
Total Synthesis of (+)-Penicyclone A and Evaluation of Biological Activity Including Intermediate Compounds

 , , and
, , and
Abstract

1. Introduction
2. Results and Discussion
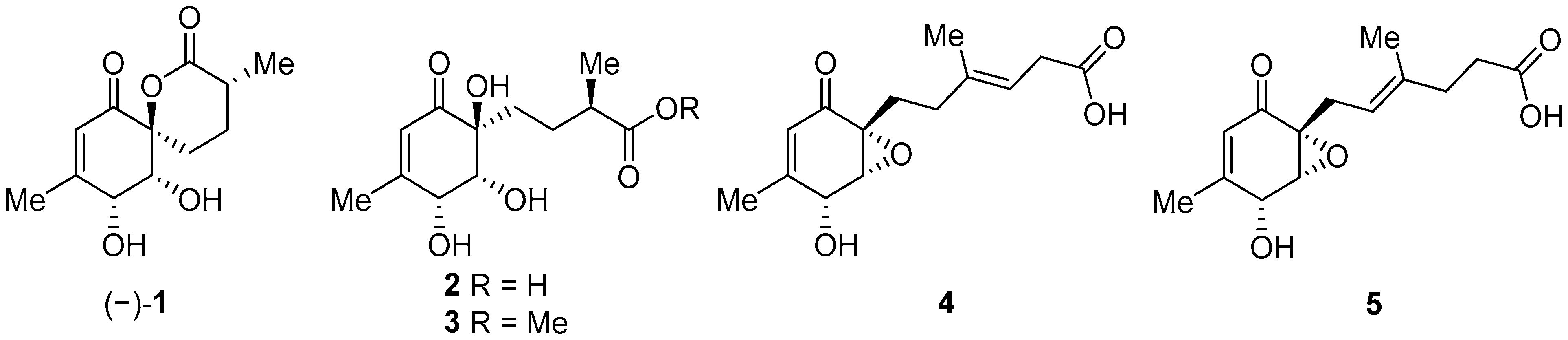
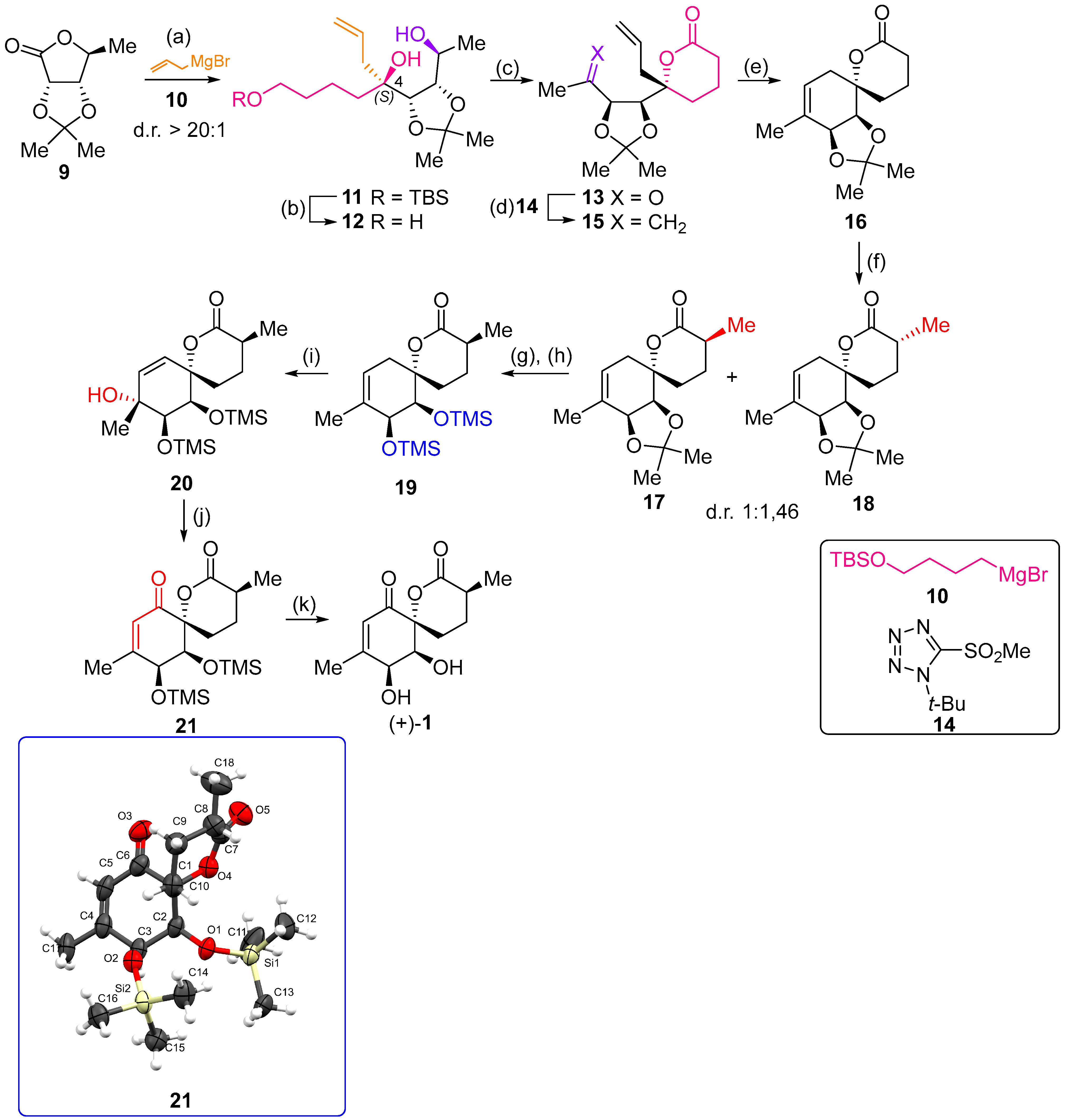
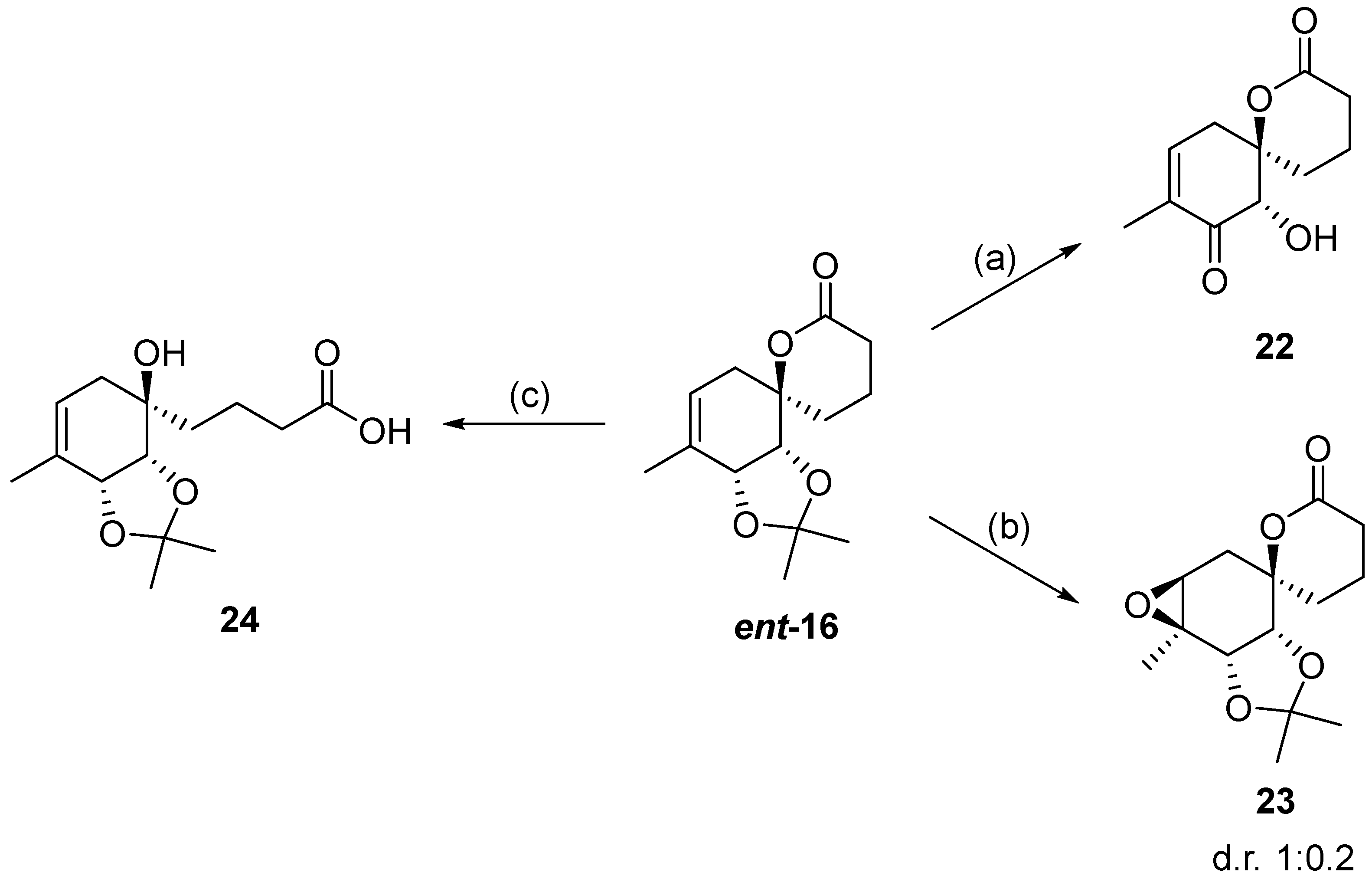
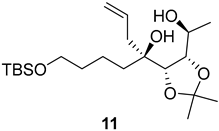
2.1. Synthesis of (+)-Penicyclone A and Selected Derivatives
2.2. Biological Evaluation
3. Materials and Methods
3.1. Chemicals
3.2. Methods
3.2.1. The In Vitro Cytotoxicity Assay
3.2.2. The In Vitro Antibacterial Activity Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| DMP | Dess–Martin periodinane |
| HRMS | High-resolution mass spectrometry |
| MIC | Minimum inhibitory concentration |
| NMR | Nuclear magnetic resonance |
| NRPS | Non-ribosomal peptide synthetase |
| OSMAC | One-strain-many-compounds |
| PYG | Peptone yeast glucose |
| SCXRD | Single-crystal X-ray diffraction |
| TBAF | Tetrabutylammonium fluoride |
| TBS | tert-butyldimethylsilyl protective group |
| TEMPO | (2,2,6,6-Tetramethylpiperidin-1-yl)oxyl |
| THF | Tetrahydrofuran |
| TMS | Trimethylsilyl protective group |
References
- Haque, N.; Parveen, S.; Tang, T.; Wei, J.; Huang, Z. Marine Natural Products in Clinical Use. Mar. Drugs 2022, 20, 528. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Luo, D.; Luesch, H. Advances in Exploring the Therapeutic Potential of Marine Natural Products. Pharmacol. Res. 2019, 147, 104373. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, Z.; Zhu, T.; Gu, Q.; Li, D. Penicyclones A-E, Antibacterial Polyketides from the Deep-Sea-Derived Fungus Penicillium Sp. F23-2. J. Nat. Prod. 2015, 78, 2699–2703. [Google Scholar] [CrossRef]
- Du, L.; Li, D.; Zhu, T.; Cai, S.; Wang, F.; Xiao, X.; Gu, Q. New Alkaloids and Diterpenes from a Deep Ocean Sediment Derived Fungus Penicillium Sp. Tetrahedron 2009, 65, 1033–1039. [Google Scholar] [CrossRef]
- Guo, W.; Peng, J.; Zhu, T.; Gu, Q.; Keyzers, R.A.; Li, D. Sorbicillamines A–E, Nitrogen-Containing Sorbicillinoids from the Deep-Sea-Derived Fungus Penicillium Sp. F23–2. J. Nat. Prod. 2013, 76, 2106–2112. [Google Scholar] [CrossRef]
- Romano, S.; Jackson, S.A.; Patry, S.; Dobson, A.D.W. Extending the “One Strain Many Compounds” (OSMAC) Principle to Marine Microorganisms. Mar. Drugs 2018, 16, 244. [Google Scholar] [CrossRef]
- Pinedo-Rivilla, C.; Aleu, J.; Durán-Patrón, R. Cryptic Metabolites from Marine-Derived Microorganisms Using OSMAC and Epigenetic Approaches. Mar. Drugs 2022, 20, 84. [Google Scholar] [CrossRef]
- Pan, R.; Bai, X.; Chen, J.; Zhang, H.; Wang, H. Exploring Structural Diversity of Microbe Secondary Metabolites Using OSMAC Strategy: A Literature Review. Front. Microbiol. 2019, 10, 294. [Google Scholar] [CrossRef]
- Quintavalla, A. Spirolactones: Recent Advances in Natural Products, Bioactive Compounds and Synthetic Strategies. Curr. Med. Chem. 2018, 25, 917–962. [Google Scholar] [CrossRef]
- Talajić, G.; Topić, E.; Meštrović, J.; Cindro, N. Total Synthesis of Penicyclone A Using a Double Grignard Reaction. J. Org. Chem. 2022, 87, 16054–16062. [Google Scholar] [CrossRef]
- Bitchagno, G.T.M.; Nchiozem-Ngnitedem, V.A.; Melchert, D.; Fobofou, S.A. Demystifying Racemic Natural Products in the Homochiral World. Nat. Rev. Chem. 2022, 6, 806–822. [Google Scholar] [CrossRef]
- Nakata, M.; Arai, M.; Tomooka, K.; Ohsawa, N.; Kinoshita, M. Total Synthesis of Erythronolide A. Bull. Chem. Soc. Jpn. 1989, 62, 2618–2635. [Google Scholar] [CrossRef]
- Subrahmanyam, A.V.; Palanichamy, K.; Kaliappan, K.P. Application of an Enyne Metathesis/Diels–Alder Cycloaddition Sequence: A New Versatile Approach to the Syntheses of C-Aryl Glycosides and Spiro-C-Aryl Glycosides. Chem. A Eur. J. 2010, 16, 8545–8556. [Google Scholar] [CrossRef]
- Hansen, T.M.; Florence, G.J.; Lugo-Mas, P.; Chen, J.; Abrams, J.N.; Forsyth, C.J. Highly Chemoselective Oxidation of 1,5-Diols to δ-Lactones with TEMPO/BAIB. ChemInform 2003, 34, 57–59. [Google Scholar] [CrossRef]
- Aïssa, C. Improved Julia−Kocienski Conditions for the Methylenation of Aldehydes and Ketones. J. Org. Chem. 2006, 71, 360–363. [Google Scholar] [CrossRef]
- Paquette, L.A.; Wang, T.Z.; Huu, V.N. Access to Naturally Occurring Cyclooctanoids by Two-Carbon Intercalation. Total Synthesis of (+)-Ceroplastol I. J. Am. Chem. Soc. 1993, 115, 1676–1683. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (Resazurin) Fluorescent Dye for the Assessment of Mammalian Cell Cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing. In 15th Informational Supplement, CLSI Document M100 S15; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2005. [Google Scholar]
- Rigaku Oxford Diffraction. CrysAlisPro Software System, Version 1.171.42.49; Rigaku Oxford Diffraction: Oxford, UK, 2020.
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Lourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Spek, A.L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Song, W.S.; Liu, S.-X.; Chang, C.-C. Synthesis of l-Deoxyribonucleosides from d-Ribose. J. Org. Chem. 2018, 83, 14923–14932. [Google Scholar] [CrossRef]
- Zeng, J.; Vedachalam, S.; Xiang, S.; Liu, X.-W. Direct C-Glycosylation of Organotrifluoroborates with Glycosyl Fluorides and Its Application to the Total Synthesis of (+)-Varitriol. Org. Lett. 2011, 13, 42–45. [Google Scholar] [CrossRef]
- Jana, S.; Sarpe, V.A.; Kulkarni, S.S. Total Synthesis of Emmyguyacins A and B, Potential Fusion Inhibitors of Influenza Virus. Org. Lett. 2018, 20, 6938–6942. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC (µg/mL) | IC50 (µM) | ||||
|---|---|---|---|---|---|---|
| S. aureus | E. faecalis | M. catarrhalis | E. coli | MES-OV | MDA-MB-468 | |
 | >128 | >128 | >128 | >128 | >10 | >10 |
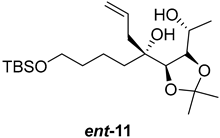 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
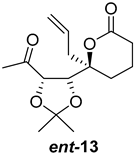 | >128 | >128 | >128 | >128 | >10 | >10 |
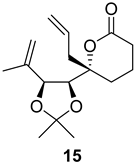 | >128 | >128 | >128 | >128 | >10 | >10 |
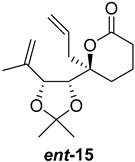 | >128 | >128 | >128 | >128 | >10 | >10 |
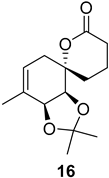 | >128 | >128 | >128 | >128 | >10 | >10 |
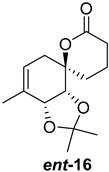 | >128 | >128 | >128 | >128 | >10 | >10 |
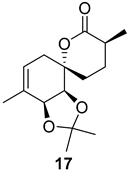 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
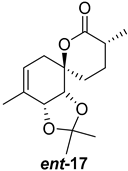 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
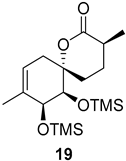 | >128 | >128 | >128 | >128 | >10 | >10 |
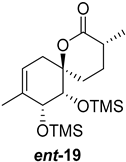 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
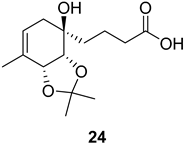 | >128 | >128 | >128 | >128 | >10 | >10 |
 | >128 | >128 | >128 | >128 | >10 | >10 |
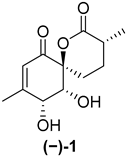 | >128 | >128 | >128 | >128 | >10 | >10 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duvnjak, M.; Talajić, G.; Baranašić, J.; Baus Topić, N.; Čipčić Paljetak, H.; Cindro, N. Total Synthesis of (+)-Penicyclone A and Evaluation of Biological Activity Including Intermediate Compounds. Int. J. Mol. Sci. 2025, 26, 6643. https://doi.org/10.3390/ijms26146643
Duvnjak M, Talajić G, Baranašić J, Baus Topić N, Čipčić Paljetak H, Cindro N. Total Synthesis of (+)-Penicyclone A and Evaluation of Biological Activity Including Intermediate Compounds. International Journal of Molecular Sciences. 2025; 26(14):6643. https://doi.org/10.3390/ijms26146643
Chicago/Turabian StyleDuvnjak, Mirko, Gregor Talajić, Jurica Baranašić, Nea Baus Topić, Hana Čipčić Paljetak, and Nikola Cindro. 2025. "Total Synthesis of (+)-Penicyclone A and Evaluation of Biological Activity Including Intermediate Compounds" International Journal of Molecular Sciences 26, no. 14: 6643. https://doi.org/10.3390/ijms26146643
APA StyleDuvnjak, M., Talajić, G., Baranašić, J., Baus Topić, N., Čipčić Paljetak, H., & Cindro, N. (2025). Total Synthesis of (+)-Penicyclone A and Evaluation of Biological Activity Including Intermediate Compounds. International Journal of Molecular Sciences, 26(14), 6643. https://doi.org/10.3390/ijms26146643