 193, 509–519. [Google Scholar] [CrossRef]
- Vilhardt, F.; Plastre, O.; Sawada, M.; Suzuki, K.; Wiznerowicz, M.; Kiyokawa, E.; Trono, D.; Krause, K.H. The HIV-1 Nef protein and phagocyte NADPH oxidase activation. J. Biol. Chem. 2002, 277, 42136–42143. [Google Scholar] [CrossRef]
- Salmen, S.; Colmenares, M.; Peterson, D.L.; Reyes, E.; Rosales, J.D.; Berrueta, L. HIV-1 Nef associates with p22-phox, a component of the NADPH oxidase protein complex. Cell. Immunol. 2010, 263, 166–171. [Google Scholar] [CrossRef]
- Olivetta, E.; Mallozzi, C.; Ruggieri, V.; Pietraforte, D.; Federico, M.; Sanchez, M. HIV-1 Nef induces p47(phox) phosphorylation leading to a rapid superoxide anion release from the U937 human monoblastic cell line. J. Cell. Biochem. 2009, 106, 812–822. [Google Scholar] [CrossRef]
- Fackler, O.T.; Luo, W.; Geyer, M.; Alberts, A.S.; Peterlin, B.M. Activation of Vav by Nef induces cytoskeletal rearrangements and downstream effector functions. Mol. Cell. 1999, 3, 729–739. [Google Scholar] [CrossRef]
- Xia, C.; Zhang, X.; Harypursat, V.; Ouyang, J.; Chen, Y. The role of pyroptosis in incomplete immune reconstitution among people living with HIV: Potential therapeutic targets. Pharmacol. Res. 2023, 197, 106969. [Google Scholar] [CrossRef]
- Le Rouzic, E.; Benichou, S. The Vpr protein from HIV-1: Distinct roles along the viral life cycle. Retrovirology 2005, 2, 11. [Google Scholar] [CrossRef]
- Deniaud, A.; Brenner, C.; Kroemer, G. Mitochondrial membrane permeabilization by HIV-1 Vpr. Mitochondrion 2004, 4, 223–233. [Google Scholar] [CrossRef] [PubMed]
- Jacotot, E.; Ravagnan, L.; Loeffler, M.; Ferri, K.F.; Vieira, H.L.; Zamzami, N.; Costantini, P.; Druillennec, S.; Hoebeke, J.; Briand, J.P.; et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 2000, 191, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Muthumani, K.; Hwang, D.S.; Desai, B.M.; Zhang, D.; Dayes, N.; Green, D.R.; Weiner, D.B. HIV-1 Vpr induces apoptosis through caspase 9 in T cells and peripheral blood mononuclear cells. J. Biol. Chem. 2002, 277, 37820–37831. [Google Scholar] [CrossRef] [PubMed]
- Roumier, T.; Vieira, H.L.; Castedo, M.; Ferri, K.F.; Boya, P.; Andreau, K.; Druillennec, S.; Joza, N.; Penninger, J.M.; Roques, B.; et al. The C-terminal moiety of HIV-1 Vpr induces cell death via a caspase-independent mitochondrial pathway. Cell Death Differ. 2002, 9, 1212–1219. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, Y.; Fu, J.; Sun, Z.; Li, H.; Xiao, W.; E, J.; Lo, B.Y.; Wang, N.; Zhang, W.; et al. Tubular-specific expression of HIV protein Vpr leads to severe tubulointerstitial damage accompanied by progressive fibrosis and cystic development. Kidney Int. 2023, 103, 529–543. [Google Scholar] [CrossRef]
- Na, H.; Acharjee, S.; Jones, G.; Vivithanaporn, P.; Noorbakhsh, F.; McFarlane, N.; Maingat, F.; Ballanyi, K.; Pardo, C.A.; Cohen, E.A.; et al. Interactions between human immunodeficiency virus (HIV)-1 Vpr expression and innate immunity influence neurovirulence. Retrovirology 2011, 8, 44. [Google Scholar] [CrossRef]
- Ferrucci, A.; Nonnemacher, M.R.; Wigdahl, B. Human immunodeficiency virus viral protein R as an extracellular protein in neuropathogenesis. Adv. Virus Res. 2011, 81, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Alsauskas, Z.C.; Leventhal, J.S.; Rosenstiel, P.E.; Gong, P.; Chan, J.J.; Barley, K.; He, J.C.; Klotman, M.E.; Ross, M.J.; et al. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS 2010, 24, 1107–1119. [Google Scholar] [CrossRef]
- Averill-Bates, D.A. The antioxidant glutathione. Vitam. Horm. 2023, 121, 109–141. [Google Scholar] [CrossRef]
- Priya Dharshini, L.C.; Vishnupriya, S.; Sakthivel, K.M.; Rasmi, R.R. Oxidative stress responsive transcription factors in cellular signalling transduction mechanisms. Cell. Signal. 2020, 72, 109670. [Google Scholar] [CrossRef]
- Sandoval, C.; Nisson, K.; Fregoso, O.I. HIV-1 Vpr-induced DNA damage activates NF-kappaB through ATM-NEMO independent of cell cycle arrest. mBio 2024, 15, e0024024. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Tolomeo, F.; Cascio, A. The Complex Interactions Between HIV-1 and Human Host Cell Genome: From Molecular Mechanisms to Clinical Practice. Int. J. Mol. Sci. 2025, 26, 3184. [Google Scholar] [CrossRef]
- Israel, N.; Gougerot-Pocidalo, M.A.; Aillet, F.; Virelizier, J.L. Redox status of cells influences constitutive or induced NF-kappa B translocation and HIV long terminal repeat activity in human T and monocytic cell lines. J. Immunol. 1992, 149, 3386–3393. [Google Scholar] [CrossRef]
- Isaguliants, M.; Smirnova, O.; Ivanov, A.V.; Kilpelainen, A.; Kuzmenko, Y.; Petkov, S.; Latanova, A.; Krotova, O.; Engstrom, G.; Karpov, V.; et al. Oxidative stress induced by HIV-1 reverse transcriptase modulates the enzyme’s performance in gene immunization. Hum. Vaccin. Immunother. 2013, 9, 2111–2119. [Google Scholar] [CrossRef] [PubMed]
- Bayurova, E.; Jansons, J.; Skrastina, D.; Smirnova, O.; Mezale, D.; Kostyusheva, A.; Kostyushev, D.; Petkov, S.; Podschwadt, P.; Valuev-Elliston, V.; et al. HIV-1 Reverse Transcriptase Promotes Tumor Growth and Metastasis Formation via ROS-Dependent Upregulation of Twist. Oxid. Med. Cell. Longev. 2019, 2019, 6016278. [Google Scholar] [CrossRef]
- Haughey, N.J.; Cutler, R.G.; Tamara, A.; McArthur, J.C.; Vargas, D.L.; Pardo, C.A.; Turchan, J.; Nath, A.; Mattson, M.P. Perturbation of sphingolipid metabolism and ceramide production in HIV-dementia. Ann. Neurol. 2004, 55, 257–267. [Google Scholar] [CrossRef]
- Breitzig, M.; Bhimineni, C.; Lockey, R.; Kolliputi, N. 4-Hydroxy-2-nonenal: A critical target in oxidative stress? Am. J. Physiol. Cell Physiol. 2016, 311, C537–C543. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhao, T.; Li, J.; Xia, M.; Li, Y.; Wang, X.; Liu, C.; Zheng, T.; Chen, R.; Kan, D.; et al. Oxidative Stress and 4-hydroxy-2-nonenal (4-HNE): Implications in the Pathogenesis and Treatment of Aging-related Diseases. J. Immunol. Res. 2022, 2022, 2233906. [Google Scholar] [CrossRef]
- Sindhu, S.; Leung, Y.H.; Arefanian, H.; Madiraju, S.R.M.; Al-Mulla, F.; Ahmad, R.; Prentki, M. Neutral sphingomyelinase-2 and cardiometabolic diseases. Obes. Rev. 2021, 22, e13248. [Google Scholar] [CrossRef] [PubMed]
- Truman, J.P.; Al Gadban, M.M.; Smith, K.J.; Hammad, S.M. Acid sphingomyelinase in macrophage biology. Cell. Mol. Life Sci. 2011, 68, 3293–3305. [Google Scholar] [CrossRef]
- Shamseddine, A.A.; Airola, M.V.; Hannun, Y.A. Roles and regulation of neutral sphingomyelinase-2 in cellular and pathological processes. Adv. Biol. Regul. 2015, 57, 24–41. [Google Scholar] [CrossRef]
- Waheed, A.A.; Zhu, Y.; Agostino, E.; Naing, L.; Hikichi, Y.; Soheilian, F.; Yoo, S.W.; Song, Y.; Zhang, P.; Slusher, B.S.; et al. Neutral sphingomyelinase 2 is required for HIV-1 maturation. Proc. Natl. Acad. Sci. USA 2023, 120, e2219475120. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.W.; Waheed, A.A.; Deme, P.; Tohumeken, S.; Rais, R.; Smith, M.D.; DeMarino, C.; Calabresi, P.A.; Kashanchi, F.; Freed, E.O.; et al. Inhibition of neutral sphingomyelinase 2 impairs HIV-1 envelope formation and substantially delays or eliminates viral rebound. Proc. Natl. Acad. Sci. USA 2023, 120, e2219543120. [Google Scholar] [CrossRef]
- McGraw, A.; Hillmer, G.; Choi, J.; Narayan, K.; Mehedincu, S.M.; Marquez, D.; Tibebe, H.; DeCicco-Skinner, K.L.; Izumi, T. Evaluating HIV-1 Infectivity and Virion Maturation across Varied Producer Cells with a Novel FRET-Based Detection and Quantification Assay. Int. J. Mol. Sci. 2024, 25, 6396. [Google Scholar] [CrossRef]
- Hui, L.; Ye, Y.; Soliman, M.L.; Lakpa, K.L.; Miller, N.M.; Afghah, Z.; Geiger, J.D.; Chen, X. Antiretroviral Drugs Promote Amyloidogenesis by De-Acidifying Endolysosomes. J. Neuroimmune Pharmacol. 2021, 16, 159–168. [Google Scholar] [CrossRef]
- Zulu, S.S.; Abboussi, O.; Simola, N.; Mabandla, M.V.; Daniels, W.M.U. Anti-HIV drugs promote beta-amyloid deposition and impair learning and memory in BALB/c mice. Acta Neuropsychiatr. 2020, 32, 257–264. [Google Scholar] [CrossRef]
- Sharma, I. Interrogating the impact of combination antiretroviral therapies on HIV-associated neurocognitive disorders. HIV Med. 2021, 22, 783–790. [Google Scholar] [CrossRef]
- Spooner, R.; Ranasinghe, S.; Urasa, S.; Yoseph, M.; Koipapi, S.; Mukaetova-Ladinska, E.B.; Lewis, T.; Howlett, W.; Dekker, M.; Kisoli, A.; et al. HIV-Associated Neurocognitive Disorders: The First Longitudinal Follow-Up of a cART-Treated Cohort of Older People in Sub-Saharan Africa. J. Acquir. Immune. Defic. Syndr. 2022, 90, 214–222. [Google Scholar] [CrossRef]
- Osborne, O.; Peyravian, N.; Nair, M.; Daunert, S.; Toborek, M. The Paradox of HIV Blood-Brain Barrier Penetrance and Antiretroviral Drug Delivery Deficiencies. Trends Neurosci. 2020, 43, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Velichkovska, M.; Toborek, M. Cerebral Vascular Toxicity of Antiretroviral Therapy. J. Neuroimmune Pharmacol. 2021, 16, 74–89. [Google Scholar] [CrossRef]
- Ingersoll, K.S.; Cohen, J. The impact of medication regimen factors on adherence to chronic treatment: A review of literature. J. Behav. Med. 2008, 31, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Yuan, N.Y.; Kaul, M. Beneficial and Adverse Effects of cART Affect Neurocognitive Function in HIV-1 Infection: Balancing Viral Suppression against Neuronal Stress and Injury. J. Neuroimmune Pharmacol. 2021, 16, 90–112. [Google Scholar] [CrossRef]
- Adrian, M.D.; Alan, G.H. Why antioxidant therapies have failed in clinical trials. J. Theor. Biol. 2018, 457, 1–5. [Google Scholar] [CrossRef]
- Kolson, D.L. Developments in Neuroprotection for HIV-Associated Neurocognitive Disorders (HAND). Curr. HIV/AIDS Rep. 2022, 19, 344–357. [Google Scholar] [CrossRef]
- Louboutin, J.P.; Strayer, D. Role of Oxidative Stress in HIV-1-Associated Neurocognitive Disorder and Protection by Gene Delivery of Antioxidant Enzymes. Antioxidants 2014, 3, 770–797. [Google Scholar] [CrossRef]
- Agrawal, L.; Louboutin, J.P.; Strayer, D.S. Preventing HIV-1 Tat-induced neuronal apoptosis using antioxidant enzymes: Mechanistic and therapeutic implications. Virology 2007, 363, 462–472. [Google Scholar] [CrossRef]
- Manda, K.R.; Banerjee, A.; Banks, W.A.; Ercal, N. Highly active antiretroviral therapy drug combination induces oxidative stress and mitochondrial dysfunction in immortalized human blood-brain barrier endothelial cells. Free Radic. Biol. Med. 2011, 50, 801–810. [Google Scholar] [CrossRef]
- Teodorof-Diedrich, C.; Spector, S.A. Human Immunodeficiency Virus Type 1 and Methamphetamine-Mediated Mitochondrial Damage and Neuronal Degeneration in Human Neurons. J. Virol. 2020, 94, e00924-20. [Google Scholar] [CrossRef]
- Allard, J.P.; Aghdassi, E.; Chau, J.; Tam, C.; Kovacs, C.M.; Salit, I.E.; Walmsley, S.L. Effects of vitamin E and C supplementation on oxidative stress and viral load in HIV-infected subjects. AIDS 1998, 12, 1653–1659. [Google Scholar] [CrossRef]
- The Dana Consortium on the Therapy of HIV Dementia and Related Cognitive Disorders. Safety and tolerability of the antioxidant OPC-14117 in HIV-associated cognitive impairment. Neurology 1997, 49, 142–146. [Google Scholar] [CrossRef]
- Schifitto, G.; Zhang, J.; Evans, S.R.; Sacktor, N.; Simpson, D.; Millar, L.L.; Hung, V.L.; Miller, E.N.; Smith, E.; Ellis, R.J.; et al. A multicenter trial of selegiline transdermal system for HIV-associated cognitive impairment. Neurology 2007, 69, 1314–1321. [Google Scholar] [CrossRef]
- Scuderi, S.A.; Ardizzone, A.; Paterniti, I.; Esposito, E.; Campolo, M. Antioxidant and Anti-inflammatory Effect of Nrf2 Inducer Dimethyl Fumarate in Neurodegenerative Diseases. Antioxidants 2020, 9, 630. [Google Scholar] [CrossRef]
- Sandouka, S.; Singh, P.K.; Saadi, A.; Taiwo, R.O.; Sheeni, Y.; Zhang, T.; Deeb, L.; Guignet, M.; White, S.H.; Shekh-Ahmad, T. Repurposing dimethyl fumarate as an antiepileptogenic and disease-modifying treatment for drug-resistant epilepsy. J. Transl. Med. 2023, 21, 796. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Siliciano, R.F. In Vivo Dynamics of the Latent Reservoir for HIV-1: New Insights and Implications for Cure. Annu. Rev. Pathol. 2022, 17, 271–294. [Google Scholar] [CrossRef]
- Sengupta, S.; Siliciano, R.F. Targeting the Latent Reservoir for HIV-1. Immunity 2018, 48, 872–895. [Google Scholar] [CrossRef]
- Wang, Z.; Gurule, E.E.; Brennan, T.P.; Gerold, J.M.; Kwon, K.J.; Hosmane, N.N.; Kumar, M.R.; Beg, S.A.; Capoferri, A.A.; Ray, S.C.; et al. Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 2018, 115, E2575–E2584. [Google Scholar] [CrossRef]
- Rausch, J.W.; Parvez, S.; Pathak, S.; Capoferri, A.A.; Kearney, M.F. HIV Expression in Infected T Cell Clones. Viruses 2024, 16, 108. [Google Scholar] [CrossRef]
- Shan, L.; Siliciano, R.F. From reactivation of latent HIV-1 to elimination of the latent reservoir: The presence of multiple barriers to viral eradication. Bioessays 2013, 35, 544–552. [Google Scholar] [CrossRef]
- Ta, T.M.; Malik, S.; Anderson, E.M.; Jones, A.D.; Perchik, J.; Freylikh, M.; Sardo, L.; Klase, Z.A.; Izumi, T. Insights Into Persistent HIV-1 Infection and Functional Cure: Novel Capabilities and Strategies. Front. Microbiol. 2022, 13, 862270. [Google Scholar] [CrossRef]
- Sardo, L.; Parolin, C.; Yoshida, T.; Garzino-Demo, A.; Izumi, T. Editorial: Novel Insights Into a Functional HIV Cure. Front. Microbiol. 2021, 12, 797570. [Google Scholar] [CrossRef]
- Li, H.; McLaurin, K.A.; Illenberger, J.M.; Mactutus, C.F.; Booze, R.M. Microglial HIV-1 Expression: Role in HIV-1 Associated Neurocognitive Disorders. Viruses 2021, 13, 924. [Google Scholar] [CrossRef]
- Borrajo Lopez, A.; Penedo, M.A.; Rivera-Baltanas, T.; Perez-Rodriguez, D.; Alonso-Crespo, D.; Fernandez-Pereira, C.; Olivares, J.M.; Agis-Balboa, R.C. Microglia: The Real Foe in HIV-1-Associated Neurocognitive Disorders? Biomedicines 2021, 9, 925. [Google Scholar] [CrossRef]
- Watkins, B.A.; Dorn, H.H.; Kelly, W.B.; Armstrong, R.C.; Potts, B.J.; Michaels, F.; Kufta, C.V.; Dubois-Dalcq, M. Specific tropism of HIV-1 for microglial cells in primary human brain cultures. Science 1990, 249, 549–553. [Google Scholar] [CrossRef]
- Ling, L.; Kim, M.; Soper, A.; Kovarova, M.; Spagnuolo, R.A.; Begum, N.; Kirchherr, J.; Archin, N.; Battaglia, D.; Cleveland, D.; et al. Analysis of the effect of HDAC inhibitors on the formation of the HIV reservoir. mBio 2024, 15, e0163224. [Google Scholar] [CrossRef]
- Huang, L.; Lai, W.H.; Zhu, L.; Li, W.; Wei, L.; Lee, K.H.; Xie, L.; Chen, C.H. Elimination of HIV-1 Latently Infected Cells by Gnidimacrin and a Selective HDAC Inhibitor. ACS Med. Chem. Lett. 2018, 9, 268–273. [Google Scholar] [CrossRef]
- Zaikos, T.D.; Painter, M.M.; Sebastian Kettinger, N.T.; Terry, V.H.; Collins, K.L. Class 1-Selective Histone Deacetylase (HDAC) Inhibitors Enhance HIV Latency Reversal while Preserving the Activity of HDAC Isoforms Necessary for Maximal HIV Gene Expression. J. Virol. 2018, 92, e02110-17. [Google Scholar] [CrossRef]
- Tibebe, H.; Marquez, D.; McGraw, A.; Gagliardi, S.; Sullivan, C.; Hillmer, G.; Narayan, K.; Izumi, C.; Keating, A.; Izumi, T. Targeting Latent HIV Reservoirs: Effectiveness of Combination Therapy with HDAC and PARP Inhibitors. Viruses 2025, 17, 400. [Google Scholar] [CrossRef]
- Komirishetty, P.; Areti, A.; Yerra, V.G.; Ruby, P.K.; Sharma, S.S.; Gogoi, R.; Sistla, R.; Kumar, A. PARP inhibition attenuates neuroinflammation and oxidative stress in chronic constriction injury induced peripheral neuropathy. Life Sci. 2016, 150, 50–60. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Padovani, R.; Tognetti, F.; Proietti, D.; Pozzati, E.; Servadei, F. Extrathecal cavernous hemangioma. Surg. Neurol. 1982, 18, 463–465. [Google Scholar] [CrossRef]
- Wang, W.; Li, N.; Li, X.; Tran, M.K.; Han, X.; Chen, J. Tankyrase Inhibitors Target YAP by Stabilizing Angiomotin Family Proteins. Cell Rep. 2015, 13, 524–532. [Google Scholar] [CrossRef]
- Huang, S.M.; Mishina, Y.M.; Liu, S.; Cheung, A.; Stegmeier, F.; Michaud, G.A.; Charlat, O.; Wiellette, E.; Zhang, Y.; Wiessner, S.; et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461, 614–620. [Google Scholar] [CrossRef]
- Hong, B.K.; You, S.; Yoo, S.A.; Park, D.; Hwang, D.; Cho, C.S.; Kim, W.U. MicroRNA-143 and -145 modulate the phenotype of synovial fibroblasts in rheumatoid arthritis. Exp. Mol. Med. 2017, 49, e363. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Samikkannu, T.; Ranjith, D.; Rao, K.V.; Atluri, V.S.; Pimentel, E.; El-Hage, N.; Nair, M.P. HIV-1 gp120 and morphine induced oxidative stress: Role in cell cycle regulation. Front. Microbiol. 2015, 6, 614. [Google Scholar] [CrossRef]
- Tachiwana, H.; Shimura, M.; Nakai-Murakami, C.; Tokunaga, K.; Takizawa, Y.; Sata, T.; Kurumizaka, H.; Ishizaka, Y. HIV-1 Vpr induces DNA double-strand breaks. Cancer Res. 2006, 66, 627–631. [Google Scholar] [CrossRef]
- Murata, M.M.; Kong, X.; Moncada, E.; Chen, Y.; Imamura, H.; Wang, P.; Berns, M.W.; Yokomori, K.; Digman, M.A. NAD+ consumption by PARP1 in response to DNA damage triggers metabolic shift critical for damaged cell survival. Mol. Biol. Cell 2019, 30, 2584–2597. [Google Scholar] [CrossRef]
- Huang, P.; Chen, G.; Jin, W.; Mao, K.; Wan, H.; He, Y. Molecular Mechanisms of Parthanatos and Its Role in Diverse Diseases. Int. J. Mol. Sci. 2022, 23, 7292. [Google Scholar] [CrossRef]
- Wang, Y.; Luo, W.; Wang, Y. PARP-1 and its associated nucleases in DNA damage response. DNA Repair 2019, 81, 102651. [Google Scholar] [CrossRef]
- Xu, X.; Sun, B.; Zhao, C. Poly (ADP-Ribose) polymerase 1 and parthanatos in neurological diseases: From pathogenesis to therapeutic opportunities. Neurobiol. Dis. 2023, 187, 106314. [Google Scholar] [CrossRef]
- Chiarugi, A.; Moskowitz, M.A. Poly(ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: Implication for neurodegenerative disorders. J. Neurochem. 2003, 85, 306–317. [Google Scholar] [CrossRef]
- Choudhuri, S.; Garg, N.J. PARP1-cGAS-NF-kappaB pathway of proinflammatory macrophage activation by extracellular vesicles released during Trypanosoma cruzi infection and Chagas disease. PLoS Pathog. 2020, 16, e1008474. [Google Scholar] [CrossRef]
- Alano, C.C.; Garnier, P.; Ying, W.; Higashi, Y.; Kauppinen, T.M.; Swanson, R.A. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J. Neurosci. 2010, 30, 2967–2978. [Google Scholar] [CrossRef]
- d’Avila, J.C.; Lam, T.I.; Bingham, D.; Shi, J.; Won, S.J.; Kauppinen, T.M.; Massa, S.; Liu, J.; Swanson, R.A. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J. Neuroinflamm. 2012, 9, 31. [Google Scholar] [CrossRef]
- Raghunatha, P.; Vosoughi, A.; Kauppinen, T.M.; Jackson, M.F. Microglial NMDA receptors drive pro-inflammatory responses via PARP-1/TRMP2 signaling. Glia 2020, 68, 1421–1434. [Google Scholar] [CrossRef]
- Kauppinen, T.M.; Suh, S.W.; Higashi, Y.; Berman, A.E.; Escartin, C.; Won, S.J.; Wang, C.; Cho, S.H.; Gan, L.; Swanson, R.A. Poly(ADP-ribose)polymerase-1 modulates microglial responses to amyloid beta. J. Neuroinflamm. 2011, 8, 152. [Google Scholar] [CrossRef]
- Mavian, C.; Ramirez-Mata, A.S.; Dollar, J.J.; Nolan, D.J.; Cash, M.; White, K.; Rich, S.N.; Magalis, B.R.; Marini, S.; Prosperi, M.C.F.; et al. Brain tissue transcriptomic analysis of SIV-infected macaques identifies several altered metabolic pathways linked to neuropathogenesis and poly (ADP-ribose) polymerases (PARPs) as potential therapeutic targets. J. Neurovirol. 2021, 27, 101–115. [Google Scholar] [CrossRef]
- Sun, J.; Liu, J.; Gao, C.; Zheng, J.; Zhang, J.; Ding, Y.; Gong, W.; Yang, M.; Li, Z.; Wang, Y.; et al. Corrigendum to ‘Targeted delivery of PARP inhibitors to neuronal mitochondria via biomimetic engineered nanosystems in a mouse model of traumatic brain injury’ [Acta Biomaterialia 140 (2022) 573–585]. Acta Biomater. 2023, 157, 734–736. [Google Scholar] [CrossRef]
- Balko, R.; Hurley, R.; Jatoi, A. Poly (ADP-Ribose) Polymerase Inhibition for Chemotherapy-Induced Peripheral Neuropathy: A Meta-Analysis of Placebo-Controlled Trials. J. Palliat. Med. 2019, 22, 977–980. [Google Scholar] [CrossRef]
- Nakajima, H.; Kakui, N.; Ohkuma, K.; Ishikawa, M.; Hasegawa, T. A newly synthesized poly(ADP-ribose) polymerase inhibitor, DR2313 [2-methyl-3,5,7,8-tetrahydrothiopyrano[4,3-d]-pyrimidine-4-one]: Pharmacological profiles, neuroprotective effects, and therapeutic time window in cerebral ischemia in rats. J. Pharmacol. Exp. Ther. 2005, 312, 472–481. [Google Scholar] [CrossRef]
- Tharamelveliyil Rajendran, A.; Dheeraj Rajesh, G.; Kumar, P.; Shivam Raju Dwivedi, P.; Shashidhara Shastry, C.; Narayanan Vadakkepushpakath, A. Selection of potential natural compounds for poly-ADP-ribose polymerase (PARP) inhibition in glioblastoma therapy by in silico screening methods. Saudi J. Biol. Sci. 2023, 30, 103698. [Google Scholar] [CrossRef]
- Wing, E.J. HIV and aging. Int. J. Infect. Dis. 2016, 53, 61–68. [Google Scholar] [CrossRef]
- Donoso, M.; D’Amico, D.; Valdebenito, S.; Hernandez, C.A.; Prideaux, B.; Eugenin, E.A. Identification, Quantification, and Characterization of HIV-1 Reservoirs in the Human Brain. Cells 2022, 11, 2379. [Google Scholar] [CrossRef]
- Sajja, R.K.; Rahman, S.; Cucullo, L. Drugs of abuse and blood-brain barrier endothelial dysfunction: A focus on the role of oxidative stress. J. Cereb. Blood Flow Metab. 2016, 36, 539–554. [Google Scholar] [CrossRef]
- Yamamoto, B.K.; Moszczynska, A.; Gudelsky, G.A. Amphetamine toxicities: Classical and emerging mechanisms. Ann. NY Acad. Sci. 2010, 1187, 101–121. [Google Scholar] [CrossRef]
- Ferris, M.J.; Mactutus, C.F.; Booze, R.M. Neurotoxic profiles of HIV, psychostimulant drugs of abuse, and their concerted effect on the brain: Current status of dopamine system vulnerability in NeuroAIDS. Neurosci. Biobehav. Rev. 2008, 32, 883–909. [Google Scholar] [CrossRef]
- Sanchez, A.B.; Kaul, M. Neuronal Stress and Injury Caused by HIV-1, cART and Drug Abuse: Converging Contributions to HAND. Brain Sci. 2017, 7, 25. [Google Scholar] [CrossRef]
- Borgmann, K.; Ghorpade, A. Methamphetamine Augments Concurrent Astrocyte Mitochondrial Stress, Oxidative Burden, and Antioxidant Capacity: Tipping the Balance in HIV-Associated Neurodegeneration. Neurotox. Res. 2018, 33, 433–447. [Google Scholar] [CrossRef]
- Jones, J.D. Potential of Glial Cell Modulators in the Management of Substance Use Disorders. CNS Drugs 2020, 34, 697–722. [Google Scholar] [CrossRef]
- Sil, S.; Thangaraj, A.; Chivero, E.T.; Niu, F.; Kannan, M.; Liao, K.; Silverstein, P.S.; Periyasamy, P.; Buch, S. HIV-1 and drug abuse comorbidity: Lessons learned from the animal models of NeuroHIV. Neurosci. Lett. 2021, 754, 135863. [Google Scholar] [CrossRef]
- Mahajan, S.D.; Aalinkeel, R.; Parikh, N.U.; Jacob, A.; Cwiklinski, K.; Sandhu, P.; Le, K.; Loftus, A.W.; Schwartz, S.A.; Quigg, R.J.; et al. Immunomodulatory Role of Complement Proteins in the Neuropathology Associated with Opiate Abuse and HIV-1 Co-Morbidity. Immunol. Investig. 2017, 46, 816–832. [Google Scholar] [CrossRef]
- Chambers, D.W. The Argument From Perfection. J. Calif. Dent. Assoc. 2017, 45, 65. [Google Scholar]
- Sil, S.; Niu, F.; Chivero, E.T.; Singh, S.; Periyasamy, P.; Buch, S. Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging. Cells 2020, 9, 1857. [Google Scholar] [CrossRef]
- Fiellin, D.A. Substance use of disorders in HIV-infected patients: Impact and new treatment strategies. Top. HIV Med. 2004, 12, 77–82. [Google Scholar]
- Kadry, H.; Noorani, B.; Bickel, U.; Abbruscato, T.J.; Cucullo, L. Comparative assessment of in vitro BBB tight junction integrity following exposure to cigarette smoke and e-cigarette vapor: A quantitative evaluation of the protective effects of metformin using small-molecular-weight paracellular markers. Fluids Barriers CNS 2021, 18, 28. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Tachibana, K.; Kondoh, M. Tight junction modulators for drug delivery to the central nervous system. Drug Discov. Today 2020, 25, 1477–1486. [Google Scholar] [CrossRef]
- Li, H.; Walker, M.; Ji, H.; Sikirzhytskaya, A.; Aksenova, M.; Shtutman, M.; Sikirzhytski, V.; Mactutus, C.F.; Booze, R.M. Identification of EcoHIV-Infected Cells in Microglia-Manipulated Transgenic Mice. J. Vis. Exp. 2024, e67150. [Google Scholar] [CrossRef]
- Gu, C.J.; Borjabad, A.; Hadas, E.; Kelschenbach, J.; Kim, B.H.; Chao, W.; Arancio, O.; Suh, J.; Polsky, B.; McMillan, J.; et al. EcoHIV infection of mice establishes latent viral reservoirs in T cells and active viral reservoirs in macrophages that are sufficient for induction of neurocognitive impairment. PLoS Pathog. 2018, 14, e1007061. [Google Scholar] [CrossRef]
- Kim, B.H.; Chao, W.; Hadas, E.; Borjabad, A.; Potash, M.J.; Volsky, D.J. EcoHIV Infection of Primary Murine Brain Cell Cultures to Model HIV Replication and Neuropathogenesis. Viruses 2024, 16, 693. [Google Scholar] [CrossRef]
- Li, H.; McLaurin, K.A.; Mactutus, C.F.; Booze, R.M. A Rat Model of EcoHIV Brain Infection. J. Vis. Exp. 2021, e62137. [Google Scholar] [CrossRef]
- Nedelcovych, M.T.; Kim, B.H.; Zhu, X.; Lovell, L.E.; Manning, A.A.; Kelschenbach, J.; Hadas, E.; Chao, W.; Prchalova, E.; Dash, R.P.; et al. Correction to: Glutamine Antagonist JHU083 Normalizes Aberrant Glutamate Production and Cognitive Deficits in the EcoHIV Murine Model of HIV-Associated Neurocognitive Disorders. J. Neuroimmune Pharmacol. 2021, 16, 693. [Google Scholar] [CrossRef]
- Jones, L.D.; Jackson, J.W.; Maggirwar, S.B. Modeling HIV-1 Induced Neuroinflammation in Mice: Role of Platelets in Mediating Blood-Brain Barrier Dysfunction. PLoS ONE 2016, 11, e0151702. [Google Scholar] [CrossRef]
- Surnar, B.; Shah, A.S.; Park, M.; Kalathil, A.A.; Kamran, M.Z.; Ramirez Jaime, R.; Toborek, M.; Nair, M.; Kolishetti, N.; Dhar, S. Correction to “Brain-Accumulating Nanoparticles for Assisting Astrocytes to Reduce Human Immunodeficiency Virus and Drug Abuse-Induced Neuroinflammation and Oxidative Stress”. ACS Nano 2024, 18, 22608. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | Mechanism | Status | Reference |
|---|---|---|---|
| NACA | Reduced neuronal death, preserved mitochondrial membrane potential, inhibited oxidative damage | Preclinical in vitro study | [130,132] |
| Vitamin E and Vitamin C | Reduced oxidative stress via decreased lipid peroxidation (measured by breath pentane, plasma lipid peroxides, and malondialdehyde) | Randomized, double-blind, placebo-controlled trial | [133] |
| OPC-14117 | Scavenges superoxide anion radicals; hypothesized to reduce oxidative neurotoxicity from HIV-infected macrophage-neuron interactions | Phase II—completed and discontinued | [134] |
| STS | Inhibits monoamine oxidase B (MAO-B); reduces oxidative stress and may promote neurotrophic activity | FDA-approved for major depressive disorder, but not for HAND | [135] |
| DMF | Activates Nrf2 pathway, reduces oxidative stress, and suppresses microglial-mediated neuroinflammation | No clinical trials in HAND yet; preclinical evidence only | [136,137] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gagliardi, S.; Hotchkin, T.; Hillmer, G.; Engelbride, M.; Diggs, A.; Tibebe, H.; Izumi, C.; Sullivan, C.; Cropp, C.; Lantz, O.; et al. Oxidative Stress in HIV-Associated Neurodegeneration: Mechanisms of Pathogenesis and Therapeutic Targets. Int. J. Mol. Sci. 2025, 26, 6724. https://doi.org/10.3390/ijms26146724
Gagliardi S, Hotchkin T, Hillmer G, Engelbride M, Diggs A, Tibebe H, Izumi C, Sullivan C, Cropp C, Lantz O, et al. Oxidative Stress in HIV-Associated Neurodegeneration: Mechanisms of Pathogenesis and Therapeutic Targets. International Journal of Molecular Sciences. 2025; 26(14):6724. https://doi.org/10.3390/ijms26146724
Chicago/Turabian StyleGagliardi, Sophia, Tristan Hotchkin, Grace Hillmer, Maeve Engelbride, Alexander Diggs, Hasset Tibebe, Coco Izumi, Cailyn Sullivan, Cecelia Cropp, Olive Lantz, and et al. 2025. "Oxidative Stress in HIV-Associated Neurodegeneration: Mechanisms of Pathogenesis and Therapeutic Targets" International Journal of Molecular Sciences 26, no. 14: 6724. https://doi.org/10.3390/ijms26146724
APA StyleGagliardi, S., Hotchkin, T., Hillmer, G., Engelbride, M., Diggs, A., Tibebe, H., Izumi, C., Sullivan, C., Cropp, C., Lantz, O., Marquez, D., Chang, J., Ezaki, J., Zestos, A. G., Riley, A. L., & Izumi, T. (2025). Oxidative Stress in HIV-Associated Neurodegeneration: Mechanisms of Pathogenesis and Therapeutic Targets. International Journal of Molecular Sciences, 26(14), 6724. https://doi.org/10.3390/ijms26146724