

A Novel Chemotherapy Combination to Enhance Proteotoxic Cell Death in Hepatocellular Carcinoma Experimental Models Without Killing Non-Cancer Cells
 , ,
, , 
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
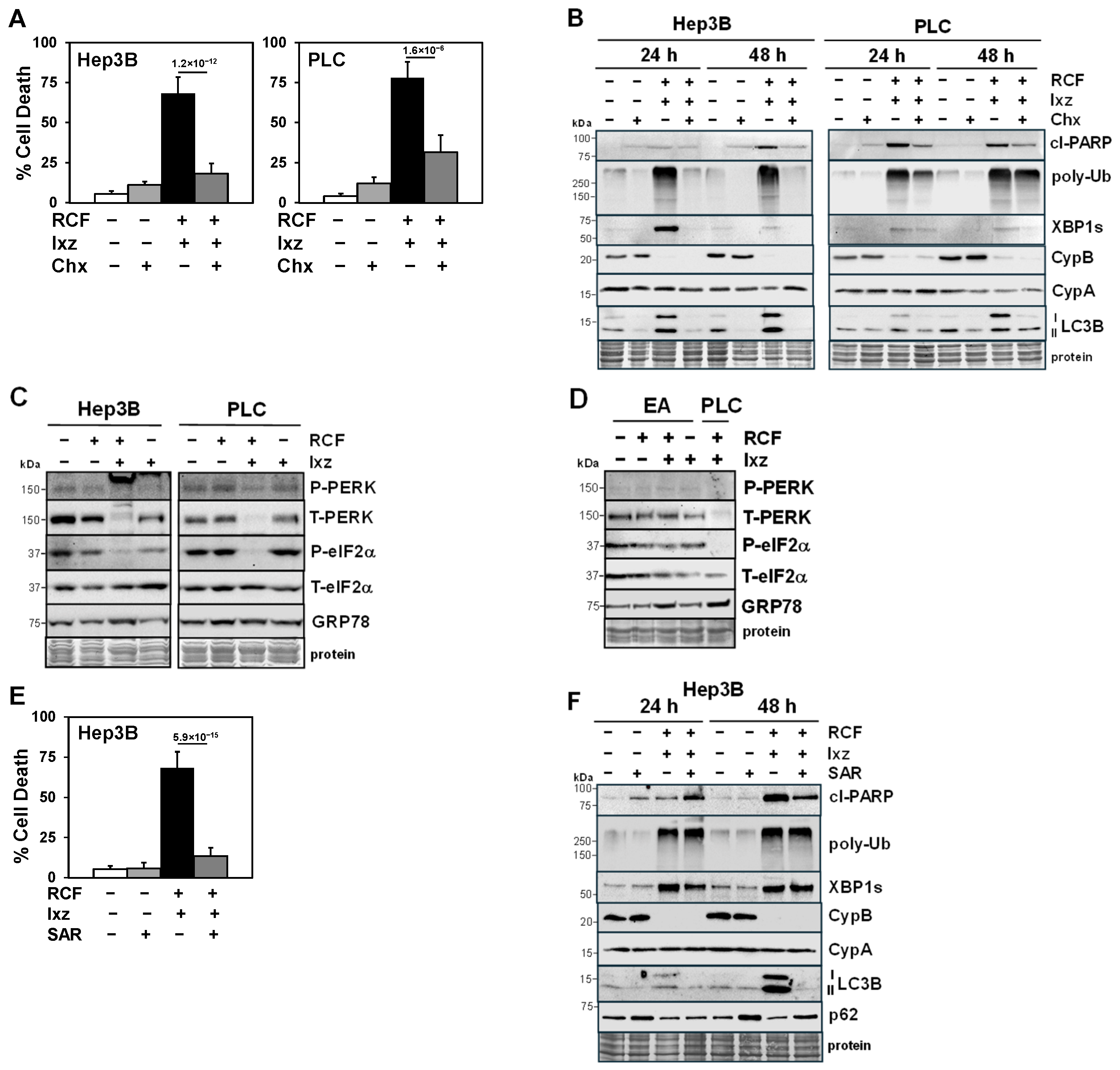
2.1. The RCF + Ixz Combination Enhances Apoptotic Cell Death in HCC Cells but Has Less of an Effect on Non-Cancer Cells
2.2. RCF + Ixz Increases Proteotoxic Stress and Alters Autophagy in HCC Cells
2.3. Maintenance of Protein Synthesis by Blocking PERK/P-eIF2α Increases RCF + Ixz-Induced Proteotoxic Cell Death in HCC Cells
2.4. Inhibition of Autophagy Blocks the RCF + Ixz-Induced Increase in Apoptotic Cell Death in HCC Cells
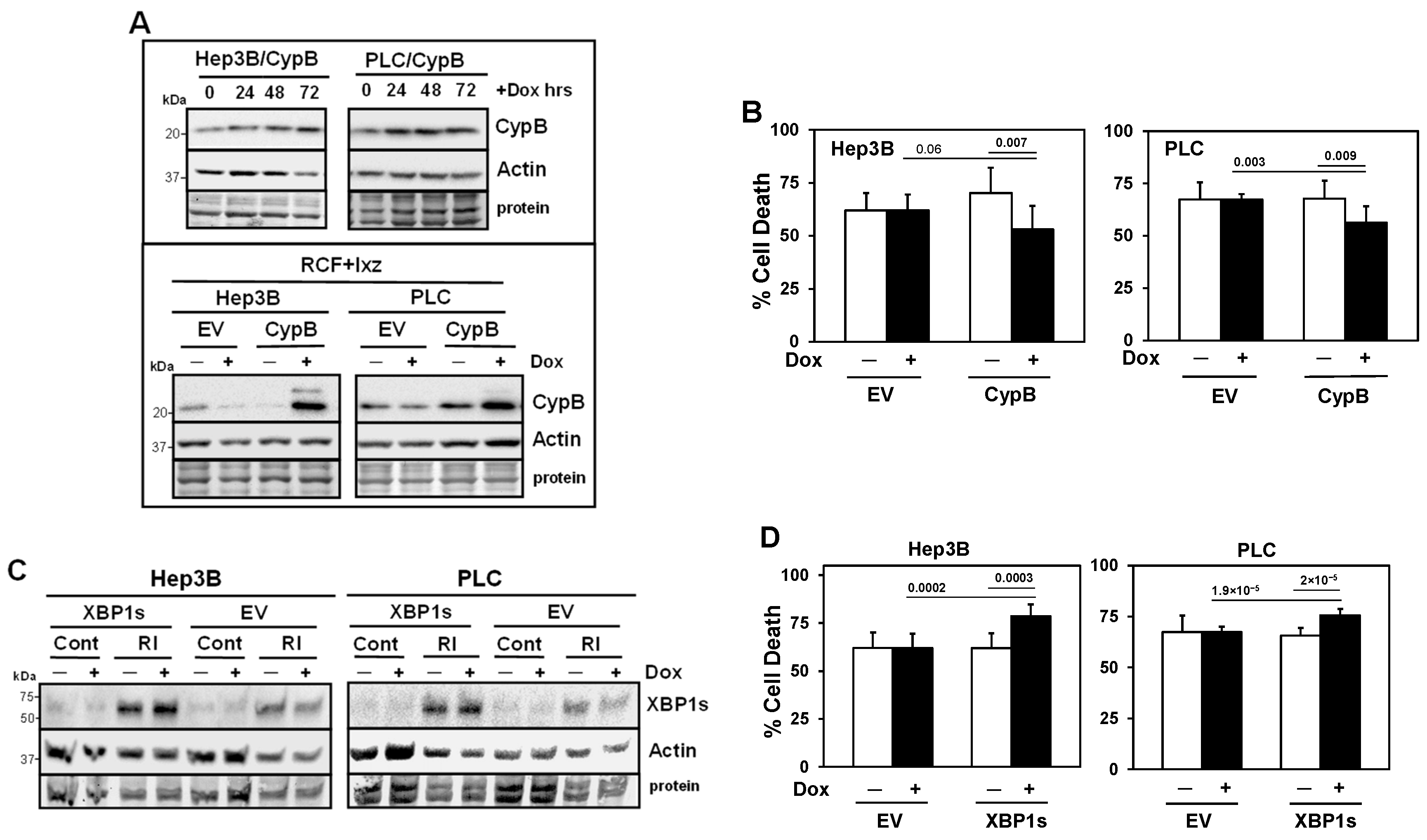
2.5. Inducible Knockdown of the RCF Targets CypA and CypB Supports Pro-Survival Roles in Ixz- and RCF + Ixz-Treated HCC Cells
2.6. Inducible Knockdown of XBP1s Supports a Pro-Survival Role Early but a Pro-Death Role Later
2.7. Inducible Expression of CypB Decreases and Inducible Expression of XBP1s Increases Cell Death in RCF + Ixz-Treated HCC Cells
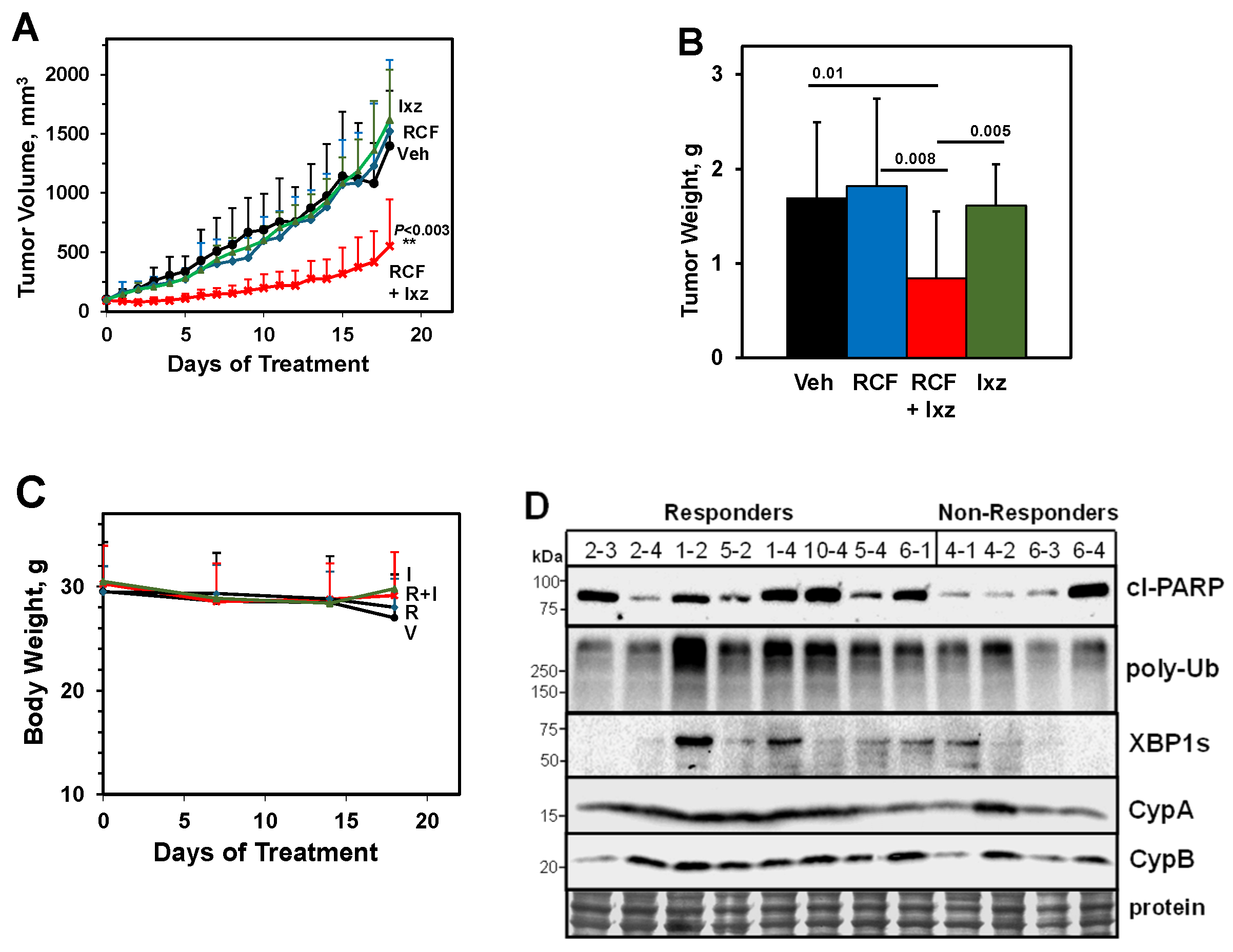
2.8. An Orally Bioavailable RCF + Ixz Combination Reduces Hep3B Xenograft HCC Tumors In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture
4.3. Drug Treatments
4.4. Trypan Blue Exclusion Assay to Measure Total Cell Death
4.5. Cell Proliferation Assay and Determination of the Synergy Combination Index
4.6. Western Blot Analysis
4.7. Quantitative Real-Time Polymerase Chain Reaction (qPCR)
4.8. Inducible Knockdown of CypA, CypB, and XBP1s with Lentivirus Transduction
4.9. Inducible Expression of CypB and XBP1s with Lentivirus Transduction
4.10. Treatment of Hep3B Xenograft-Bearing Mice with Orally Bioavailable RCF + Ixz
4.11. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Chx | Cycloheximide |
| CI | Combination Index |
| CsA | Cyclosporin A |
| Cyp | Cyclophilin |
| Dox | Doxycycline |
| EA | EA.hy926 Umbilical Vein |
| ER | Endoplasmic Reticulum |
| EV | Empty Vector |
| FA | Fraction Affected |
| HCC | Hepatocellular Carcinoma |
| HDF | Human Dermal Fibroblast |
| Ixz | Ixazomib |
| kDa | Kilo Daltons |
| P | Phospho |
| RCF | Rencofilstat |
| Scr | Scrambled |
| T | Total |
| TG | Thapsigargin |
| Ub | Ubiquitinated |
| UPR | Unfolded Protein Response |
| UPS | Ubiquitin–Proteasome System |
References
- Luo, J.; Solimini, N.L.; Elledge, S.J. Principles of cancer therapy: Oncogene and non-oncogene addiction. Cell 2009, 136, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Guang, M.H.Z.; Kavanagh, E.L.; Dunne, L.P.; Dowling, P.; Zhang, L.; Lindsay, S.; Bazou, D.; Goh, C.Y.; Hanley, C.; Bianchi, G.; et al. Targeting proteotoxic stress in cancer: A review of the role that protein quality control pathways play in oncogenesis. Cancers 2019, 11, 66. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.Q.; Shen, T.; Fang, S.J.; Sun, X.M.; Li, G.Y.; Li, Y.F. Protein homeostasis in aging and cancer. Front. Cell Dev. Biol. 2023, 11, 1143532. [Google Scholar] [CrossRef]
- Livneh, I.; Cohen-Kaplan, V.; Cohen-Rosenzweig, C.; Avni, N.; Ciechanover, A. The life cycle of the 26S proteasome: From birth, through regulation and function, and onto its death. Cell Res. 2016, 26, 869–885. [Google Scholar] [CrossRef]
- Teicher, B.A.; Tomaszewski, J.E. Proteasome inhibitors. Biochem. Pharmacol. 2015, 96, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Manasanch, E.E.; Orlowski, R.Z. Proteasome inhibitors in cancer therapy. Nat. Rev. Clin. Oncol. 2017, 14, 417–433. [Google Scholar] [CrossRef]
- Kim, G.P.; Mahoney, M.R.; Szydlo, D.; Mok, T.S.; Marshke, R.; Holen, K.; Picus, J.; Boyer, M.; Pitot, H.C.; Rubin, J.; et al. An international, multicenter phase II trial of bortezomib in patients with hepatocellular carcinoma. Investig. New Drugs 2012, 30, 387–394. [Google Scholar] [CrossRef]
- Sawyers, C.L.; Abate-Shen, C.; Anderson, K.C.; Barker, A.; Baselga, J.; Berger, N.A.; Foti, M.; Jemal, A.; Lawrence, T.S.; Li, C.I.; et al. AACR cancer progress report 2013. Clin. Cancer Res. 2013, 19 (Suppl. 20), S1–S98. [Google Scholar] [CrossRef]
- Llovet, J.M.; Villanueva, A.; Lachenmayer, A.; Finn, R.S. Advances in targeted therapies for hepatocellular carcinoma in the genomic era. Nat. Rev. Clin. Oncol. 2015, 12, 408–424. [Google Scholar] [CrossRef]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic reticulum stress and the hallmarks of cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef]
- Oakes, S.A. Endoplasmic reticulum stress signaling in cancer cells. Am. J. Pathol. 2020, 190, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Iwakoshi, N.N.; Anderson, K.C.; Glimcher, L.H. Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc. Natl. Acad. Sci. USA 2003, 100, 9946–9951. [Google Scholar] [CrossRef]
- Nawrocki, S.T.; Carew, J.S.; Pino, M.S.; Highshaw, R.A.; Dunner, K.; Huang, P.; Abbruzzese, J.L.; McConkey, D.J. Bortezomib sensitizes pancreatic cells to endoplasmic stress-mediated apoptosis. Cancer Res. 2005, 65, 11658–11666. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Carlson, L.M.; Gutman, D.M.; Harrington, W.J.; Lee, K.P.; Boise, L.H. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 2006, 107, 4907–4916. [Google Scholar] [CrossRef]
- Zhu, K.; Dunner, K.; McConkey, D.J. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene 2010, 29, 451–462. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef]
- Song, S.; Tan, J.; Miao, Y.; Li, M.; Zhang, Q. Crosstalk of autophagy and apoptosis: Involvement of the dual role of autophagy under ER stress. J. Cell. Physiol. 2017, 232, 2977–2984. [Google Scholar] [CrossRef] [PubMed]
- Bhat, T.A.; Chaudhary, A.K.; Kumar, S.; O’Malley, J.; Inigo, J.R.; Kumar, R.; Yadav, N.; Chandra, D. Endoplasmic reticulum-mediated unfolded protein response and mitochondrial apoptosis in cancer. Biochim. Biophys. Acta 2017, 1867, 58–66. [Google Scholar] [CrossRef]
- Schenk, E.; Hendrickson, A.E.; Northfelt, D.; Toft, D.O.; Ames, M.M.; Menefee, M.; Satele, D.; Qin, R.; Erlichman, C. Phase I study of tanespimycin in combination with bortezomib in patients with advanced solid malignancies. Investig. New Drugs 2013, 31, 1251–1256. [Google Scholar] [CrossRef]
- San-Miguel, J.F.; Hungria, V.T.; Yoon, S.S.; Beksac, M.; Dimopoulos, M.A.; Elghandour, A.; Jedrzejczak, W.W.; Günther, A.; Nakorn, T.N.; Siritanaratkul, N.; et al. Overall survival of patients with relapsed multiple myeloma treated with panobinostat or placebo plus bortezomib and dexamethasone (the PANORAMA 1 trial): A randomised, placebo-controlled, phase 3 trial. Lancet Haematol. 2016, 3, e506–e515. [Google Scholar] [CrossRef]
- Kuo, J.; Bobardt, M.; Chatterji, U.; Mayo, P.R.; Trepanier, D.J.; Foster, R.T.; Gallay, P.; Ure, D.R. A pan-cyclophilin inhibitor, CRV431, decreases fibrosis and tumor development in chronic liver disease models. J. Pharmacol. Exp. Ther. 2019, 371, 231–241. [Google Scholar] [CrossRef]
- Stauffer, W.; Bobardt, M.; Ure, D.; Foster, R.; Gallay, P. The cyclophilin inhibitor rencofilstat decreases HCV-induced hepatocellular carcinoma independently of its antiviral activity. Viruses 2023, 15, 2099. [Google Scholar] [CrossRef]
- Moreau, P.; Masszi, T.; Grzasko, N.; Bahlis, N.J.; Hansson, M.; Pour, L.; Sandhu, I.; Ganly, P.; Baker, B.W.; Jackson, S.R.; et al. Oral ixazomib, lenalidomide, and dexamethasone for multiple myeloma. N. Engl. J. Med. 2016, 374, 1621–1634. [Google Scholar] [CrossRef]
- Al-Salama, Z.T.; Garnock-Jones, K.P.; Scott, L.J. Ixazomib: A review in relapsed and/or refractory multiple myeloma. Target. Oncol. 2017, 12, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Lavin, P.T.; McGee, M.M. Cyclophilin function in cancer: Lessons from virus replication. Curr. Mol. Pharmacol. 2015, 9, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M. Extracellular cyclophilins in health and disease. Biochim. Biophys. Acta 2015, 1850, 2087–2095. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, W.T.; Goodman, A.Z.; Gallay, P.A. Cyclophilin inhibition as a strategy for the treatment of human disease. Front. Pharmacol. 2024, 15, 1417945. [Google Scholar] [CrossRef]
- Chu, M.Y.; Huang, H.C.; Li, E.M.; Xu, L.Y. CypA: A potential target of tumor radiotherapy and/or chemotherapy. Curr. Med. Chem. 2021, 28, 3787–3802. [Google Scholar] [CrossRef]
- Choi, J.W.; Schroeder, M.A.; Sarkaria, J.N.; Bram, R.J. Cyclophilin B supports Myc and mutant p53-dependent survival of glioblastoma multiforme cells. Cancer Res. 2014, 74, 484–496. [Google Scholar] [CrossRef]
- Cohen, Y.C.; Zada, M.; Wang, S.Y.; Bornstein, C.; David, E.; Moshe, A.; Li, B.; Shlomi-Loubaton, S.; Gatt, M.E.; Gur, C.; et al. Identification of resistance pathways and therapeutic targets in relapsed multiple myeloma patients through single-cell sequencing. Nat. Med. 2021, 27, 491–503. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Price, E.R.; Jin, M.; Lim, D.; Pati, S.; Walsh, C.T.; McKeon, F.D. Cyclophilin B trafficking through the secretory pathway is altered by binding of cyclosporin A. Proc. Natl. Acad. Sci. USA 1994, 91, 3931–3935. [Google Scholar] [CrossRef] [PubMed]
- Fearon, P.; Lonsdale-Eccles, A.A.; Ross, O.K.; Todd, C.; Sinha, A.; Allain, F.; Reynolds, N.J. Keratinocyte secretion of cyclophilin B via the constitutive pathway is regulated through its cyclosporin-binding site. J. Investig. Dermatol. 2011, 131, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- Ronan, B.; Flamand, O.; Vescovi, L.; Dureuil, C.; Durand, L.; Fassy, F.; Bachelot, M.F.; Lamberton, A.; Mathieu, M.; Bertrand, T.; et al. A highly potent and selective Vps34 inhibitor alters vesicle trafficking and autophagy. Nat. Chem. Biol. 2014, 10, 1013–1019. [Google Scholar] [CrossRef]
- Zeng, L.; Zampetaki, A.; Margariti, A.; Pepe, A.E.; Alam, S.; Martin, D.; Xiao, Q.; Wang, W.; Jin, Z.G.; Cockerill, G.; et al. Sustained activation of XBP1s splicing leads to endothelial apoptosis and atherosclerosis development in response to disturbed flow. Proc. Natl. Acad. Sci. USA 2009, 106, 8326–8331. [Google Scholar] [CrossRef]
- Allagnat, F.; Christulia, F.; Ortis, F.; Pirot, P.; Lortz, S.; Lenzen, S.; Eizirik, D.L.; Cardozo, A.K. Sustained production of spliced X-box binding protein 1 (XBP1) induces pancreatic beta cell dysfunction and apoptosis. Diabetologia 2010, 53, 1120–1130. [Google Scholar] [CrossRef]
- Valentine, C.D.; Anderson, M.O.; Papa, F.R.; Haggie, P.M. X-box binding protein 1 (XBP1s) is a critical determinant of Pseudomonas aeruginosa homoserine lactone-mediated apoptosis. PloS Pathog. 2013, 9, e1003576. [Google Scholar] [CrossRef]
- Fink, E.E.; Moparthy, S.; Bagati, A.; Bianchi-Smiraglia, A.; Lipchick, B.C.; Wolff, D.W.; Roll, M.V.; Wang, J.; Liu, S.; Bakin, A.V.; et al. XBP1-KLF9 axis acts as a molecular rheostat to control the transition from adaptive to cytotoxic unfolded protein response. Cell Rep. 2018, 25, 212–223. [Google Scholar] [CrossRef]
- Chen, Y.T.; Kung, J.T. Rapid death of follicular B cells and Burkitt lymphoma cells effectuated by Xbp1s. J. Immunol. 2020, 204, 3236–3247. [Google Scholar] [CrossRef]
- Xu, Q.; Zhang, H.; Wang, S.; Qin, C.; Lu, Y. Constitutive expression of spliced XBP1 causes perinatal lethality in mice. Genesis 2021, 59, e23420. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Q.; Kolls, B.J.; Mace, B.; Yu, S.; Li, X.; Liu, W.; Chaparro, E.; Shen, Y.; Dang, L.; et al. Sustained overexpression of spliced X-box-binding protein-1 in neurons leads to spontaneous seizures and sudden death in mice. Commun. Biol. 2023, 6, 252. [Google Scholar] [CrossRef] [PubMed]
- Philippe, C.; Jaud, M.; Féral, K.; Gay, A.; Van Den Berghe, L.; Farce, M.; Bousquet, M.; Pyronnet, S.; Mazzolini, L.; Rouault-Pierre, K.; et al. Pivotal role of the endoplasmic reticulum stress-related XBP1s/miR-22/SIRT1 axis in acute myeloid leukemia apoptosis and response to chemotherapy. Leukemia 2024, 38, 1764–1776. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.J.; Hooi, L.; Dan, Y.Y.; Bonney, G.K.; Zhou, L.; Chow, P.K.; Chee, C.E.; Toh, T.B.; Chow, E.K. Rational drug combination design in patient-derived avatars reveals effective inhibition of hepatocellular carcinoma with proteasome and CDK inhibitors. J. Exp. Clin. Cancer Res. 2022, 41, 249. [Google Scholar]
- Yang, H.; Liu, Y.; Zhang, N.; Tao, F.; Yin, G. Therapeutic advances in hepatocellular carcinoma: An update from the 2024 ASCO annual meeting. Front. Oncol. 2024, 14, 1453412. [Google Scholar] [CrossRef] [PubMed]
- Konda, P.; Garinet, S.; Van Allen, E.M.; Viswanathan, S.R. Genome-guided discovery of cancer therapeutic targets. Cell Rep. 2023, 42, 112978. [Google Scholar] [CrossRef]
- Haslam, A.; Kim, M.S.; Prasad, V. Updated estimates of eligibility for and response to genome-targeted oncology drugs among US cancer patients, 2006–2020. Ann. Oncol. 2021, 32, 926–932. [Google Scholar] [CrossRef]
- Colgan, J.; Asmal, M.; Neagu, M.; Yu, B.; Schneidkraut, J.; Lee, Y.; Sokolskaja, E.; Andreotti, A.; Luban, J. Cyclophilin A regulates TCR signal strength in CD4+ T cells via a proline-directed conformational switch in Itk. Immunity 2004, 21, 189–201. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Choi, J.W.; Sutor, S.L.; Lindquist, L.; Evans, G.L.; Madden, B.J.; Bergen, H.R.; Hefferan, T.E.; Yaszemski, M.J.; Bram, R.J. Severe osteogenesis imperfecta in cyclophilin B-deficient mice. PloS Genet. 2009, 5, e1000750. [Google Scholar] [CrossRef]
- Voss, A.K.; Thomas, T.; Gruss, P. Mice lacking HSP90 beta fail to develop a placental labyrinth. Development 2000, 127, 1–11. [Google Scholar] [CrossRef]
- Witt, O.; Deubzer, H.E.; Milde, T.; Oehme, I. HDAC family: What are the cancer relevant targets? Cancer Lett. 2009, 277, 8–21. [Google Scholar] [CrossRef]
- Schubert, A.; Grimm, S. Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 2004, 64, 85–93. [Google Scholar] [CrossRef]
- Machida, K.; Ohta, Y.; Osada, H. Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J. Biol. Chem. 2006, 281, 14314–14320. [Google Scholar] [CrossRef]
- Eliseev, R.A.; Malecki, J.; Lester, T.; Zhang, Y.; Humphrey, J.; Gunter, T.E. Cyclophilin D interacts with Bcl2 and exerts an anti-apoptotic effect. J. Biol. Chem. 2009, 284, 9692–9699. [Google Scholar] [CrossRef]
- Bigi, A.; Beltrami, E.; Trinel, M.; Stendardo, M.; Pelicci, P.G.; Giorgio, M. Cyclophilin D counteracts p53-mediated growth arrest and promotes Ras tumorigenesis. Oncogene 2016, 35, 5132–5143. [Google Scholar] [CrossRef] [PubMed]
- Stauffer, W.T.; Bobardt, M.; Ure, D.R.; Foster, R.T.; Gallay, P. Cyclophilin D knockout significantly prevents HCC development in a streptozotocin-induced mouse model of diabetes-linked NASH. PloS ONE 2024, 19, e0301711. [Google Scholar] [CrossRef] [PubMed]
- Kuo, J.; Serrano, S.S.; Grönberg, A.; Massoumi, R.; Hansson, M.J.; Gallay, P. Cyclophilin inhibitor NV556 reduces fibrosis and hepatocellular carcinoma development in mice with non-alcoholic steatohepatitis. Front. Pharmacol. 2019, 10, 1129. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.S.; Tavecchio, M.; Grönberg, A.; Sime, W.; Jemaà, M.; Moss, S.; Gregory, M.A.; Gallay, P.; Elmér, E.; Hansson, M.J.; et al. Novel cyclophilin inhibitor decreases cell proliferation and tumor growth in models of hepatocellular carcinoma. Cancers 2021, 13, 3041. [Google Scholar] [CrossRef]
- Reiner, T.; Parrondo, R.; de Las Pozas, A.; Palenzuela, D.; Perez-Stable, C. Betulinic acid selectively increases protein degradation and enhances prostate cancer-specific apoptosis: Possible role for inhibition of deubiquitinase activity. PloS ONE 2013, 8, e56234. [Google Scholar] [CrossRef]
- Gomez, L.A.; de las Pozas, A.; Perez-Stable, C. Sequential combination of flavopiridol and docetaxel reduces the levels of XIAP and AKT proteins and stimulates apoptosis in human LNCaP prostate cancer cells. Mol. Cancer Ther. 2006, 5, 1216–1226. [Google Scholar] [CrossRef]
- Parrondo, R.; de las Pozas, A.; Reiner, T.; Rai, P.; Perez-Stable, C. NF-κB activation enhances cell death by antimitotic drugs in human prostate cancer cells. Mol. Cancer 2010, 9, 182. [Google Scholar] [CrossRef]
- Wang, X.; Spandidos, A.; Wang, H.; Seed, B. PrimerBank: PCR primer database for quantitative gene expression analysis, 2012 update. Nucl. Acids Res. 2012, 40, D1144–D1149. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Wiederschain, D.; Wee, S.; Chen, L.; Loo, A.; Yang, G.; Huang, A.; Chen, Y.; Caponigro, G.; Yao, Y.M.; Lengauer, C.; et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 2009, 8, 498–504. [Google Scholar] [CrossRef]
- Reiner, T.; de las Pozas, A.; Parrondo, R.; Palenzuela, D.; Cayuso, W.; Rai, P.; Perez-Stable, C. Mcl-1 protects prostate cancer cells from cell death mediated by chemotherapy-induced DNA damage. Oncoscience 2015, 2, 703–715. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Durinck, S.; Stawiski, E.W.; Poirier, J.T.; Modrusan, Z.; Shames, D.S.; Bergbower, E.A.; Guan, Y.; Shin, J.; Guillory, J.; et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat. Genet. 2012, 44, 1111–1116. [Google Scholar] [CrossRef]
- Barger, C.J.; Branick, C.; Chee, L.; Karpf, A.R. Pan-cancer analyses reveal genomic features of FOXM1 overexpression in cancer. Cancers 2019, 11, 251. [Google Scholar] [CrossRef] [PubMed]
- Trepanier, D.J.; Ure, D.R.; Foster, R.T. Development, characterization, and pharmacokinetic evaluation of a CRV431 loaded self-microemulsifying drug delivery system. J. Pharm. Pharm. Sci. 2018, 21, 335s–348s. [Google Scholar] [CrossRef]
- Harrison, S.A.; Mayo, P.R.; Hobbs, T.M.; Canizares, C.; Foster, E.P.; Zhao, C.; Ure, D.R.; Trepanier, D.J.; Greytok, J.A.; Foster, R.T. Rencofilstat, a cyclophilin inhibitor: A phase 2a, multicenter, single-blind, placebo-controlled study in F2/F3 NASH. Hepatol. Commun. 2022, 6, 3379–3392. [Google Scholar] [CrossRef]
- Harrison, S.A.; Mayo, P.; Hobbs, T.; Zhao, C.; Canizares, C.; Foster, R.; McRae, M.P.; Helmke, S.M.; Everson, G.T. Rencofilstat treatment improves liver function in MASH with advanced fibrosis as quantified by HepQuant DuO. Liver Int. 2025, 45, e70036. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perez-Stable, C.; de las Pozas, A.; Reiner, T.; Gomez, J.; Nagarajan, M.; Foster, R.T.; Ure, D.R.; Wangpaichitr, M. A Novel Chemotherapy Combination to Enhance Proteotoxic Cell Death in Hepatocellular Carcinoma Experimental Models Without Killing Non-Cancer Cells. Int. J. Mol. Sci. 2025, 26, 6699. https://doi.org/10.3390/ijms26146699
Perez-Stable C, de las Pozas A, Reiner T, Gomez J, Nagarajan M, Foster RT, Ure DR, Wangpaichitr M. A Novel Chemotherapy Combination to Enhance Proteotoxic Cell Death in Hepatocellular Carcinoma Experimental Models Without Killing Non-Cancer Cells. International Journal of Molecular Sciences. 2025; 26(14):6699. https://doi.org/10.3390/ijms26146699
Chicago/Turabian StylePerez-Stable, Carlos, Alicia de las Pozas, Teresita Reiner, Jose Gomez, Manojavan Nagarajan, Robert T. Foster, Daren R. Ure, and Medhi Wangpaichitr. 2025. "A Novel Chemotherapy Combination to Enhance Proteotoxic Cell Death in Hepatocellular Carcinoma Experimental Models Without Killing Non-Cancer Cells" International Journal of Molecular Sciences 26, no. 14: 6699. https://doi.org/10.3390/ijms26146699
APA StylePerez-Stable, C., de las Pozas, A., Reiner, T., Gomez, J., Nagarajan, M., Foster, R. T., Ure, D. R., & Wangpaichitr, M. (2025). A Novel Chemotherapy Combination to Enhance Proteotoxic Cell Death in Hepatocellular Carcinoma Experimental Models Without Killing Non-Cancer Cells. International Journal of Molecular Sciences, 26(14), 6699. https://doi.org/10.3390/ijms26146699