
Causes of and Solutions to Mitochondrial Disorders: A Literature Review
 , , , and
, , , and
Abstract
1. Introduction
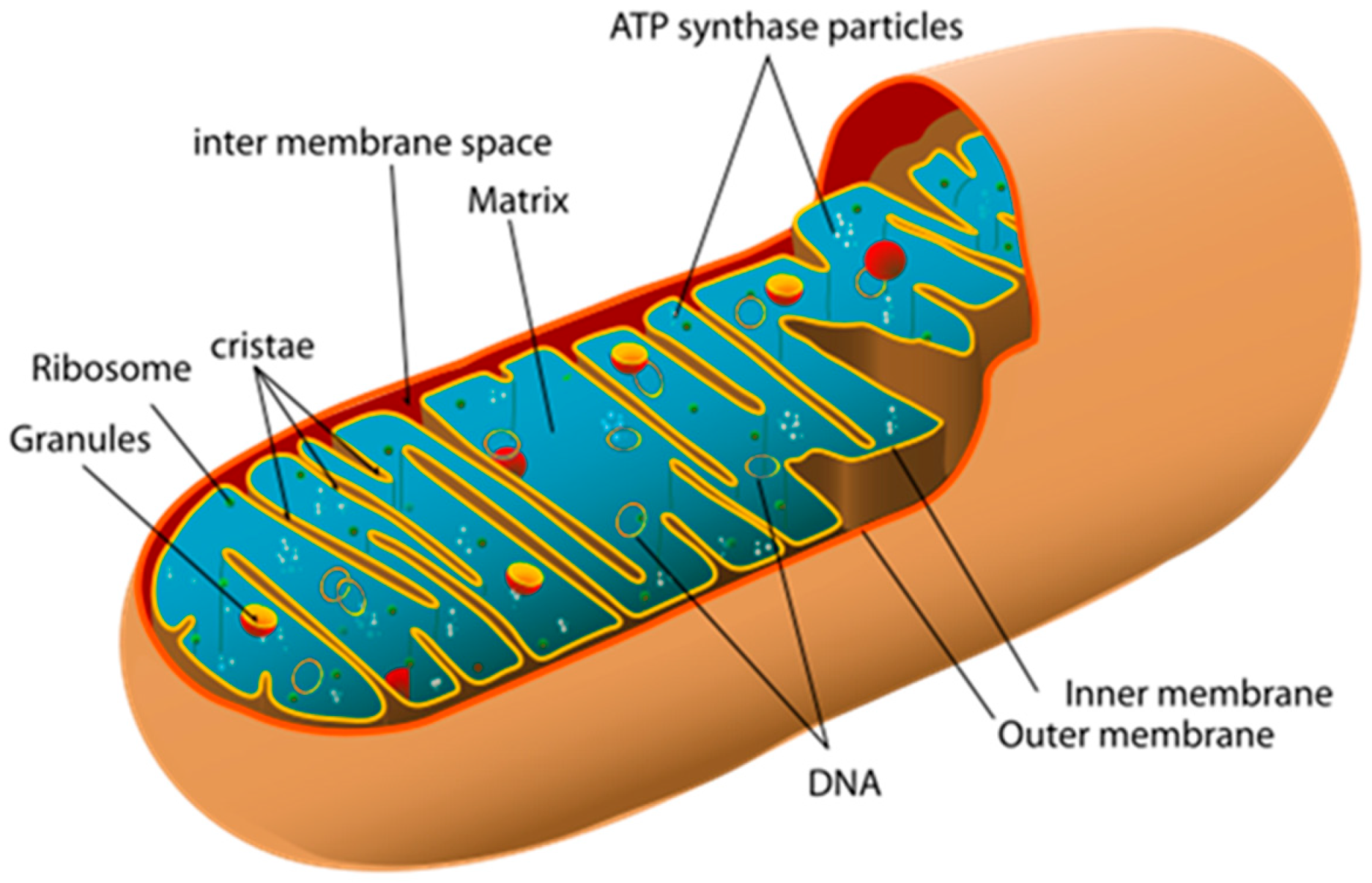
2. Mitochondria: Structure and Function
3. Mitochondrial DNA
4. Diagnostics of Mitochondrial Disorders
5. Mitochondrial Therapy
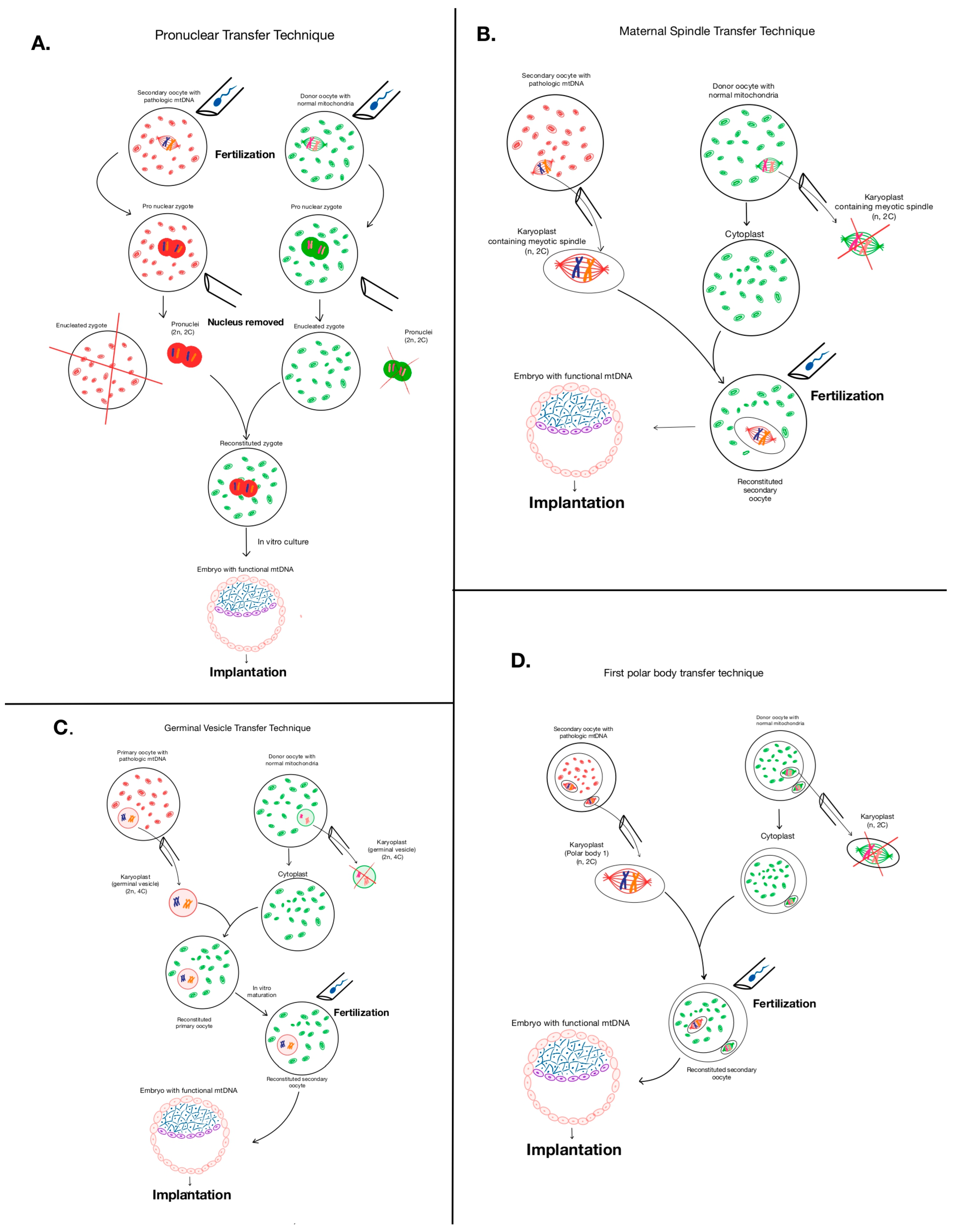
5.1. Mitochondrial Replacement Therapy—The Heterologous Approach
5.1.1. Pronuclear Transfer Method
5.1.2. Maternal Spindle Transfer Method (MST)
5.1.3. Polar Body Genome Transfer (PBT)
5.1.4. Germinal Vesicle Transfer (GVT)
5.2. Risks Associated with Mitochondrial Replacement Therapies (The Heterologous Approach)
5.3. Results of Mitochondrial Replacement Therapy (The Heterologous Approach)
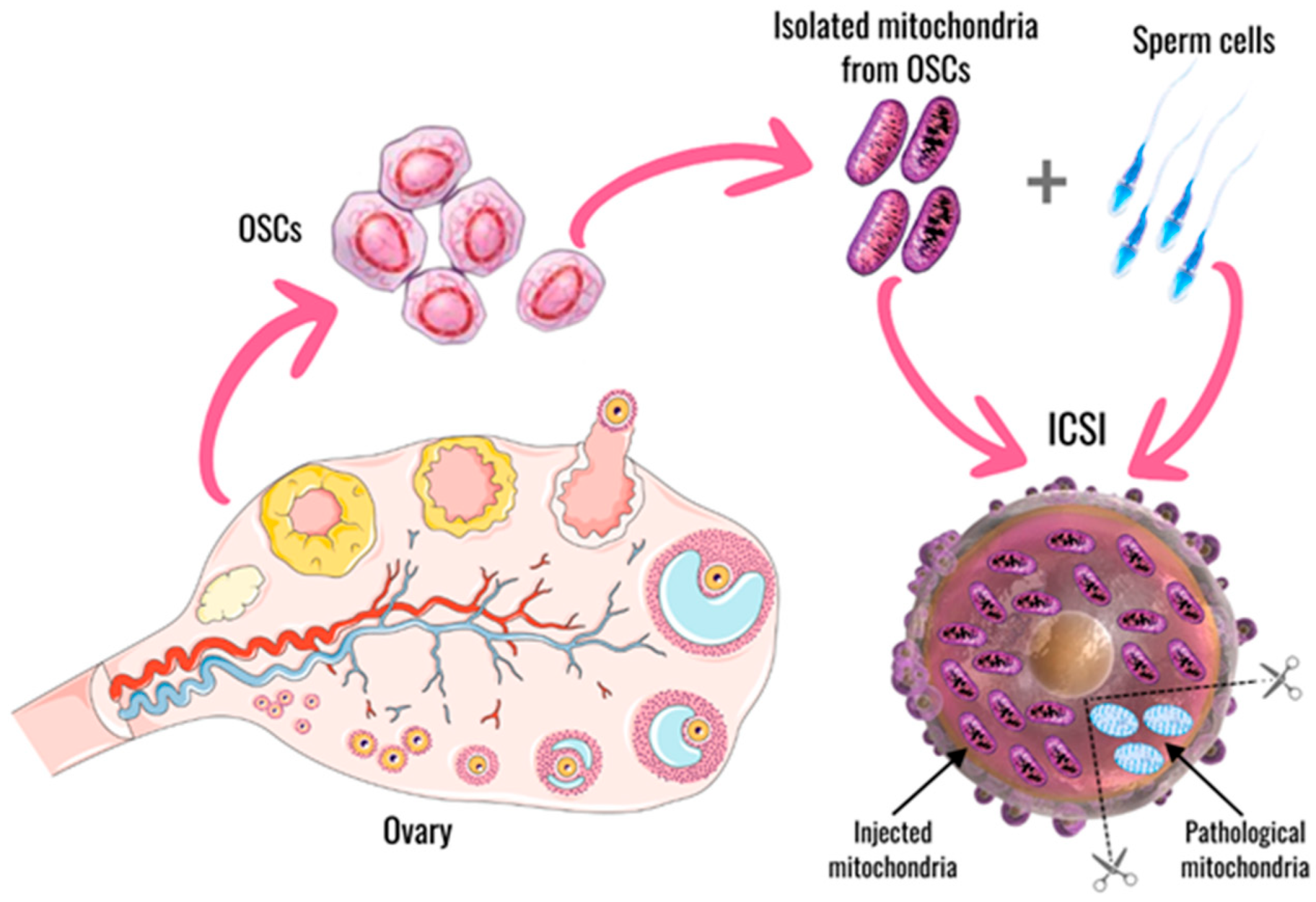
5.4. Mitochondrial Replacement Therapy—The Autologous Approach
6. Mitochondrial Transplantation Therapy
6.1. Sources of Mitochondria
6.2. Mitochondria Transplantation in Brain Disorders
6.3. Mitochondria Transplantation in Heart Diseases
6.4. Mitochondrial Transplantation in Humans
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DNA | Deoxyribonucleic acid |
| mtDNA | Mitochondrial DNA |
| MRT | Mitochondrial Replacement Therapy |
| MTT | Mitochondrial Transplantation Therapy |
| PNT | Pronuclear transfer technique |
| MST | Maternal spindle transfer |
| PBT | Polar body genome transfer |
| GVT | Germinal vesicle transfer |
| AUGMENT | Autologous germline mitochondrial energy transfer |
| ICSI | Intracytoplasmic sperm injection |
| ATP | Adenosine triphosphate |
| AIF | Apoptosis-inducing factor |
| ROS | Reactive oxygen species |
| OS | Oxidative stress |
| CNS | Central nervous system |
| RNA | Ribonucleic acid |
| iPSCs | Induced pluripotent stem cells |
| FDA | Food and Drug Administration |
| IOM | Institute of Medicine |
| OSCs | Oogonial stem cells |
| IVF | In vitro fertilization |
| MT | Mitochondrial transfer |
| TNTs | Tunneling nanotubes |
| GJs | Gap junctions |
| EVs | Extracellular vesicles |
| MSCs | Mesenchymal stromal/stem cells |
| NDs | Neurodegenerative diseases |
| AD | Alzheimer’s disease |
| PD | Parkinson’s disease |
| OPCs | Oligodendrocyte progenitor cells |
| ICH | Intracerebral hemorrhage |
| TBI | Traumatic brain injury |
| hiPSC-CMs | Human induced pluripotent stem cell-derived cardiomyocytes |
| IRI | Ischemia-reperfusion injury |
| IHD | Ischemic heart disease |
| ECMO | Extracorporeal membrane oxygenation |
| STEMI | ST-elevation myocardial infarction |
| LVEF | Left ventricular ejection fraction |
| IIM | Idiopathic inflammatory myopathy |
References
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K., 2nd. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Caldarazzo Ienco, E.; Rossi, A.; Siciliano, G.; Mancuso, M. Mitochondrial Syndromes Revisited. J. Clin. Med. 2021, 10, 1249. [Google Scholar] [CrossRef]
- Kang, E.; Wu, J.; Gutierrez, N.M.; Koski, A.; Tippner-Hedges, R.; Agaronyan, K.; Platero-Luengo, A.; Martinez-Redondo, P.; Ma, H.; Lee, Y.; et al. Mitochondrial replacement in human oocytes carrying pathogenic mitochondrial DNA mutations. Nature 2016, 540, 270–275, Erratum in: Nature 2019, 567, E5–E9. [Google Scholar] [CrossRef] [PubMed]
- Tavare, A. Scientists are to investigate “three parent IVF” for preventing mitochondrial diseases. BMJ 2012, 344, e540. [Google Scholar] [CrossRef] [PubMed]
- Pozdnyakova, A.A.; Volodina, M.A.; Rshtuni, S.D.; Marchenko, L.A.; Vysokih, M.Y. Mitohondrial’naya disfunkciya kak odna iz vozmozhnyh prichinnarusheniya follikulo—I steroidogeneza pri prezhdevremennoj nedostatochnosti yaichnikov. Akusherstvo. Ginekol. Reprodukciya 2015, 9, 55–65. Available online: https://cyberleninka.ru/article/n/mitohondrialnaya-disfunktsiya-kak-odna-iz-vozmozhnyh-prichinnarusheniya-follikulo-i-steroidogeneza-pri-prezhdevremennoy (accessed on 18 May 2025). [CrossRef]
- Chen, S.; Li, Q.; Shi, H.; Li, F.; Duan, Y.; Guo, Q. New insights into the role of mitochondrial dynamics in oxidative stress-induced diseases. Biomed. Pharmacother. 2024, 178, 117084. [Google Scholar] [CrossRef] [PubMed]
- Craven, L.; Tang, M.X.; Gorman, G.S.; De Sutter, P.; Heindryckx, B. Novel reproductive technologies to prevent mitochondrial disease. Hum. Reprod. Update 2017, 23, 501–519. [Google Scholar] [CrossRef] [PubMed]
- Fragouli, E.; McCaffrey, C.; Ravichandran, K.; Spath, K.; Grifo, J.A.; Munné, S.; Wells, D. Clinical implications of mitochondrial DNA quantification on pregnancy outcomes: A blinded prospective non-selection study. Hum. Reprod. 2017, 32, 2340–2347, Erratum in: Hum. Reprod. 2019, 34, 782. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.Y.; Zhao, X.R.; Zhang, N.; Yang, Y.; Yi, Y.; Shao, Q.H.; Liu, M.X.; Zhang, X.L. Mitochondrial dysfunction as a therapeutic strategy for neurodegenerative diseases: Current insights and future directions. Ageing Res. Rev. 2024, 102, 102577. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Shi, R. Weigh and wait: The prospect of mitochondrial gene replacement. Hum. Fertil. 2016, 19, 222–229. [Google Scholar] [CrossRef] [PubMed]
- DiMauro, S. A Brief History of Mitochondrial Pathologies. Int. J. Mol. Sci. 2019, 20, 5643. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rebolledo-Jaramillo, B.; Su, M.S.; Stoler, N.; McElhoe, J.A.; Dickins, B.; Blankenberg, D.; Korneliussen, T.S.; Chiaromonte, F.; Nielsen, R.; Holland, M.M.; et al. Maternal age effect and severe germ-line bottleneck in the inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 2014, 111, 15474–15479. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mitalipov, S.; Amato, P.; Parry, S.; Falk, M.J. Limitations of preimplantation genetic diagnosis for mitochondrial DNA diseases. Cell Rep. 2014, 7, 935–937. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Molnar, T.; Lehoczki, A.; Fekete, M.; Varnai, R.; Zavori, L.; Erdo-Bonyar, S.; Simon, D.; Berki, T.; Csecsei, P.; Ezer, E. Mitochondrial dysfunction in long COVID: Mechanisms, consequences, and potential therapeutic approaches. Geroscience 2024, 46, 5267–5286. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rebrikov, D.V. Human Genome Editing; Bulletin of Russian State Medical University: Moscow, Russia, 2016; pp. 4–15. [Google Scholar]
- Bhai, S.; Hirano, M. Diagnosis of Primary Mitochondrial Diseases. Muscle Nerve 2025, 71, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, M.; Papadopoulou, M.T.; Ng, Y.S.; Ardissone, A.; Bellusci, M.; Bertini, E.; Di Vito, L.; Evangelista, T.; Fons, C.; Hikmat, O.; et al. Management of seizures in patients with primary mitochondrial diseases: Consensus statement from the InterERNs Mitochondrial Working Group. Eur. J. Neurol. 2024, 31, e16275. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wen, H.; Deng, H.; Li, B.; Chen, J.; Zhu, J.; Zhang, X.; Yoshida, S.; Zhou, Y. Mitochondrial diseases: From molecular mechanisms to therapeutic advances. Signal Transduct. Target. Ther. 2025, 10, 9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Selvanathan, A.; Teo, J.; Parayil Sankaran, B. Hematologic Manifestations in Primary Mitochondrial Diseases. J. Pediatr. Hematol. Oncol. 2024, 46, e338–e347. [Google Scholar] [CrossRef] [PubMed]
- Heiduschka, S.; Prigione, A. iPSC models of mitochondrial diseases. Neurobiol. Dis. 2025, 207, 106822. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.P.; Mitalipov, N.; Mitalipov, S. Mitochondrial replacement therapy in reproductive medicine. Trends Mol. Med. 2015, 21, 68–76. [Google Scholar] [CrossRef]
- Haimes, E.; Taylor, K. Sharpening the cutting edge: Additional considerations for the UK debates on embryonic interventions for mitochondrial diseases. Life Sci. Soc. Policy. 2017, 13, 1. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sharma, H.; Singh, D.; Mahant, A.; Sohal, S.K.; Kesavan, A.K. Samiksha Development of mitochondrial replacement therapy: A review. Heliyon 2020, 6, e04643. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Subirá, J.; Soriano, M.J.; Del Castillo, L.M.; de Los Santos, M.J. Mitochondrial replacement techniques to resolve mitochondrial dysfunction and ooplasmic deficiencies: Where are we now? Hum. Reprod. 2025, 40, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Nagpal, M. Recent advancement in human reproduction three-parent babies: A technique to neutralize mitochondrial disease load- A boon or a bane for society? Curry Trends Diagn. Treat. 2017, 1, 100–103. [Google Scholar]
- Labarta, E.; Santos, M.J.d.L.; Escriba, M.J.; Pellicer, A.; Herriaz, S. Mitochondria as a tool for oocyte rejuvenation. Fertil. Steril. 2019, 111, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Jose, F.; Lekshmi, S.; Lal, S.; Jjiju, V.; Abraham, E. Three parent child: A review. Int. J. Nov. Trends Pharm. Sci. 2017, 7, 2277–2782. [Google Scholar]
- Schmerler, S.; Wessel, G.M. Polar bodies—More a lack of understanding than a lack of respect. Mol. Reprod. Dev. 2011, 78, 3–8. [Google Scholar] [CrossRef]
- Wang, T.; Sha, H.; Ji, D.; Zhang, H.L.; Chen, D.; Cao, Y.; Zhu, J. Polar body genome transfer for preventing the transmission of inherited mitochondrial diseases. Cell 2014, 157, 1591–1604. [Google Scholar] [CrossRef] [PubMed]
- Neupane, J.; Vandewoestyne, M.; Ghimire, S.; Lu, Y.; Qian, C.; Van Coster, R.; Gerris, J.; DeRoo, T.; Deforce, D.; De Sutter, P.; et al. Assessment of nuclear transfer techniques to prevent the transmission of heritable mitochondrial disorders without compromising embryonic development competence in mice. Mitochondrion 2014, 18, 27–33. [Google Scholar] [CrossRef]
- Sendra, L.; García-Mares, A.; Herrero, M.J.; Aliño, S.F. Mitochondrial DNA Replacement Techniques to Prevent Human Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 551. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, A.A.; Makova, K.D. lnvestigating mitonuclear interactions in human admixed populations. Nat. Ecol. Evol. 2019, 3, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.fda.gov/vaccines-blood-biologics/cellular-gene-therapy-products/therapeutic-cloning-and-genome-modification (accessed on 16 March 2018).
- Zhang, J. World’s First Baby Born from New Procedure Using DNA of Three People; New Hope Fertility Center: New York, NY, USA, 2016; Available online: https://www.theguardian.com/science/2016/sep/27/worlds-first-baby-born-using-dna-from-three-parents (accessed on 10 March 2017).
- Reardon, S. Genetic details of controversial ‘three-parent baby’ revealed. Nature 2017, 544, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Choudhary, A.; Munshi, A. Epigenetic reprogramming of mtDNA and its etiology in mitochondrial diseases. J. Physiol. Biochem. 2024, 80, 727–741. [Google Scholar] [CrossRef] [PubMed]
- Hamzelou, J. ‘3-parent baby’ Success. New Sci. 2016, 232, 8–9. [Google Scholar] [CrossRef]
- Woods, D.C.; Tilly, J.L. Autologous Germline Mitochondrial Energy Transfer (AUGMENT) in Human Assisted Reproduction. Semin. Reprod. Med. 2015, 33, 410–421. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, L.; Clevers, H. Coexistence of quiescent and active adult stem cells in mammals. Science 2010, 327, 542–545. [Google Scholar] [CrossRef]
- Johnson, J.; Canning, J.; Kaneko, T.; Pru, J.K.; Tilly, J.L. Germline stem cells and follicular renewal in the postnatal mammalian ovary. Nature 2004, 428, 145–150. [Google Scholar] [CrossRef]
- Zou, K.; Yuan, Z.; Yang, Z.; Luo, H.; Sun, K.; Zhou, L.; Xiang, J.; Shi, L.; Yu, Q.; Zhang, Y.; et al. Production of offspring from a germline stem cell line derived from neonatal ovaries. Nat. Cell Biol. 2009, 11, 631–636. [Google Scholar] [CrossRef]
- Grieve, K.M.; McLaughlin, M.; Dunlop, C.E.; Telfer, E.E.; Anderson, R.A. The controversial existence and functional potential of oogonial stem cells. Maturitas 2015, 82, 278–281. [Google Scholar] [CrossRef]
- Silvestris, E.; D’Oronzo, S.; Cafforio, P.; DAmato, G.; Loverro, G. Perspective in infertility: The ovarian stem cells. J. Ovarian Res. 2015, 8, 55. [Google Scholar] [CrossRef] [PubMed]
- 31st Annual Meeting of the European Society of Human Reproduction and Embryology (ESHRE), Lisbon, Portugal, 14–17 July 2015; Proceedings from the Workshop on Experts in Egg Health Advancing Fertility Care; ESHRE: Strombeek-Bever, Belgium, 2015.
- Labarta, E.; de Los Santos, M.J.; Herraiz, S.; Escribá, M.J.; Marzal, A.; Buigues, A.; Pellicer, A. Autologous mitochondrial transfer as a complementary technique to intracytoplasmic sperm injection to improve embryo quality in patients undergoing in vitro fertilization—A randomized pilot study. Fertil. Steril. 2019, 111, 86–96. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Cozzolino, M.; Marin, D.; Sisti, G. New frontiers in IVF: mtDNA and autologous germline mitochondrial energy transfer. Reprod. Biol. Endocrinol. 2019, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Langford, D.; Natarajaseenivasan, K. Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res. Rev. 2020, 62, 101128. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Iorio, R.; Petricca, S.; Mattei, V.; Delle Monache, S. Horizontal mitochondrial transfer as a novel bioenergetic tool for mesenchymal stromal/stem cells: Molecular mechanisms and therapeutic potential in a variety of diseases. J. Transl. Med. 2024, 22, 491. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, X.; Guan, Y.; Li, C.; Cheng, H.; Bai, J.; Zhao, J.; Wang, Y.; Peng, J. Recent advances in mitochondrial transplantation to treat disease. Biomater. Transl. 2025, 6, 4–23. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramirez-Barbieri, G.; Moskowitzova, K.; Shin, B.; Blitzer, D.; Orfany, A.; Guariento, A.; Iken, K.; Friehs, I.; Zurakowski, D.; Del Nido, P.J.; et al. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion 2019, 46, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Koch, R.E.; Josefson, C.C.; Hill, G.E. Mitochondrial function, ornamentation, and immunocompetence. Biol. Rev. Camb. Philos. Soc. 2017, 92, 1459–1474. [Google Scholar] [CrossRef]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T cells with dysfunctional mitochondria induce multimorbidity and premature senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Hou, Y.; Zhou, W.; Zhao, Z.; Liu, Z.; Fu, A. The effect of mitochondrial transplantation therapy from different gender on inhibiting cell proliferation of malignant melanoma. Int. J. Biol. Sci. 2021, 17, 2021–2033. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Irie, K.; Matsuo, K.; Mishima, K.; Nakamura, Y. Molecular and cellular mechanisms of mitochondria transfer in models of central nervous system disease. J. Cereb. Blood Flow. Metab. 2024, 271678X241300223. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bamshad, C.; Habibi Roudkenar, M.; Abedinzade, M.; Yousefzadeh Chabok, S.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; Sato, T.; Tomita, K.; Jahanian-Najafabadi, A.; Feizkhah, A.; et al. Human umbilical cord-derived mesenchymal stem cells-harvested mitochondrial transplantation improved motor function in TBI models through rescuing neuronal cells from apoptosis and alleviating astrogliosis and microglia activation. Int. Immunopharmacol. 2023, 118, 110106. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Lo, E.H.; Hayakawa, K. Placental Mitochondria Therapy for Cerebral Ischemia-Reperfusion Injury in Mice. Stroke 2020, 51, 3142–3146. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pekkurnaz, G.; Wang, X. Mitochondrial heterogeneity and homeostasis through the lens of a neuron. Nat. Metab. 2022, 4, 802–812. [Google Scholar] [CrossRef]
- Tian, H.; Chen, X.; Liao, J.; Yang, T.; Cheng, S.; Mei, Z.; Ge, J. Mitochondrial quality control in stroke: From the mechanisms to therapeutic potentials. J. Cell. Mol. Med. 2022, 26, 1000–1012. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, T.; Zhu, Y.; Jia, J.; Meng, H.; Xu, C.; Xian, P.; Li, Z.; Tang, Z.; Wu, Y.; Liu, Y. Mitochondrial Transplantation Promotes Remyelination and Long-Term Locomotion Recovery following Cerebral Ischemia. Mediat. Inflamm. 2022, 2022, 1346343. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Norat, P.; Sokolowski, J.D.; Gorick, C.M.; Soldozy, S.; Kumar, J.S.; Chae, Y.; Yagmurlu, K.; Nilak, J.; Sharifi, K.A.; Walker, M.; et al. Intraarterial Transplantation of Mitochondria After Ischemic Stroke Reduces Cerebral Infarction. Stroke Vasc. Interv. Neurol. 2023, 3, e000644. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hayashida, K.; Takegawa, R.; Endo, Y.; Yin, T.; Choudhary, R.C.; Aoki, T.; Nishikimi, M.; Murao, A.; Nakamura, E.; Shoaib, M.; et al. Exogenous mitochondrial transplantation improves survival and neurological outcomes after resuscitation from cardiac arrest. BMC Med. 2023, 21, 56. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tashiro, R.; Bautista-Garrido, J.; Ozaki, D.; Sun, G.; Obertas, L.; Mobley, A.S.; Kim, G.S.; Aronowski, J.; Jung, J.E. Transplantation of Astrocytic Mitochondria Modulates Neuronal Antioxidant Defense and Neuroplasticity and Promotes Functional Recovery after Intracerebral Hemorrhage. J. Neurosci. 2022, 42, 7001–7014. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Li, S.J.; Zheng, Q.W.; Zheng, J.; Zhang, J.B.; Liu, H.; Tie, J.J.; Zhang, K.L.; Wu, F.F.; Li, X.D.; Zhang, S.; et al. Mitochondria transplantation transiently rescues cerebellar neurodegeneration improving mitochondrial function and reducing mitophagy in mice. Nat. Commun. 2025, 16, 2839. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sun, X.; Chen, H.; Gao, R.; Huang, Y.; Qu, Y.; Yang, H.; Wei, X.; Hu, S.; Zhang, J.; Wang, P.; et al. Mitochondrial transplantation ameliorates doxorubicin-induced cardiac dysfunction via activating glutamine metabolism. iScience 2023, 26, 107790. [Google Scholar] [CrossRef] [PubMed]
- Blitzer, D.; Guariento, A.; Doulamis, I.P.; Shin, B.; Moskowitzova, K.; Barbieri, G.R.; Orfany, A.; Del Nido, P.J.; McCully, J.D. Delayed transplantation of autologous mitochondria for cardioprotection in a porcine model. Ann. Thorac. Surg. 2020, 109, 711–719. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Chen, L.; Guo, W.; Wang, J.; Ni, H.; Liu, J.; Jiang, W.; Shen, J.; Mao, C.; Zhou, M.; et al. Oral mitochondrial transplantation using nanomotors to treat ischaemic heart disease. Nat. Nanotechnol. 2024, 19, 1375–1385, Erratum in: Nat. Nanotechnol. 2024, 19, 1236. [Google Scholar] [CrossRef] [PubMed]
- Shin, B.; Saeed, M.Y.; Esch, J.J.; Guariento, A.; Blitzer, D.; Moskowitzova, K.; Ramirez-Barbieri, G.; Orfany, A.; Thedsanamoorthy, J.K.; Cowan, D.B.; et al. A novel biological strategy for myocardial protection by intracoronary delivery of mitochondria:safety and efficacy. JACC Basic Transl. Sci. 2019, 4, 871–888. [Google Scholar] [CrossRef]
- Mokhtari, B.; Delkhah, M.; Badalzadeh, R.; Ghaffari, S. Mitochondrial transplantation combined with mitoquinone and melatonin: A survival strategy against myocardial reperfusion injury in aged rats. Exp. Physiol. 2025, 110, 844–856. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rossi, A.; Asthana, A.; Riganti, C.; Sedrakyan, S.; Byers, L.N.B.; Robertson, J.V.; Senger, R.S.; Montali, F.; Grange, C.; Dalmasso, A.; et al. Mitochondria transplantation mitigates damage in an in vitro model of renal tubular injury and in an ex vivo model of DCD renal transplantation. Ann. Surg. 2023, 278, e1313–e1326. [Google Scholar] [CrossRef]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Kido, T.; Orfany, A.; Saeed, M.Y.; Weixler, V.H.; Blitzer, D.; Shin, B.; Snay, E.R.; et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 319, F403–F413. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pang, Y.L.; Fang, S.Y.; Cheng, T.T.; Huang, C.C.; Lin, M.W.; Lam, C.F.; Chen, K.B. Viable allogeneic mitochondria transplantation improves gas exchange and alveolar-capillary permeability in rats with endotoxin-induced acute lung injuries. Int. J. Med. Sci. 2022, 19, 1036–1046. [Google Scholar] [CrossRef]
- Zhong, G.; Liu, W.; Venkatesan, J.K.; Wang, D.; Madry, H.; Cucchiarini, M. Autologous transplantation of mitochondria/rAAV IGF-I platforms in human osteoarthritic articular chondrocytes to treat osteoarthritis. Mol. Ther. 2025, 33, 2900–2912. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, Z.; Hou, Y.; Zhou, W.; Keerthiga, R.; Fu, A. Mitochondrial transplantation therapy inhibit carbon tetrachloride-induced liver injury through scavenging free radicals and protecting hepatocytes. Bioeng. Transl. Med. 2021, 6, e10209. [Google Scholar] [CrossRef]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Baharvand, F.; Habibi Roudkenar, M.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; Feizkhah, A.; Bashiri Aliabadi, S.; Salari, A.; Mohammadi Roushandeh, A. Safety and efficacy of platelet-derived mitochondrial transplantation in ischaemic heart disease. Int. J. Cardiol. 2024, 410, 132227. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Kang, Y.C.; Kim, M.J.; Kim, S.U.; Kang, H.R.; Yeo, J.S.; Kim, Y.; Yu, S.-H.; Song, B.; Hwang, J.W.; et al. Mitochondrial transplantation as a novel therapeutic approach in idiopathic inflammatory myopathy. Ann. Rheum. Dis. 2025, 84, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Fan, X.Y.; Guo, L.; Chen, L.N.; Yin, S.; Wen, J.; Li, S.; Ma, J.Y.; Jing, T.; Jiang, M.X.; Sun, X.H.; et al. Reduction of mtDNA heteroplasmy in mitochondrial replacement therapy by inducing forced mitophagy. Nat. Biomed. Eng. 2022, 6, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.G.; Adashi, E.Y.; Gerke, S.; Palacios-González, C.; Ravitsky, V. The Regulation of Mitochondrial Replacement Techniques Around the World. Annu. Rev. Genom. Hum. Genet. 2020, 21, 565–586. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mitochondrial Disorder | Relative Frequency | Typical Feature(s) | Associated Feature(s) | Inheritance | Most Frequent Genetic Findings | Treatment of Choice |
|---|---|---|---|---|---|---|
| Alpers syndrome | Very rare | Childhood myocerebrohepatopathy | Autosomal recessive | POLG mutations with secondary mtDNA depletion | Symptomatic (avoid valproate) | |
| Autosomal dominant optic atrophy (ADOA) | Rare | Optic neuropathy (blindness) | Autosomal dominant | OPA1 mutations | Symptomatic | |
| Coenzyme Q10 deficiency | Very rare | Ataxia or myopathy or multi-system disease | Autosomal recessive | Various nuclear genes | Coenzyme Q10 | |
| Kearns–Sayre syndrome (KSS) | Frequent | Ocular myopathy (ptosis, ophthalmoparesis) | Ataxia, cardiac conduction defects | Sporadic | Single large-scale deletion of mtDNA | Symptomatic |
| Leber hereditary optic neuropathy (LHON) | Very frequent | Optic neuropathy (blindness) | Maternal (low penetrance, higher in male smokers) | Various mtDNA mutations | Idebenone | |
| Leigh syndrome | Frequent | Severe pediatric encephalopathy | Autosomal recessive, X-linked or maternal | Various nuclear or mtDNA mutations (e.g., m.8993T>G) | Symptomatic | |
| Mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS) | Frequent | Stroke-like episodes | Cardiac involvement, hearing loss, diabetes | Maternal | m.3243A>G | Symptomatic |
| Myoclonic encephalopathy with ragged-red fiber (MERRF) | Frequent | Myoclonus | Ataxia, myopathy | Maternal | m.8344A>G | Symptomatic (e.g., Levetiracetam) |
| Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) | Very rare | Gastrointestinal dysmotility | Leukodystrophy, ocular myopathy, peripheral neuropathy | Autosomal recessive | TYMP mutations | Liver transplantation |
| Neuropathy, ataxia, retinitis pigmentosa (NARP) | Rare | Ataxia | Neuropathy, retinitis pigmentosa | Maternal | m.8993T>G | Symptomatic |
| Non syndromic hearing loss (NSHL) | Frequent | Hearing loss | Maternal | m.1555A>G | Symptomatic (avoid aminoglycosides) | |
| Progressive external ophthalmoplegia (PEO) | Very frequent | Ocular myopathy | Myopathy | Autosomal dominant, recessive, maternal, or sporadic | Various nuclear genes with secondary mtDNA multiple deletions, various mtDNA point mutations, and mtDNA single large-scale deletions | Symptomatic |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belousova, V.; Ignatko, I.; Bogomazova, I.; Sosnova, E.; Pesegova, S.; Samusevich, A.; Zarova, E.; Kardanova, M.; Skorobogatova, O.; Maltseva, A. Causes of and Solutions to Mitochondrial Disorders: A Literature Review. Int. J. Mol. Sci. 2025, 26, 6645. https://doi.org/10.3390/ijms26146645
Belousova V, Ignatko I, Bogomazova I, Sosnova E, Pesegova S, Samusevich A, Zarova E, Kardanova M, Skorobogatova O, Maltseva A. Causes of and Solutions to Mitochondrial Disorders: A Literature Review. International Journal of Molecular Sciences. 2025; 26(14):6645. https://doi.org/10.3390/ijms26146645
Chicago/Turabian StyleBelousova, Vera, Irina Ignatko, Irina Bogomazova, Elena Sosnova, Svetlana Pesegova, Anastasia Samusevich, Evdokiya Zarova, Madina Kardanova, Oxana Skorobogatova, and Anna Maltseva. 2025. "Causes of and Solutions to Mitochondrial Disorders: A Literature Review" International Journal of Molecular Sciences 26, no. 14: 6645. https://doi.org/10.3390/ijms26146645
APA StyleBelousova, V., Ignatko, I., Bogomazova, I., Sosnova, E., Pesegova, S., Samusevich, A., Zarova, E., Kardanova, M., Skorobogatova, O., & Maltseva, A. (2025). Causes of and Solutions to Mitochondrial Disorders: A Literature Review. International Journal of Molecular Sciences, 26(14), 6645. https://doi.org/10.3390/ijms26146645