_Kim.png)
LightSpot Fluorescent Conjugates as Highly Efficient Tools for Lysosomal P-gp Quantification in Olaparib-Treated Triple-Negative Breast Cancer Cells

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
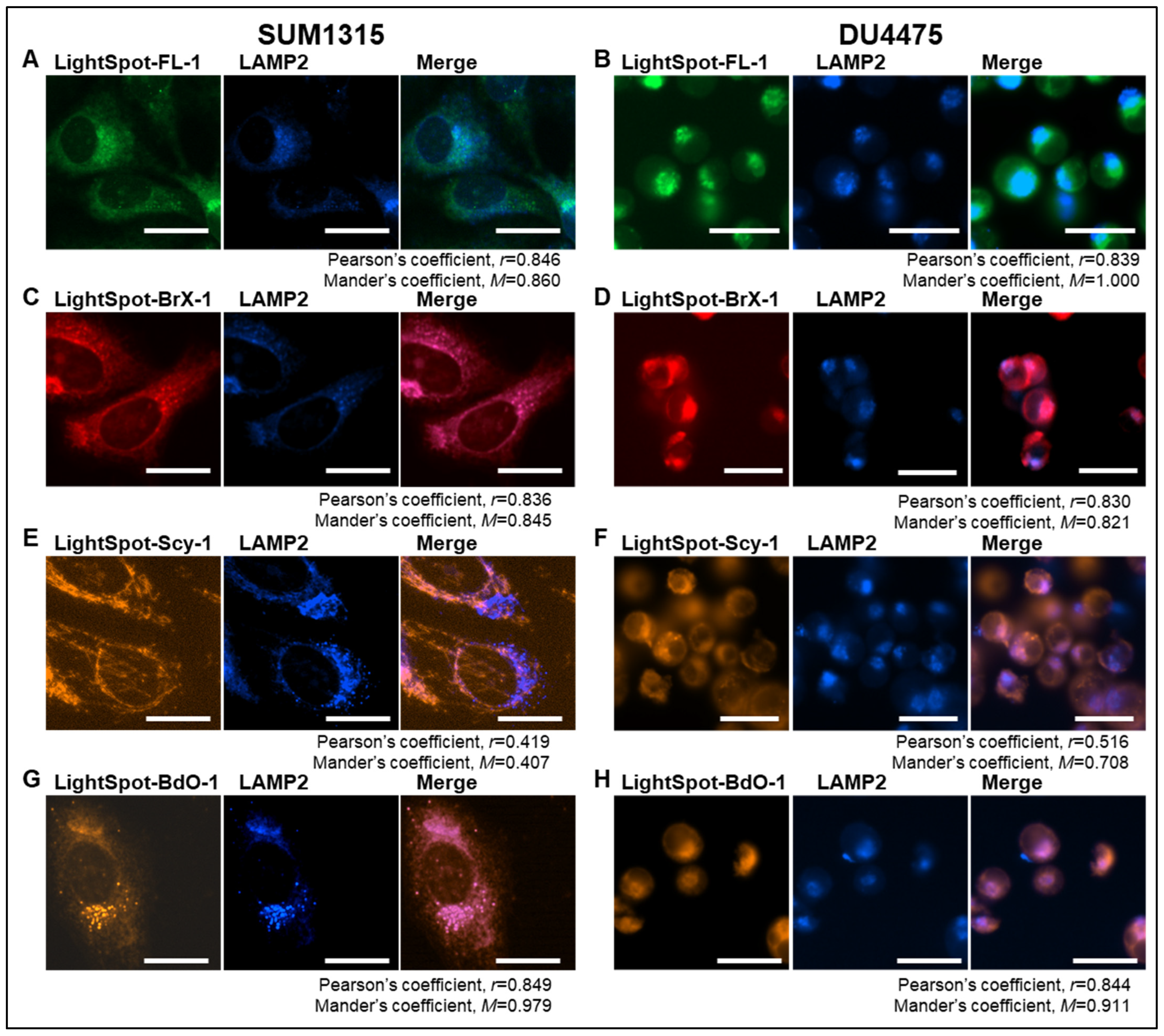
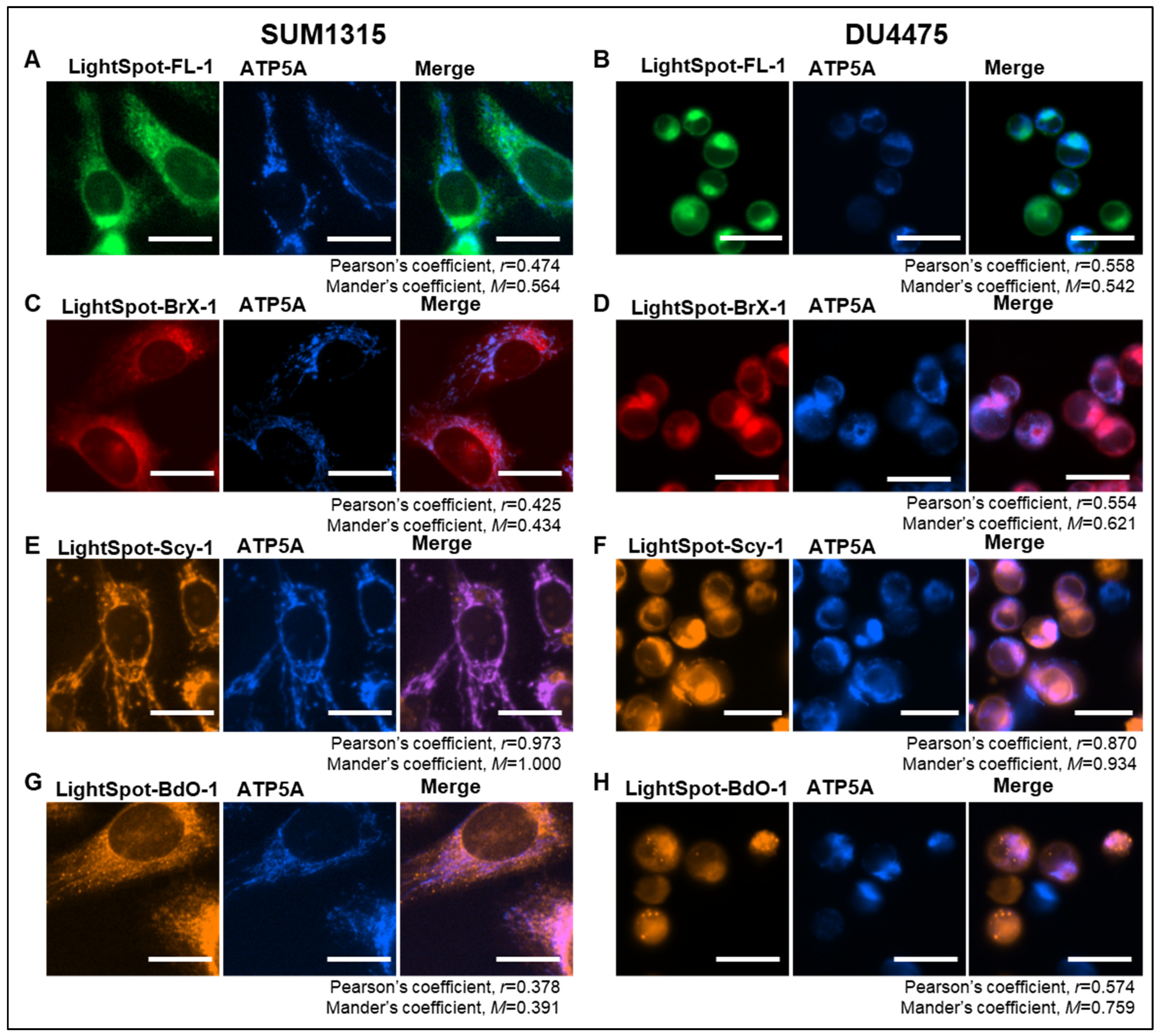
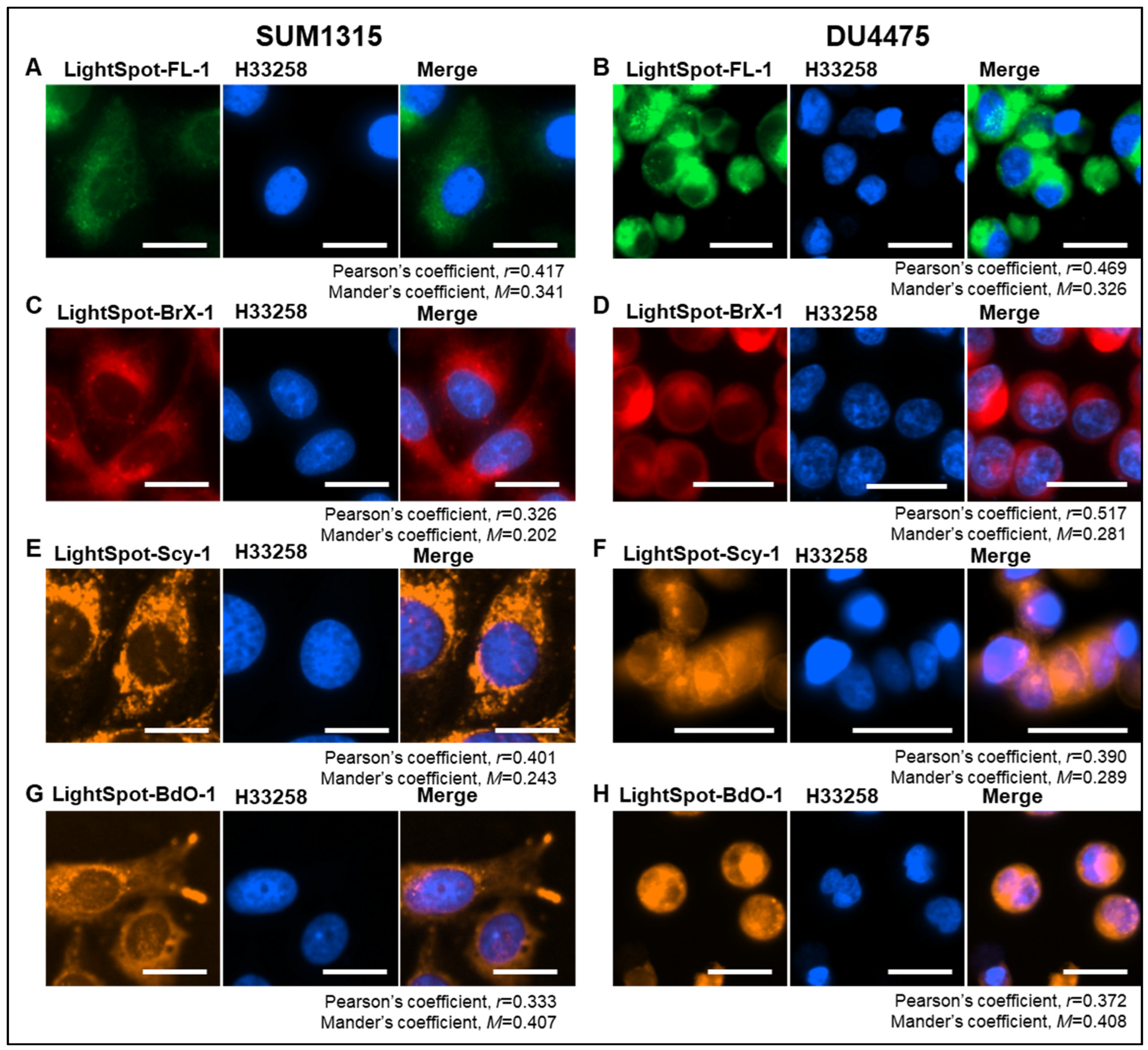
2.1. LightSpot Fluorescent Conjugate Intracellular Staining Distribution in SUM1315 and DU4475 Cell Lines
2.2. Selective P-gp Targeting Efficiency Study by the Four LightSpot Fluorescent Conjugates in SUM1315 and DU4475 Cell Lines
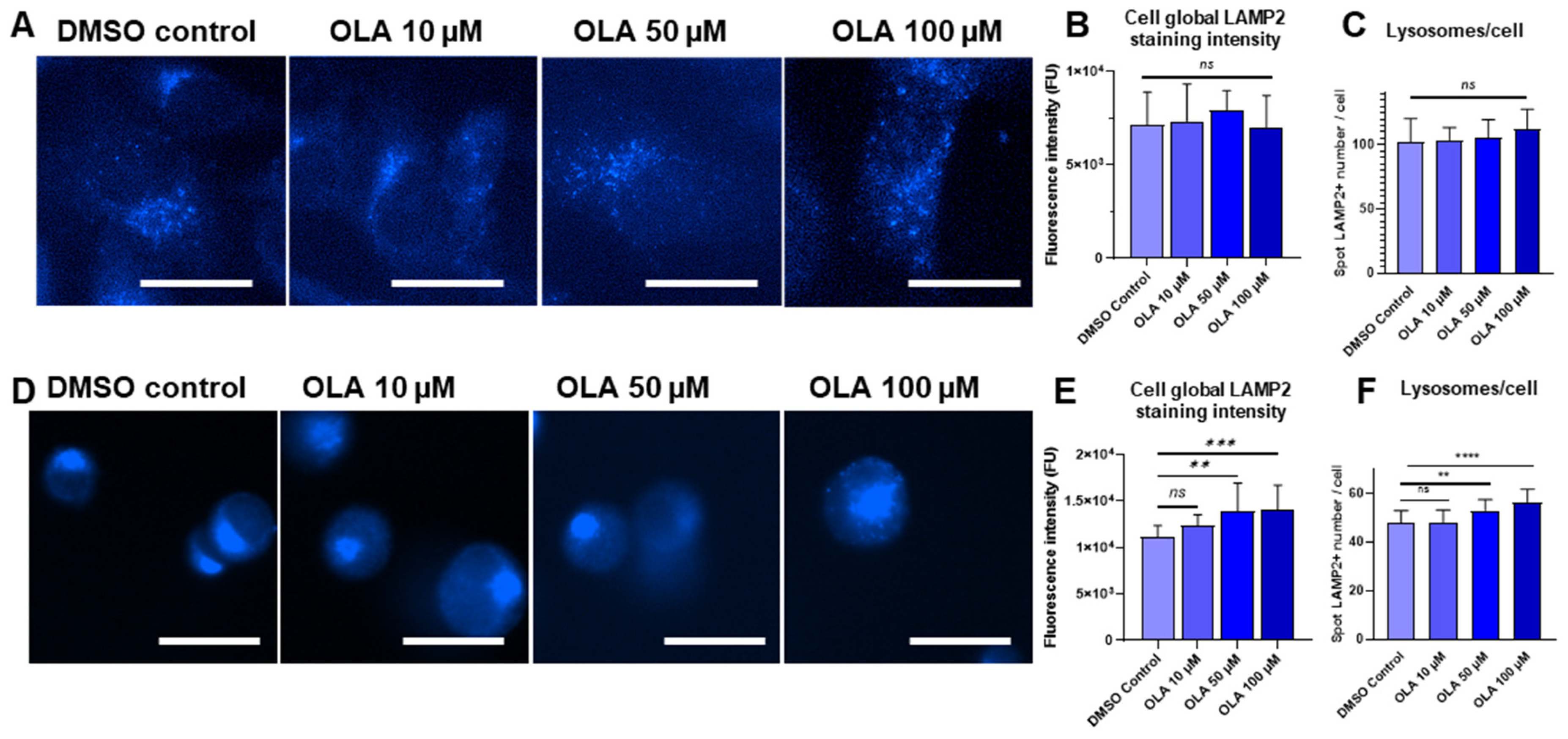
2.3. Increased PARP-1 Inhibitor OLA Concentrations Impact on Lysosome Number in SUM1315 and DU4475 Cell Lines
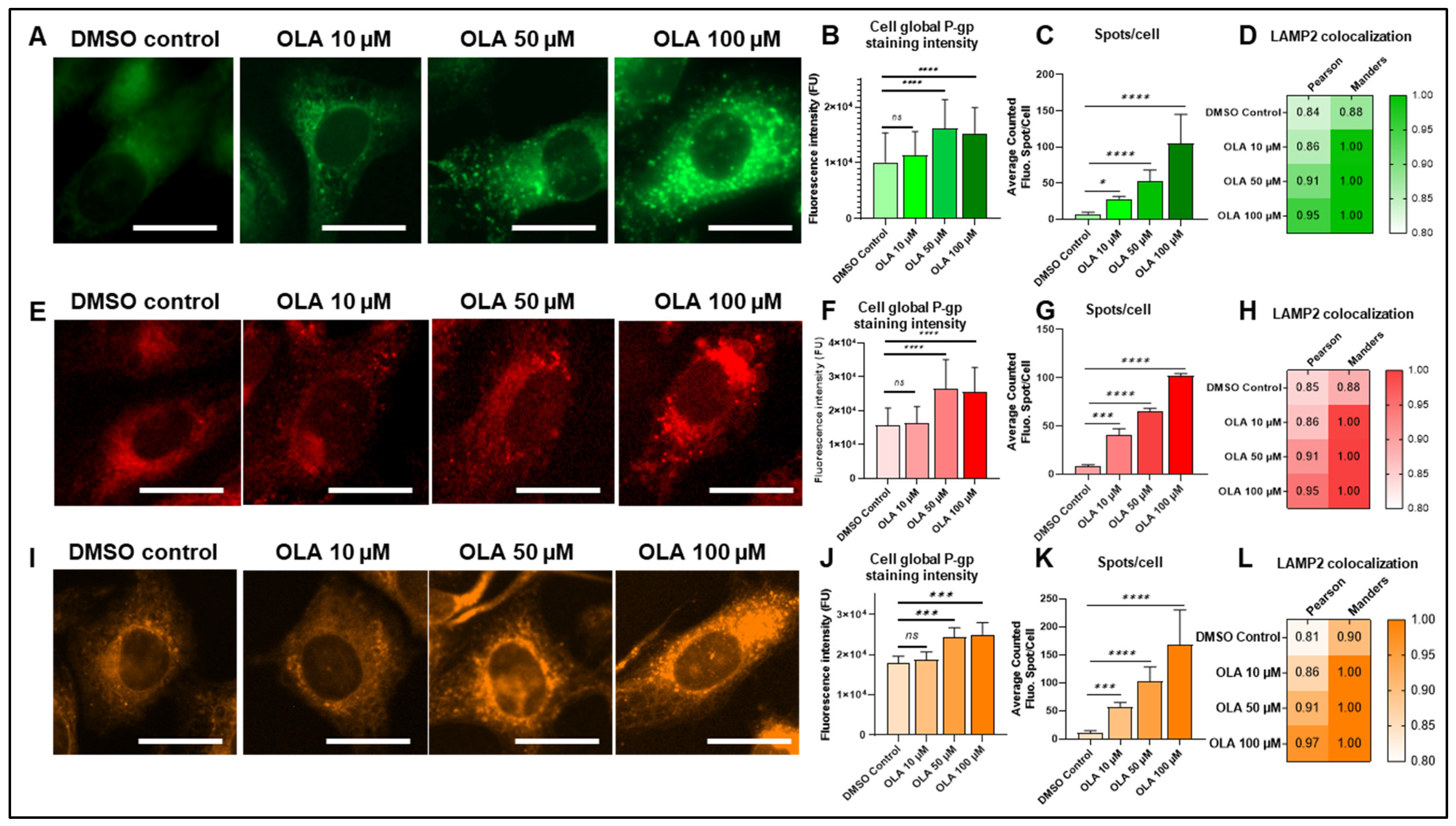
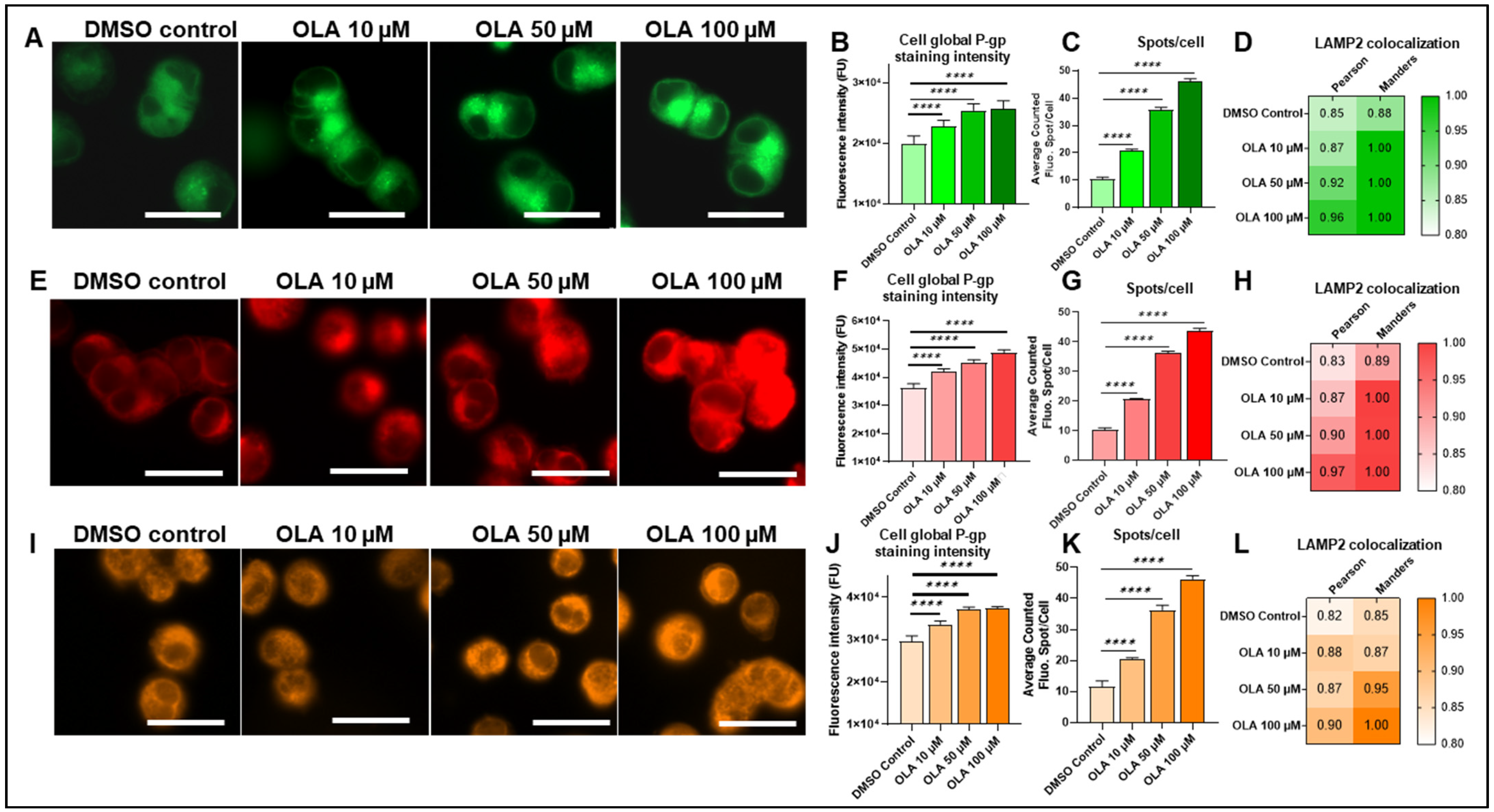
2.4. Lysosomal-P-gp-Mediated Resistance in SUM1315 and DU4475 Cells After OLA Treatment
3. Discussion
4. Materials and Methods
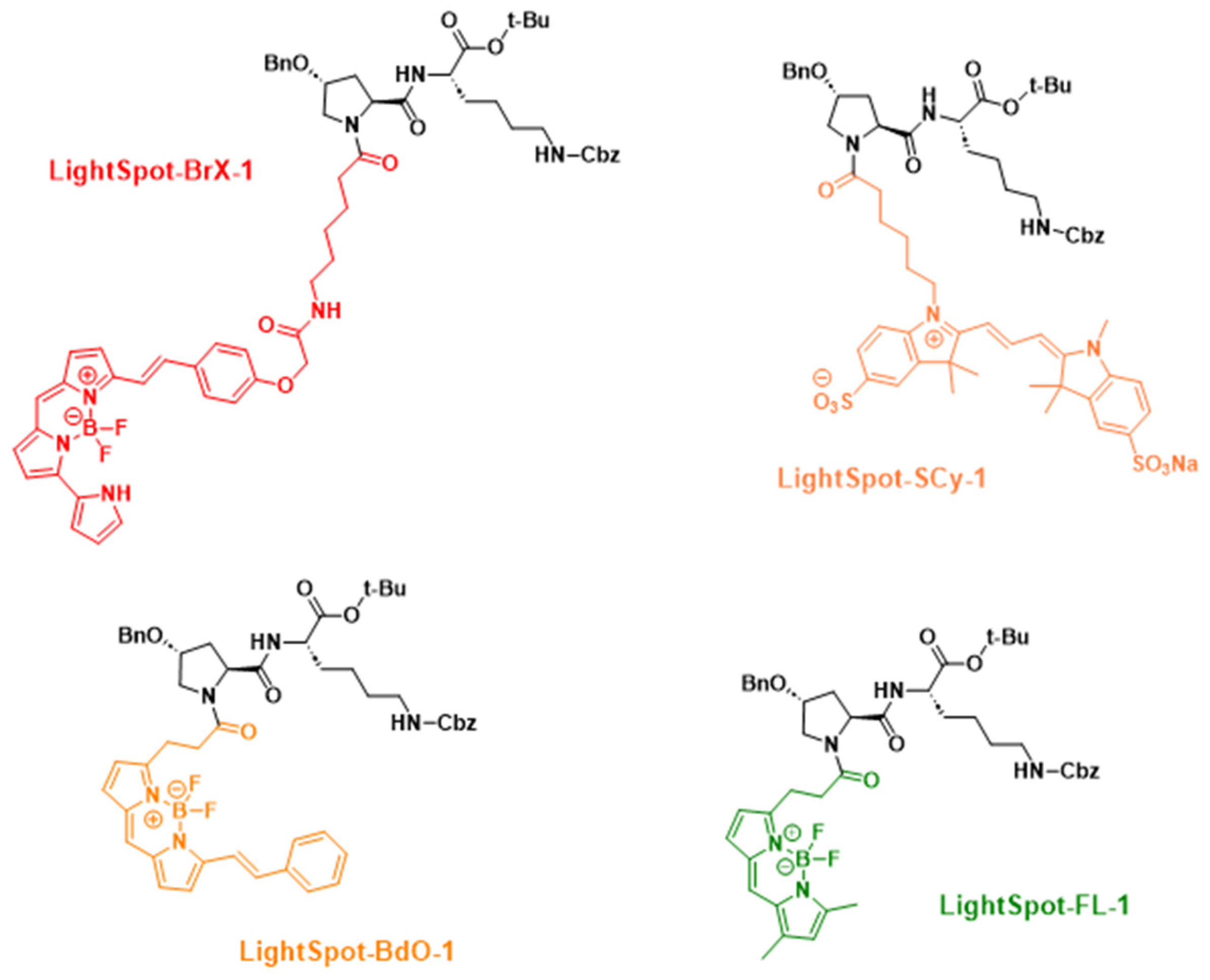
4.1. Fluorescent Compound Chemical Synthesis and Characterization
4.2. Cell Culture
4.3. Fluorescent Compound Cellular Staining
4.4. P-gp Knockdown Using Specific siRNAs
4.5. Subcellular and Colocalization Studies
4.6. OLA Exposure and P-gp Induction Analysis
4.7. Statistical Analysis
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP Binding Cassette |
| BSA | Bovine Serum Albumin |
| FCS | Fetal Calf Serum |
| FU | Fluorescence Unit |
| HPLC-UV-DAD | High Performance Liquid Chromatography-Ultra Violet-Diode Array Detector |
| MDR | Multidrug Resistance |
| OLA | Olaparib |
| PFA | Paraformaldehyde |
| P-gp | Permeability Glycoprotein |
| TNBC | Triple-Negative Breast Cancer |
References
- Bukowski, K.; Kciuk, M.; Kontek, R. Mechanisms of Multidrug Resistance in Cancer Chemotherapy. Int. J. Mol. Sci. 2020, 21, 3233. [Google Scholar] [CrossRef] [PubMed]
- Sarkadi, B.; Homolya, L.; Szakács, G.; Váradi, A. Human Multidrug Resistance ABCB and ABCG Transporters: Participation in a Chemoimmunity Defense System. Physiol. Rev. 2006, 86, 1179–1236. [Google Scholar] [CrossRef] [PubMed]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the Role of ABC Transporters in Multidrug-Resistant Cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The Role of ABC Transporters in Clinical Practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-Glycoprotein in Pharmacokinetics: Clinical Implications. Clin. Pharmacokinet. 2003, 42, 59–98. [Google Scholar] [CrossRef]
- Linn, S.C.; Giaccone, G.; van Diest, P.J.; Blokhuis, W.M.D.; van der Valk, P.; van Kalken, C.K.; Kuiper, C.M.; Pinedo, H.M.; Baak, J.P.A. Prognostic Relevance of P-Glycoprotein Expression in Breast Cancer. Ann. Oncol. 1995, 6, 679–685. [Google Scholar] [CrossRef]
- Clarke, R.; Leonessa, F.; Trock, B. Multidrug Resistance/P-Glycoprotein and Breast Cancer: Review and Meta-Analysis. Semin. Oncol. 2005, 32, 9–15. [Google Scholar] [CrossRef]
- Goldstein, L.J.; Galski, H.; Fojo, A.; Willingham, M.; Lai, S.L.; Gazdar, A.; Pirker, R.; Green, A.; Crist, W.; Brodeur, G.M. Expression of a Multidrug Resistance Gene in Human Cancers. J. Natl. Cancer Inst. 1989, 81, 116–124. [Google Scholar] [CrossRef]
- Tamaki, A.; Ierano, C.; Szakacs, G.; Robey, R.W.; Bates, S.E. The Controversial Role of ABC Transporters in Clinical Oncology. Essays Biochem. 2011, 50, 209–232. [Google Scholar] [CrossRef]
- Gala, J.L.; McLachlan, J.M.; Bell, D.R.; Michaux, J.L.; Ma, D.D. Specificity and Sensitivity of Immunocytochemistry for Detecting P-Glycoprotein in Haematological Malignancies. J. Clin. Pathol. 1994, 47, 619–624. [Google Scholar] [CrossRef]
- Nedeljković, M.; Tanić, N.; Prvanović, M.; Milovanović, Z.; Tanić, N. Friend or Foe: ABCG2, ABCC1 and ABCB1 Expression in Triple-Negative Breast Cancer. Breast Cancer 2021, 28, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S.; et al. High Expression of ATP-Binding Cassette Transporter ABCC11 in Breast Tumors Is Associated with Aggressive Subtypes and Low Disease-Free Survival. Breast Cancer Res. Treat. 2013, 137, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Fu, D. Where Is It and How Does It Get There—Intracellular Localization and Traffic of P-Glycoprotein. Front. Oncol. 2013, 3, 321. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, T.; Sahni, S.; Sharp, D.M.; Arvind, A.; Jansson, P.J.; Richardson, D.R. P-Glycoprotein Mediates Drug Resistance via a Novel Mechanism Involving Lysosomal Sequestration. J. Biol. Chem. 2013, 288, 31761–31771. [Google Scholar] [CrossRef]
- Guo, W.; Dong, W.; Li, M.; Shen, Y. Mitochondria P-Glycoprotein Confers Paclitaxel Resistance on Ovarian Cancer Cells. OTT 2019, 12, 3881–3891. [Google Scholar] [CrossRef]
- Paterson, J.K.; Gottesman, M.M. P-Glycoprotein is not present in mitochondrial membranes. Exp. Cell Res. 2007, 313, 3100–3105. [Google Scholar] [CrossRef]
- Stefan, S.M.; Jansson, P.J.; Kalinowski, D.S.; Anjum, R.; Dharmasivam, M.; Richardson, D.R. The Growing Evidence for Targeting P-Glycoprotein in Lysosomes to Overcome Resistance. Future Med. Chem. 2020, 12, 473–477. [Google Scholar] [CrossRef]
- Seebacher, N.A.; Krchniakova, M.; Stacy, A.E.; Skoda, J.; Jansson, P.J. Tumour Microenvironment Stress Promotes the Development of Drug Resistance. Antioxidants 2021, 10, 1801. [Google Scholar] [CrossRef]
- Daumar, P.; Goisnard, A.; Dubois, C.; Roux, M.; Depresle, M.; Penault-Llorca, F.; Bamdad, M.; Mounetou, E. Chemical Biology Fluorescent Tools for in Vitro Investigation of the Multidrug Resistant P-Glycoprotein (P-Gp) Expression in Tumor Cells. RSC Adv. 2023, 13, 27016–27035. [Google Scholar] [CrossRef]
- Gay, E.; Dubois, M.; Roux, M.; Goisnard, A.; Depresle, M.; Bamdad, M.; Daumar, P.; Mounetou, E. Development and Validation of a High-Performance Liquid Chromatography Method with Fluorescence Detection for the Quantification of the Resistance Protein P-Gp in Cancer Cells. J. Chromatogr. B 2025, 1253, 124475. [Google Scholar] [CrossRef]
- Goisnard, A.; Daumar, P.; Dubois, C.; Aubel, C.; Roux, M.; Depresle, M.; Gauthier, J.; Vidalinc, B.; Penault-Llorca, F.; Mounetou, E.; et al. LightSpot®-FL-1 Fluorescent Probe: An Innovative Tool for Cancer Drug Resistance Analysis by Direct Detection and Quantification of the P-Glycoprotein (P-Gp) on Monolayer Culture and Spheroid Triple Negative Breast Cancer Models. Cancers 2021, 13, 4050. [Google Scholar] [CrossRef] [PubMed]
- Abd El-Aziz, Y.S.; Spillane, A.J.; Jansson, P.J.; Sahni, S. Role of ABCB1 in Mediating Chemoresistance of Triple-Negative Breast Cancers. Biosci. Rep. 2021, 41, BSR20204092. [Google Scholar] [CrossRef]
- Gregorcyk, S.; Kang, Y.; Brandt, D.; Kolm, P.; Singer, G.; Perry, R.R. P-Glycoprotein Expression as a Predictor of Breast Cancer Recurrence. Ann. Surg. Oncol. 1996, 3, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; He, Z.; Wu, Y.; He, J.; Li, Y. Clinical Significance of P-Glycoprotein Expression in Metastatic Breast Carcinoma. Chin. J. Cancer Res. 2005, 17, 71–74. [Google Scholar] [CrossRef]
- Ling, V.; Thompson, L.H. Reduced Permeability in CHO Cells as a Mechanism of Resistance to Colchicine. J. Cell Physiol. 1974, 83, 103–116. [Google Scholar] [CrossRef]
- Linton, K.J.; Higgins, C.F. Structure and Function of ABC Transporters: The ATP Switch Provides Flexible Control. Pflug. Arch. 2007, 453, 555–567. [Google Scholar] [CrossRef]
- Alfarouk, K.O.; Stock, C.-M.; Taylor, S.; Walsh, M.; Muddathir, A.K.; Verduzco, D.; Bashir, A.H.H.; Mohammed, O.Y.; Elhassan, G.O.; Harguindey, S.; et al. Resistance to Cancer Chemotherapy: Failure in Drug Response from ADME to P-Gp. Cancer Cell Int. 2015, 15, 71. [Google Scholar] [CrossRef]
- Linn, S.C.; Honkoop, A.H.; Hoekman, K.; van der Valk, P.; Pinedo, H.M.; Giaccone, G. P53 and P-Glycoprotein Are Often Co-Expressed and Are Associated with Poor Prognosis in Breast Cancer. Br. J. Cancer 1996, 74, 63–68. [Google Scholar] [CrossRef]
- Yokoyama, H.; Ishida, T.; Sugio, K.; Inoue, T.; Sugimachi, K. Immunohistochemical Evidence That P-Glycoprotein in Non-Small Cell Lung Cancers Is Associated with Shorter Survival. Surg. Today 1999, 29, 1141–1147. [Google Scholar] [CrossRef]
- Chintamani; Singh, J.P.; Mittal, M.K.; Saxena, S.; Bansal, A.; Bhatia, A.; Kulshreshtha, P. Role of P-Glycoprotein Expression in Predicting Response to Neoadjuvant Chemotherapy in Breast Cancer—A Prospective Clinical Study. World J. Surg. Oncol. 2005, 3, 61. [Google Scholar] [CrossRef]
- Baekelandt, M.M.; Holm, R.; Nesland, J.M.; Tropé, C.G.; Kristensen, G.B. P-Glycoprotein Expression Is a Marker for Chemotherapy Resistance and Prognosis in Advanced Ovarian Cancer. Anticancer Res. 2000, 20, 1061–1067. [Google Scholar] [PubMed]
- Vishnukumar, S.; Umamaheswaran, G.; Anichavezhi, D.; Indumathy, S.; Adithan, C.; Srinivasan, K.; Kadambari, D. P-Glycoprotein Expression as a Predictor of Response to Neoadjuvant Chemotherapy in Breast Cancer. Indian J. Cancer 2013, 50, 195. [Google Scholar] [CrossRef] [PubMed]
- de la Torre, M.; Larsson, R.; Nygren, P.; Lindgren, A.; Bergh, J. Expression of the Multidrug-Resistance Gene Product in Untreated Human Breast Cancer and Its Relationship to Prognostic Markers. Acta Oncol. 1994, 33, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Shimizu, C.; Shimoyama, T.; Takeda, M.; Ando, M.; Kohno, T.; Katsumata, N.; Kang, Y.-K.; Nishio, K.; Fujiwara, Y. Gene Expression Profiling of ATP-Binding Cassette (ABC) Transporters as a Predictor of the Pathologic Response to Neoadjuvant Chemotherapy in Breast Cancer Patients. Breast Cancer Res. Treat. 2006, 99, 9–17. [Google Scholar] [CrossRef]
- Demidenko, R.; Razanauskas, D.; Daniunaite, K.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. Frequent Down-Regulation of ABC Transporter Genes in Prostate Cancer. BMC Cancer 2015, 15, 683. [Google Scholar] [CrossRef]
- Yan, L.; Ding, B.; Liu, H.; Zhang, Y.; Zeng, J.; Hu, J.; Yao, W.; Yu, G.; An, R.; Chen, Z.; et al. Inhibition of SMYD2 Suppresses Tumor Progression by Down-Regulating microRNA-125b and Attenuates Multi-Drug Resistance in Renal Cell Carcinoma. Theranostics 2019, 9, 8377–8391. [Google Scholar] [CrossRef]
- Aloia, T.A.; Harpole, D.H.; Reed, C.E.; Allegra, C.; Moore, M.-B.H.; Herndon, J.E.; D’Amico, T.A. Tumor Marker Expression Is Predictive of Survival in Patients with Esophageal Cancer. Ann. Thorac. Surg. 2001, 72, 859–866. [Google Scholar] [CrossRef]
- Pilotto Heming, C.; Muriithi, W.; Wanjiku Macharia, L.; Niemeyer Filho, P.; Moura-Neto, V.; Aran, V. P-Glycoprotein and Cancer: What Do We Currently Know. Heliyon 2022, 8, e11171. [Google Scholar] [CrossRef]
- Mansoori, B.; Mohammadi, A.; Davudian, S.; Shirjang, S.; Baradaran, B. The Different Mechanisms of Cancer Drug Resistance: A Brief Review. Adv. Pharm. Bull. 2017, 7, 339–348. [Google Scholar] [CrossRef]
- Ramos, A.; Sadeghi, S.; Tabatabaeian, H. Battling Chemoresistance in Cancer: Root Causes and Strategies to Uproot Them. Int. J. Mol. Sci. 2021, 22, 9451. [Google Scholar] [CrossRef]
- Ferrao, P.; Sincock, P.; Cole, S.; Ashman, L. Intracellular P-Gp Contributes to Functional Drug Efflux and Resistance in Acute Myeloid Leukaemia. Leuk. Res. 2001, 25, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Katayama, K.; Kapoor, K.; Ohnuma, S.; Patel, A.; Swaim, W.; Ambudkar, I.S.; Ambudkar, S.V. Revealing the Fate of Cell Surface Human P-Glycoprotein (ABCB1): The Lysosomal Degradation Pathway. Biochim. Biophys. Acta 2015, 1853, 2361–2370. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, A.B.; Fox, K.; Lee, P.; Yang, Y.D.; Ling, V. Functional Intracellular P-Glycoprotein. Int. J. Cancer 1998, 76, 857–864. [Google Scholar] [CrossRef]
- Seebacher, N.; Lane, D.J.R.; Richardson, D.R.; Jansson, P.J. Turning the Gun on Cancer: Utilizing Lysosomal P-Glycoprotein as a New Strategy to Overcome Multi-Drug Resistance. Free Radic. Biol. Med. 2016, 96, 432–445. [Google Scholar] [CrossRef]
- White, E. Deconvoluting the Context-Dependent Role for Autophagy in Cancer. Nat. Rev. Cancer 2012, 12, 401–410. [Google Scholar] [CrossRef]
- Liu, W.; Meng, Y.; Zong, C.; Zhang, S.; Wei, L. Autophagy and Tumorigenesis. Adv. Exp. Med. Biol. 2020, 1207, 275–299. [Google Scholar] [CrossRef]
- Bellio, C.; Villanueva, J. Hitting the Brakes on Autophagy for Overcoming Acquired Resistance in Triple Negative Breast Cancer. Ann. Transl. Med. 2020, 8, 848. [Google Scholar] [CrossRef]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and Chemotherapy Resistance: A Promising Therapeutic Target for Cancer Treatment. Cell Death Dis. 2013, 4, e838. [Google Scholar] [CrossRef]
- Lefort, S.; Joffre, C.; Kieffer, Y.; Givel, A.-M.; Bourachot, B.; Zago, G.; Bieche, I.; Dubois, T.; Meseure, D.; Vincent-Salomon, A.; et al. Inhibition of Autophagy as a New Means of Improving Chemotherapy Efficiency in High-LC3B Triple-Negative Breast Cancers. Autophagy 2014, 10, 2122–2142. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, A.; Wang, M.; Liu, W.; Wang, C.; Xie, X.; Chen, X.; Dong, J.; Li, M. Enhanced Autophagy Reveals Vulnerability of P-Gp Mediated Epirubicin Resistance in Triple Negative Breast Cancer Cells. Apoptosis 2016, 21, 473–488. [Google Scholar] [CrossRef]
- Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer-How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef] [PubMed]
- Baldini, N.; Scotlandi, K.; Serra, M.; Shikita, T.; Zini, N.; Ognibene, A.; Santi, S.; Ferracini, R.; Maraldi, N.M. Nuclear Immunolocalization of P-Glycoprotein in Multidrug-Resistant Cell Lines Showing Similar Mechanisms of Doxorubicin Distribution. Eur. J. Cell Biol. 1995, 68, 226–239. [Google Scholar] [PubMed]
- Maraldi, N.M.; Zini, N.; Santi, S.; Scotlandi, K.; Serra, M.; Baldini, N. P-Glycoprotein Subcellular Localization and Cell Morphotype in MDR1 Gene-Transfected Human Osteosarcoma Cells. Biol. Cell 1999, 91, 17–28. [Google Scholar] [PubMed]
- Gericke, B.; Wienböker, I.; Brandes, G.; Löscher, W. Is P-Glycoprotein Functionally Expressed in the Limiting Membrane of Endolysosomes? A Biochemical and Ultrastructural Study in the Rat Liver. Cells 2022, 11, 1556. [Google Scholar] [CrossRef]
- Arun, B.; Akar, U.; Gutierrez-Barrera, A.M.; Hortobagyi, G.N.; Ozpolat, B. The PARP Inhibitor AZD2281 (Olaparib) Induces Autophagy/Mitophagy in BRCA1 and BRCA2 Mutant Breast Cancer Cells. Int. J. Oncol. 2015, 47, 262–268. [Google Scholar] [CrossRef]
- Cahuzac, M.; Langlois, P.; Péant, B.; Fleury, H.; Mes-Masson, A.-M.; Saad, F. Pre-Activation of Autophagy Impacts Response to Olaparib in Prostate Cancer Cells. Commun. Biol. 2022, 5, 251. [Google Scholar] [CrossRef]
- Zhitomirsky, B.; Assaraf, Y.G. Lysosomal Sequestration of Hydrophobic Weak Base Chemotherapeutics Triggers Lysosomal Biogenesis and Lysosome-Dependent Cancer Multidrug Resistance. Oncotarget 2015, 6, 1143–1156. [Google Scholar] [CrossRef]
- Schmitt, M.V.; Lienau, P.; Fricker, G.; Reichel, A. Quantitation of Lysosomal Trapping of Basic Lipophilic Compounds Using In Vitro Assays and In Silico Predictions Based on the Determination of the Full pH Profile of the Endo-/Lysosomal System in Rat Hepatocytes. Drug Metab. Dispos. 2019, 47, 49–57. [Google Scholar] [CrossRef]
- Kossatz, S.; Pirovano, G.; Demétrio De Souza França, P.; Strome, A.L.; Sunny, S.P.; Zanoni, D.K.; Mauguen, A.; Carney, B.; Brand, C.; Shah, V.; et al. Validation of the Use of a Fluorescent PARP1 Inhibitor for the Detection of Oral, Oropharyngeal and Oesophageal Epithelial Cancers. Nat. Biomed. Eng. 2020, 4, 272–285. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goisnard, A.; Daumar, P.; Dubois, M.; Gay, E.; Roux, M.; Depresle, M.; Penault-Llorca, F.; Mounetou, E.; Bamdad, M. LightSpot Fluorescent Conjugates as Highly Efficient Tools for Lysosomal P-gp Quantification in Olaparib-Treated Triple-Negative Breast Cancer Cells. Int. J. Mol. Sci. 2025, 26, 6675. https://doi.org/10.3390/ijms26146675
Goisnard A, Daumar P, Dubois M, Gay E, Roux M, Depresle M, Penault-Llorca F, Mounetou E, Bamdad M. LightSpot Fluorescent Conjugates as Highly Efficient Tools for Lysosomal P-gp Quantification in Olaparib-Treated Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences. 2025; 26(14):6675. https://doi.org/10.3390/ijms26146675
Chicago/Turabian StyleGoisnard, Antoine, Pierre Daumar, Maxime Dubois, Elodie Gay, Manon Roux, Marie Depresle, Frédérique Penault-Llorca, Emmanuelle Mounetou, and Mahchid Bamdad. 2025. "LightSpot Fluorescent Conjugates as Highly Efficient Tools for Lysosomal P-gp Quantification in Olaparib-Treated Triple-Negative Breast Cancer Cells" International Journal of Molecular Sciences 26, no. 14: 6675. https://doi.org/10.3390/ijms26146675
APA StyleGoisnard, A., Daumar, P., Dubois, M., Gay, E., Roux, M., Depresle, M., Penault-Llorca, F., Mounetou, E., & Bamdad, M. (2025). LightSpot Fluorescent Conjugates as Highly Efficient Tools for Lysosomal P-gp Quantification in Olaparib-Treated Triple-Negative Breast Cancer Cells. International Journal of Molecular Sciences, 26(14), 6675. https://doi.org/10.3390/ijms26146675