
Comparing DNA Methylation Landscapes in Peripheral Blood from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID Patients
 ,
,  , , , and
, , , and
Abstract
1. Introduction
2. Results
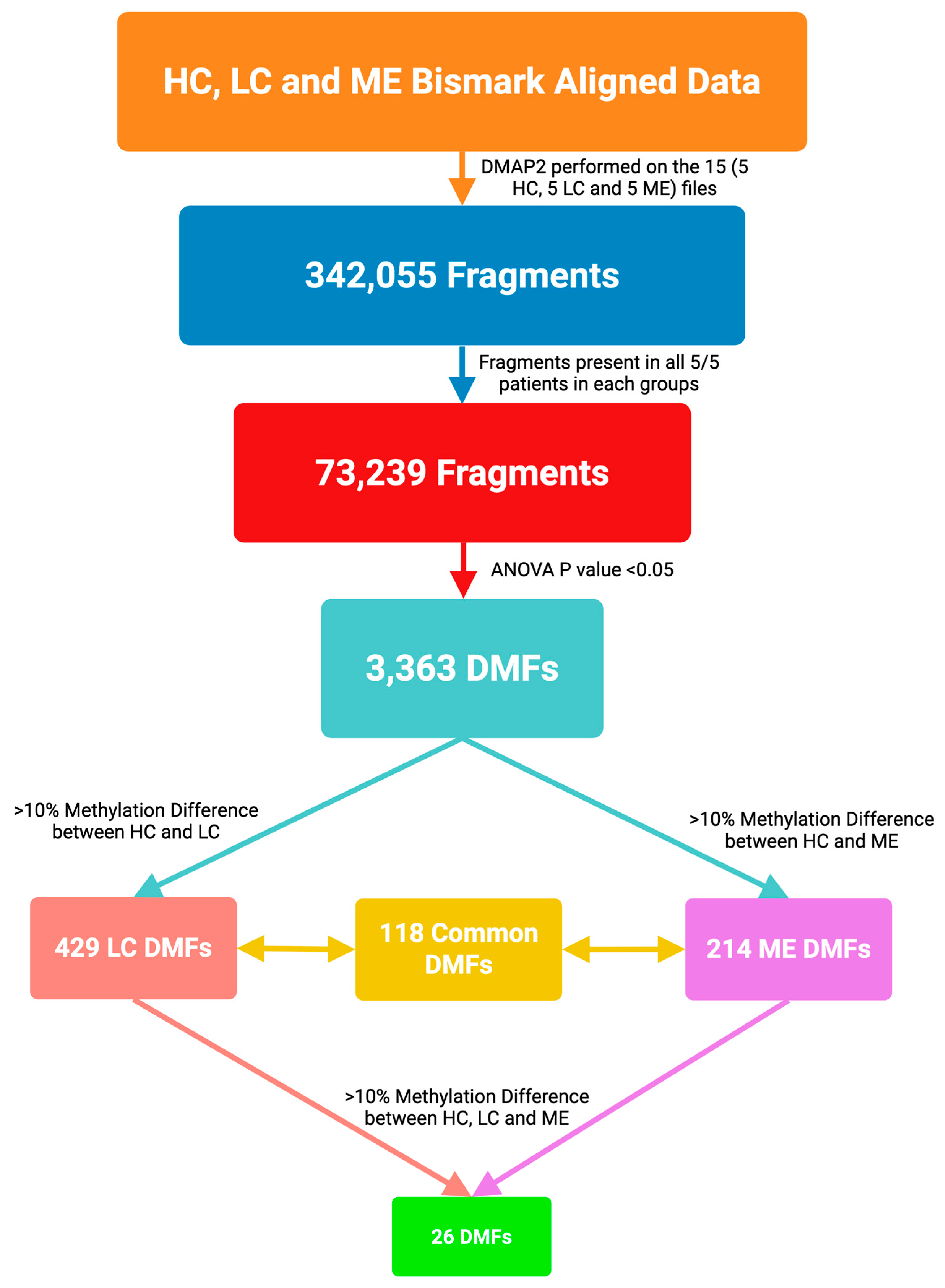
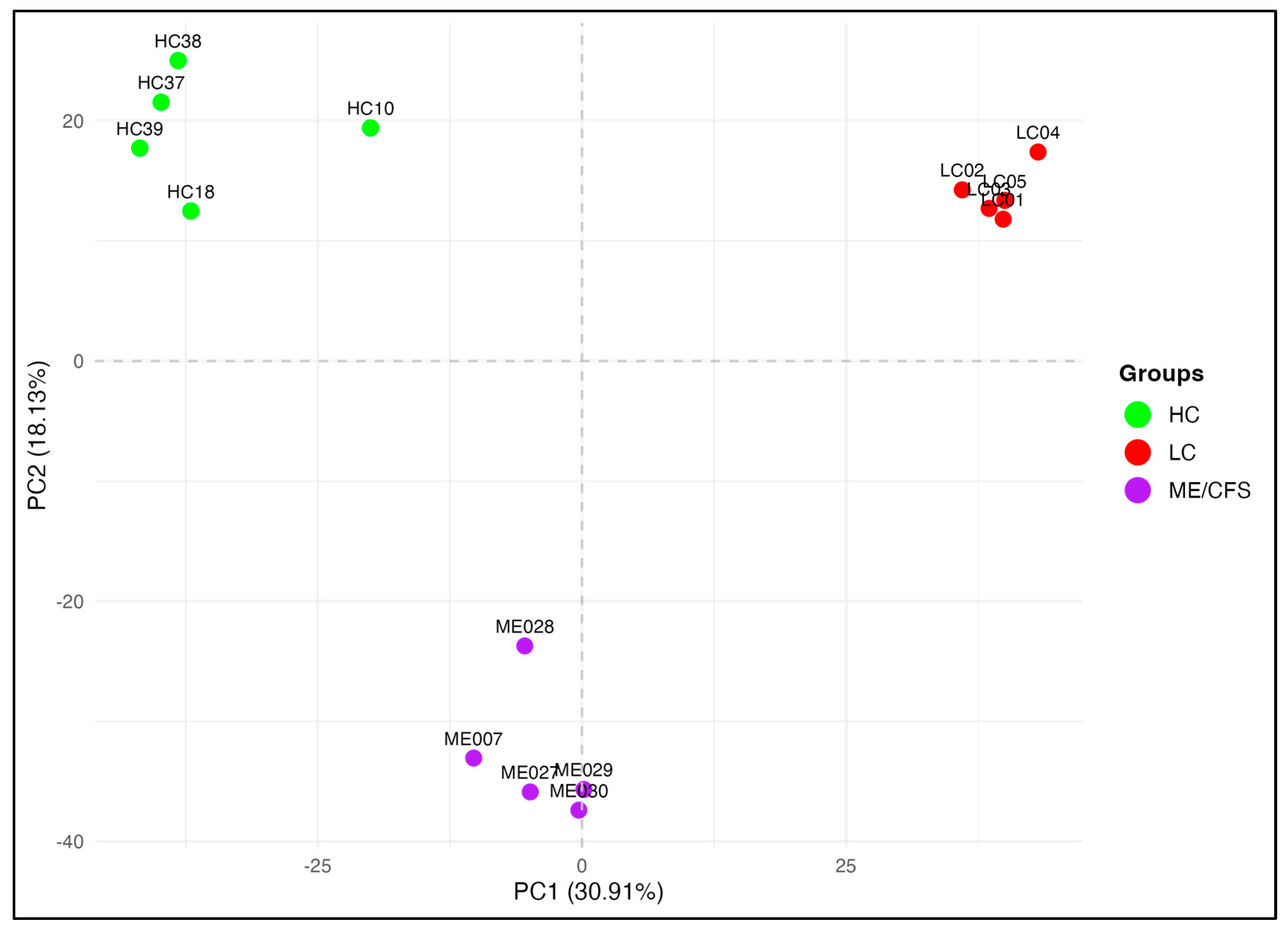
2.1. Whole-Genome DNA Methylation Patterns
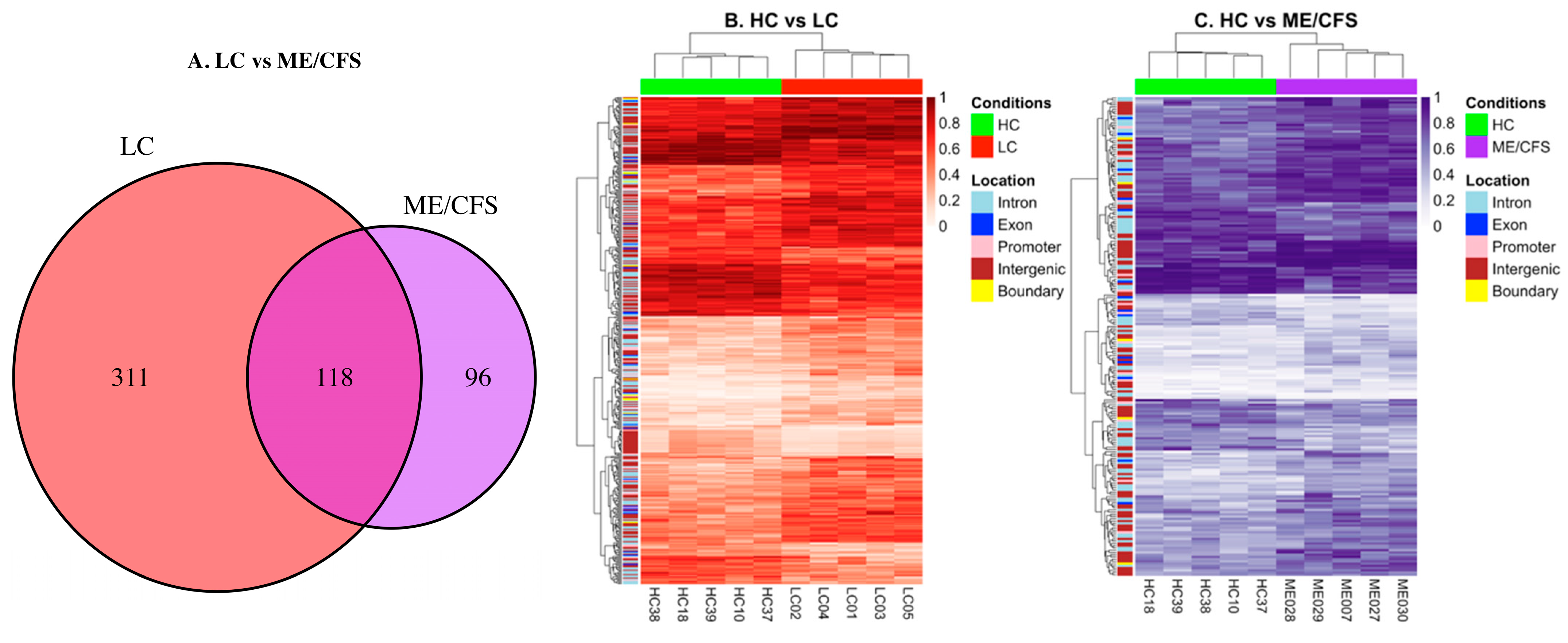
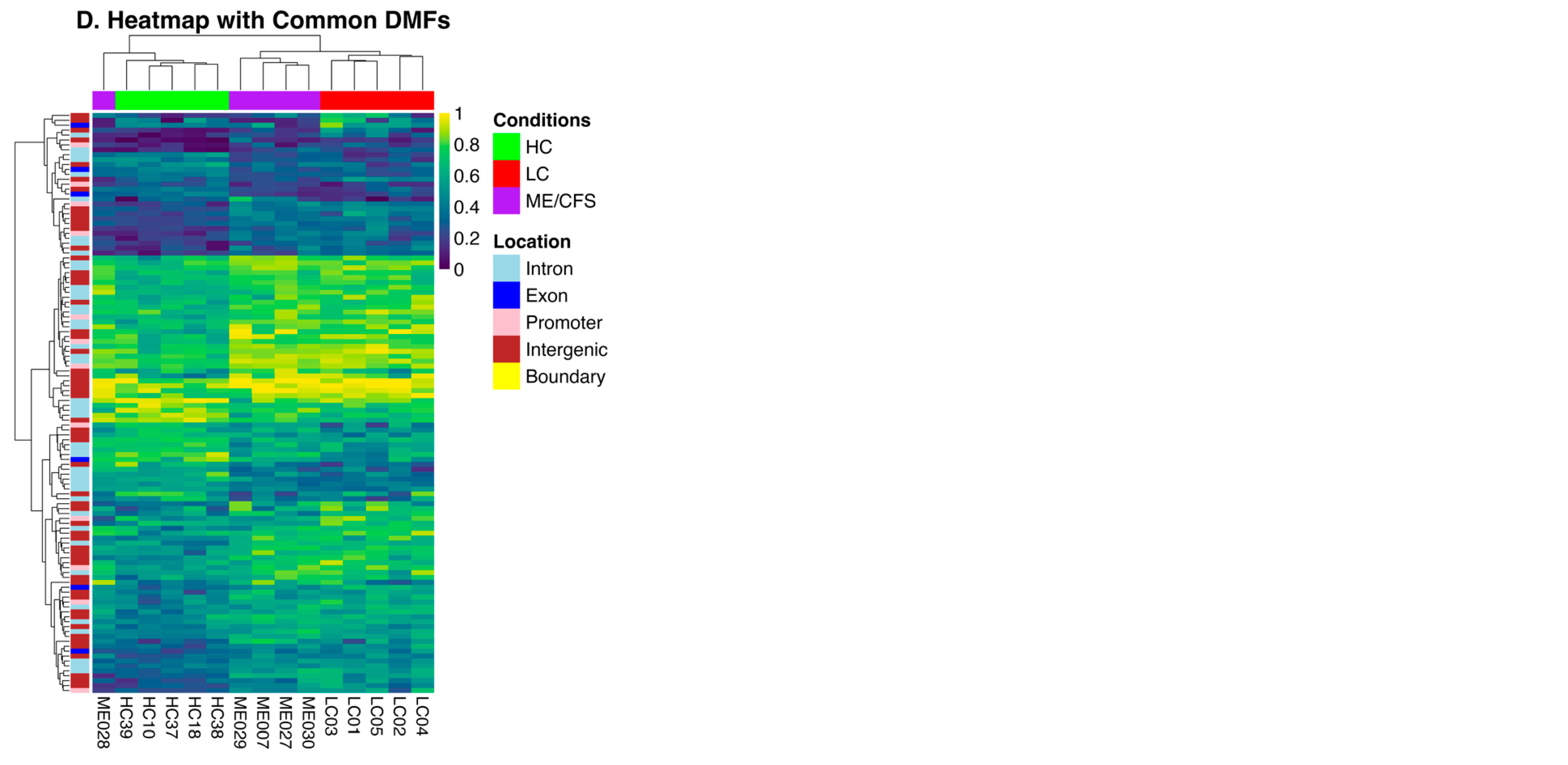
2.2. Characteristics of the Differential Methylation Changes in LC and ME/CFS
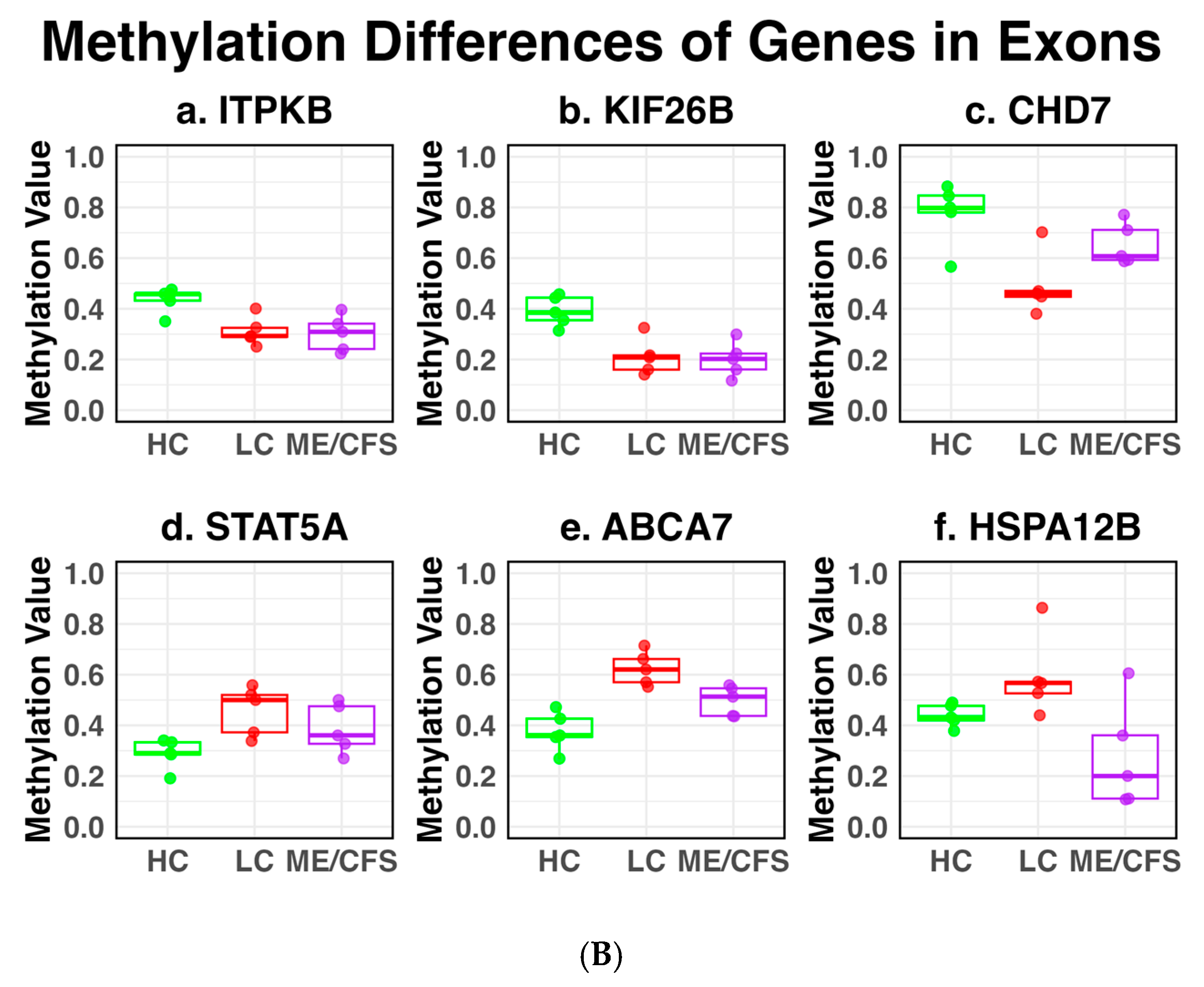
2.3. DMFs Associated with Gene Promoters and Gene Exons
2.4. Methylation Differences Between Long COVID and ME/CFS
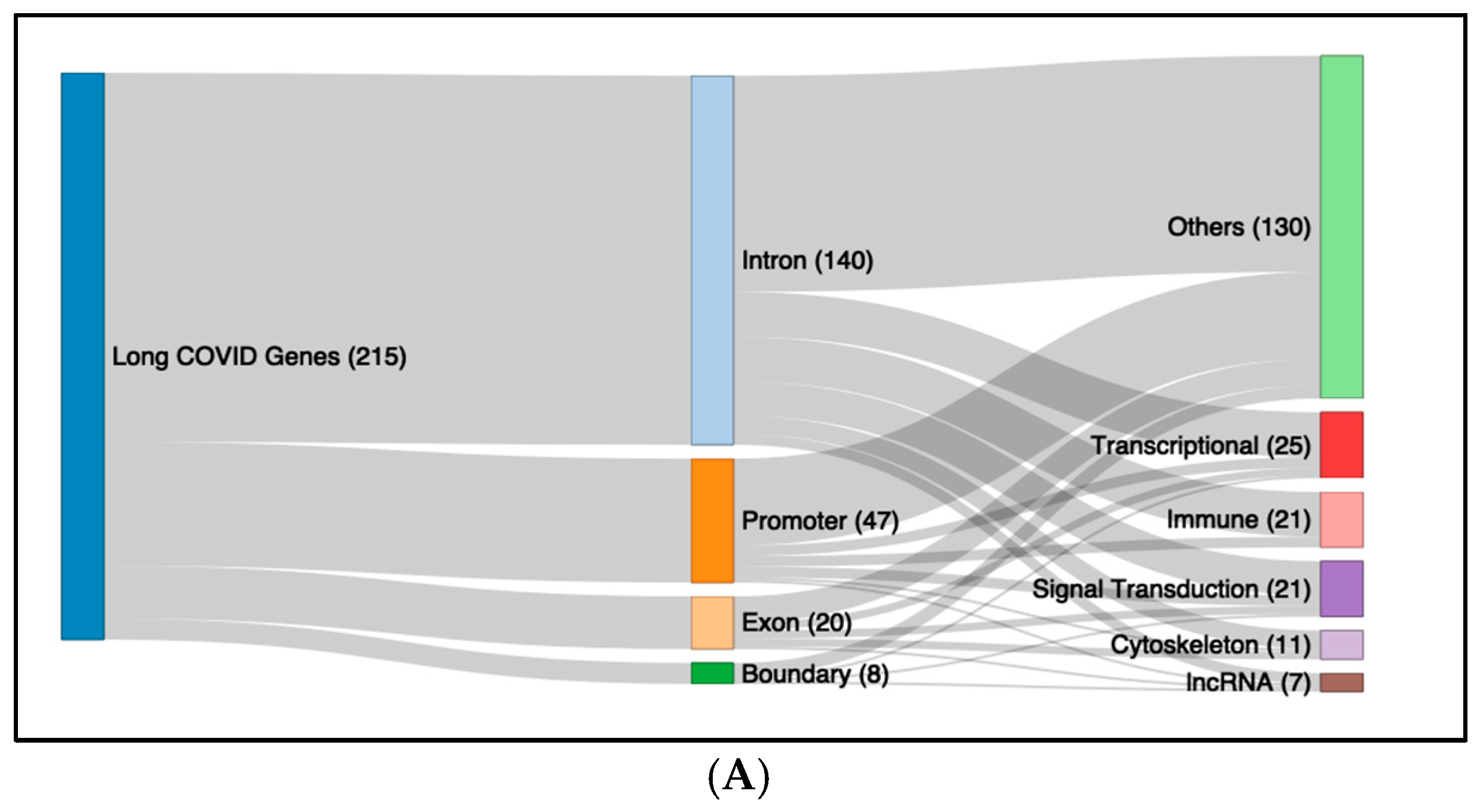
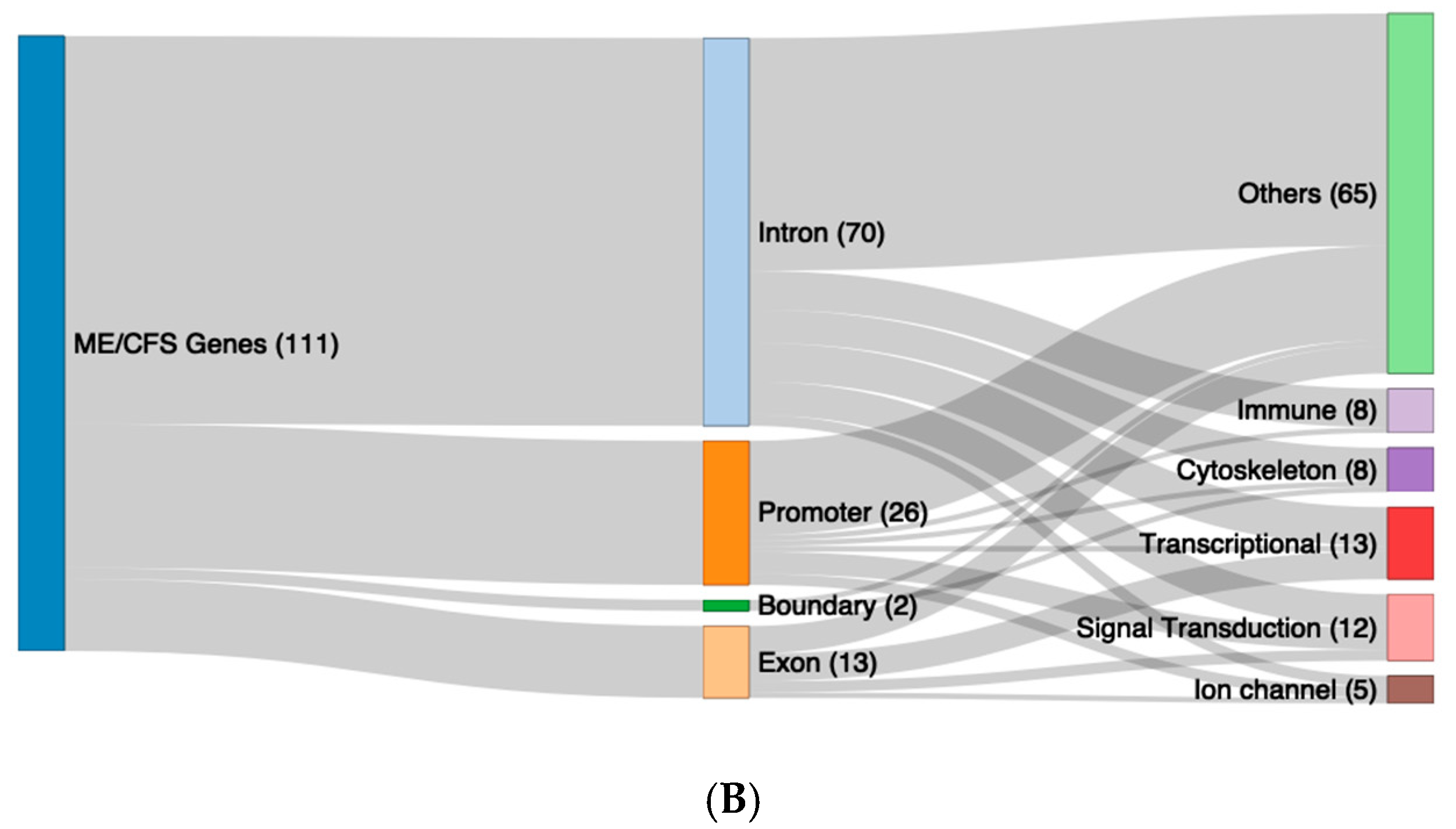
2.5. Functional Pathway Analysis of the DMFs of Long COVID and ME/CFS
3. Discussion
4. Materials and Methods
4.1. The Analysis Cohorts
4.2. PBMC Isolation
4.3. DNA Extraction
4.4. Reduced Representation Bisulphite Sequencing
4.4.1. DNA Sequencing
4.4.2. Statistical Analyses
5. Conclusions
6. Future Directions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. COVID-19 Cases, World. 2025. Available online: https://data.who.int/dashboards/covid19/cases (accessed on 3 April 2025).
- Tate, W.P.; Walker, M.O.M.; Peppercorn, K.; Blair, A.L.H.; Edgar, C.D. Towards a Better Understanding of the Complexities of Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID. Int. J. Mol. Sci. 2023, 24, 5124. [Google Scholar] [CrossRef]
- Davis, H.E.; McCorkell, L.; Vogel, J.M.; Topol, E.J. Long COVID: Major findings, mechanisms and recommendations. Nat. Rev. Microbiol. 2023, 23, 133–146. [Google Scholar] [CrossRef]
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Global Health; Board on Health Sciences Policy; Committee on Examining the Working Definition for Long COVID. A Long COVID Definition: A Chronic, Systemic Disease State with Profound Consequences; Goldowitz, I., Worku, T., Brown, L., Fineberg, H.V., Eds.; National Academies Press (US): Washington, DC, USA, 2024. [Google Scholar]
- Collins, F.S. NIH Launches New Initiative to Study “Long COVID”. 2021. Available online: https://www.nih.gov/about-nih/who-we-are/nih-director/statements/nih-launches-new-initiative-study-long-covid (accessed on 1 April 2025).
- Staff, M. An Outbreak of encephalomyelitis in the Royal Free Hospital Group, London, in 1955. Br. Med. J. 1957, 2, 895–904. [Google Scholar]
- Levine, P.H.; Jacobson, S.; Pocinki, A.G.; Cheney, P.; Peterson, D.; Connelly, R.R.; Weil, R.; Robinson, S.M.; Ablashi, D.V.; Salahuddin, S.Z.; et al. Clinical, epidemiologic, and virologic studies in four clusters of the chronic fatigue syndrome. Arch. Intern. Med. 1992, 152, 1611–1616. [Google Scholar] [CrossRef]
- Shepherd, C.; Chaudhuri, A.; Association, M.; Association, M.E.; Society, A.N.Z.M. ME/CFS/PVFS: An Exploration of the Key Clinical Issues; ME Association: Buckinghamshire, UK, 2013. [Google Scholar]
- ME-Pedia. Millions Missing Protests. 2023. Available online: https://me-pedia.org/wiki/Millions_Missing_protests (accessed on 10 May 2025).
- Tate, W.; Walker, M.; Sweetman, E.; Helliwell, A.; Peppercorn, K.; Edgar, C.; Blair, A.; Chatterjee, A. Molecular Mechanisms of Neuroinflammation in ME/CFS and Long COVID to Sustain Disease and Promote Relapses. Front. Neurol. 2022, 13, 877772. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Ahrnsbrak, R.; Rekito, A. Evidence Mounts That About 7% of US Adults Have Had Long COVID. JAMA 2024, 332, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Choi, T.; Al-Aly, Z. Postacute Sequelae of SARS-CoV-2 Infection in the Pre-Delta, Delta, and Omicron Eras. N. Engl. J. Med. 2024, 391, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Taylor, K.; Kozubek, J.; Sardell, J.; Gardner, S. Genetic risk factors for ME/CFS identified using combinatorial analysis. J. Transl. Med. 2022, 20, 598. [Google Scholar] [CrossRef]
- Taylor, K.; Pearson, M.; Das, S.; Sardell, J.; Chocian, K.; Gardner, S. Genetic risk factors for severe and fatigue dominant long COVID and commonalities with ME/CFS identified by combinatorial analysis. J. Transl. Med. 2023, 21, 775. [Google Scholar] [CrossRef]
- Gentilotti, E.; Górska, A.; Tami, A.; Gusinow, R.; Mirandola, M.; Rodríguez Baño, J.; Palacios Baena, Z.R.; Rossi, E.; Hasenauer, J.; Lopes-Rafegas, I.; et al. Clinical phenotypes and quality of life to define post-COVID-19 syndrome: A cluster analysis of the multinational, prospective ORCHESTRA cohort. eClinicalMedicine 2023, 62, 102107. [Google Scholar] [CrossRef]
- Jason, L.A.; Natelson, B.H.; Bonilla, H.; Sherif, Z.A.; Vernon, S.D.; Verduzco Gutierrez, M.; O’Brien, L.; Taylor, E. What Long COVID investigators can learn from four decades of ME/CFS research. Brain Behav. Immun. Integr. 2023, 4, 100022. [Google Scholar] [CrossRef]
- Horwitz, L.I.; Thaweethai, T.; Brosnahan, S.B.; Cicek, M.S.; Fitzgerald, M.L.; Goldman, J.D.; Hess, R.; Hodder, S.L.; Jacoby, V.L.; Jordan, M.R.; et al. Researching COVID to Enhance Recovery (RECOVER) adult study protocol: Rationale, objectives, and design. PLoS ONE 2023, 18, e0286297. [Google Scholar] [CrossRef] [PubMed]
- Komaroff, A.L.; Lipkin, W.I. Insights from myalgic encephalomyelitis/chronic fatigue syndrome may help unravel the pathogenesis of postacute COVID-19 syndrome. Trends Mol. Med. 2021, 27, 895–906. [Google Scholar] [CrossRef] [PubMed]
- Marshall-Gradisnik, S.; Eaton-Fitch, N. Understanding myalgic encephalomyelitis. Science 2022, 377, 1150–1151. [Google Scholar] [CrossRef]
- Komaroff, A.L. MECFS and Long COVID: Emerging Similarities and Why It Matters. 2022. Available online: https://www.youtube.com/watch?v=AbVMvRS-a7Yc (accessed on 7 June 2025).
- Vernon, S.D.; Hartle, M.; Sullivan, K.; Bell, J.; Abbaszadeh, S.; Unutmaz, D.; Bateman, L. Post-exertional malaise among people with long COVID compared to myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Work 2023, 74, 1179–1186. [Google Scholar] [CrossRef]
- Peppercorn, K.; Edgar, C.D.; Kleffmann, T.; Tate, W.P. A pilot study on the immune cell proteome of long COVID patients shows changes to physiological pathways similar to those in myalgic encephalomyelitis/chronic fatigue syndrome. Sci. Rep. 2023, 13, 22068. [Google Scholar] [CrossRef]
- Eaton-Fitch, N.; Rudd, P.; Er, T.; Hool, L.; Herrero, L.; Marshall-Gradisnik, S. Immune exhaustion in ME/CFS and long COVID. JCI Insight 2024, 9, e183810. [Google Scholar] [CrossRef]
- Helliwell, A.M.; Sweetman, E.C.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Changes in DNA methylation profiles of myalgic encephalomyelitis/chronic fatigue syndrome patients reflect systemic dysfunctions. Clin. Epigenetics 2020, 12, 167. [Google Scholar] [CrossRef]
- Helliwell, A.M.; Stockwell, P.A.; Edgar, C.D.; Chatterjee, A.; Tate, W.P. Dynamic Epigenetic Changes during a Relapse and Recovery Cycle in Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Int. J. Mol. Sci. 2022, 23, 11852. [Google Scholar] [CrossRef]
- Brenu, E.W.S.; Staines, D.R.; Marshall-Gradisnik, S.M. Methylation Profile of CD4+ T Cells in Chronic Fatigue Syndrome/Myalgic Encephalomyelitis. J. Clin. Cell. Immunol. 2014, 5, 228. [Google Scholar]
- de Vega, W.C.; Vernon, S.D.; McGowan, P.O. DNA methylation modifications associated with chronic fatigue syndrome. PLoS ONE 2014, 9, e104757. [Google Scholar] [CrossRef] [PubMed]
- de Vega, W.C.; McGowan, P.O. The epigenetic landscape of myalgic encephalomyelitis/chronic fatigue syndrome: Deciphering complex phenotypes. Epigenomics 2017, 9, 1337–1340. [Google Scholar] [CrossRef]
- de Vega, W.C.; Erdman, L.; Vernon, S.D.; Goldenberg, A.; McGowan, P.O. Integration of DNA methylation & health scores identifies subtypes in myalgic encephalomyelitis/chronic fatigue syndrome. Epigenomics 2018, 10, 539–557. [Google Scholar] [CrossRef]
- Herrera, S.; de Vega, W.C.; Ashbrook, D.; Vernon, S.D.; McGowan, P.O. Genome-epigenome interactions associated with Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. Epigenetics 2018, 13, 1174–1190. [Google Scholar] [CrossRef]
- Stockwell, P.A.; Rodger, E.J.; Gimenez, G.; Morison, I.M.; Chatterjee, A. DMAP2: A Pipeline for Analysis of Whole-Genome-Scale DNA Methylation Sequencing Data. Curr. Protoc. 2024, 4, e70003. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef]
- Gopalan, S.; Carja, O.; Fagny, M.; Patin, E.; Myrick, J.W.; McEwen, L.M.; Mah, S.M.; Kobor, M.S.; Froment, A.; Feldman, M.W.; et al. Trends in DNA Methylation with Age Replicate Across Diverse Human Populations. Genetics 2017, 206, 1659–1674. [Google Scholar] [CrossRef] [PubMed]
- Marcogliese, P.C.; Shashi, V.; Spillmann, R.C.; Stong, N.; Rosenfeld, J.A.; Koenig, M.K.; Martínez-Agosto, J.A.; Herzog, M.; Chen, A.H.; Dickson, P.I.; et al. IRF2BPL Is Associated with Neurological Phenotypes. Am. J. Hum. Genet. 2018, 103, 245–260. [Google Scholar] [CrossRef]
- Zang, W.; Zheng, X. Structure and functions of cellular redox sensor HSCARG/NMRAL1, a linkage among redox status, innate immunity, DNA damage response, and cancer. Free Radic. Biol. Med. 2020, 160, 768–774. [Google Scholar] [CrossRef]
- Di Gregoli, K.; Somerville, M.; Bianco, R.; Thomas, A.C.; Frankow, A.; Newby, A.C.; George, S.J.; Jackson, C.L.; Johnson, J.L. Galectin-3 Identifies a Subset of Macrophages With a Potential Beneficial Role in Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1491–1509. [Google Scholar] [CrossRef]
- Kuht, H.J.; Han, J.; Maconachie, G.D.E.; Park, S.E.; Lee, S.T.; McLean, R.; Sheth, V.; Hisaund, M.; Dawar, B.; Sylvius, N.; et al. SLC38A8 mutations result in arrested retinal development with loss of cone photoreceptor specialization. Hum. Mol. Genet. 2020, 29, 2989–3002. [Google Scholar] [CrossRef]
- Yang, J.Y.; Deng, X.Y.; Li, Y.S.; Ma, X.C.; Feng, J.X.; Yu, B.; Chen, Y.; Luo, Y.L.; Wang, X.; Chen, M.L.; et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nat. Commun. 2018, 9, 1165. [Google Scholar] [CrossRef]
- Kanno, T.; Lin, W.D.; Fu, J.L.; Matzke, A.J.M.; Matzke, M. A genetic screen implicates a CWC16/Yju2/CCDC130 protein and SMU1 in alternative splicing in Arabidopsis thaliana. Rna 2017, 23, 1068–1079. [Google Scholar] [CrossRef]
- Dreiza, C.M.; Komalavilas, P.; Furnish, E.J.; Flynn, C.R.; Sheller, M.R.; Smoke, C.C.; Lopes, L.B.; Brophy, C.M. The small heat shock protein, HSPB6, in muscle function and disease. Cell Stress Chaperones 2010, 15, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Krueger, S.; Bernhardt, A.; Kalinski, T.; Baldensperger, M.; Zeh, M.; Teller, A.; Adolf, D.; Reinheckel, T.; Roessner, A.; Kuester, D. Induction of premalignant host responses by cathepsin x/z-deficiency in Helicobacter pylori-infected mice. PLoS ONE 2013, 8, e70242. [Google Scholar] [CrossRef]
- Kumar, C.C.; Mohan, S.R.; Zavodny, P.J.; Narula, S.K.; Leibowitz, P.J. Characterization and differential expression of human vascular smooth muscle myosin light chain 2 isoform in nonmuscle cells. Biochemistry 1989, 28, 4027–4035. [Google Scholar] [CrossRef] [PubMed]
- Qin, N.; Yagel, S.; Momplaisir, M.L.; Codd, E.E.; D’Andrea, M.R. Molecular cloning and characterization of the human voltage-gated calcium channel alpha(2)delta-4 subunit. Mol. Pharmacol. 2002, 62, 485–496. [Google Scholar] [CrossRef]
- Huber, C.; Mårtensson, A.; Bokoch, G.M.; Nemazee, D.; Gavin, A.L. FGD2, a CDC42-specific exchange factor expressed by antigen-presenting cells, localizes to early endosomes and active membrane ruffles. J. Biol. Chem. 2008, 283, 34002–34012. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Calarco, J.A.; Zhang, Y. EOL-1, the homolog of the mammalian Dom3Z, regulates olfactory learning in C. elegans. J. Neurosci. 2014, 34, 13364–13370. [Google Scholar] [CrossRef]
- Apicco, D.J.; Shlevkov, E.; Nezich, C.L.; Tran, D.T.; Guilmette, E.; Nicholatos, J.W.; Bantle, C.M.; Chen, Y.; Glajch, K.E.; Abraham, N.A.; et al. The Parkinson’s disease-associated gene ITPKB protects against α-synuclein aggregation by regulating ER-to-mitochondria calcium release. Proc. Natl. Acad. Sci. USA 2021, 118, e2006476118. [Google Scholar] [CrossRef]
- Uchiyama, Y.; Sakaguchi, M.; Terabayashi, T.; Inenaga, T.; Inoue, S.; Kobayashi, C.; Oshima, N.; Kiyonari, H.; Nakagata, N.; Sato, Y.; et al. Kif26b, a kinesin family gene, regulates adhesion of the embryonic kidney mesenchyme. Proc. Natl. Acad. Sci. USA 2010, 107, 9240–9245. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.C.; Majidi, S.P.; Kong, L.; Nemera, M.; Ferguson, C.J.; Moore, M.; Goncalves, T.M.; Liu, H.K.; Fitzpatrick, J.A.J.; Zhao, G.; et al. CHARGE syndrome protein CHD7 regulates epigenomic activation of enhancers in granule cell precursors and gyrification of the cerebellum. Nat. Commun. 2021, 12, 5702. [Google Scholar] [CrossRef]
- Lin, J.X.; Leonard, W.J. The role of Stat5a and Stat5b in signaling by IL-2 family cytokines. Oncogene 2000, 19, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.S.; Weickert, C.S.; Garner, B. Role of ATP-binding cassette transporters in brain lipid transport and neurological disease. J. Neurochem. 2008, 104, 1145–1166. [Google Scholar] [CrossRef]
- Dib, S.; Pahnke, J.; Gosselet, F. Role of ABCA7 in Human Health and in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 4603. [Google Scholar] [CrossRef]
- Steagall, R.J.; Rusiñol, A.E.; Truong, Q.A.; Han, Z. HSPA12B is predominantly expressed in endothelial cells and required for angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2012–2018. [Google Scholar] [CrossRef] [PubMed]
- Przybylowicz, P.K.; Klepinowski, T.; Taryma-Leśniak, O.; Bińkowski, J.; Wojdacz, T.K. Evaluation of methylation changes in blood cells of COVID-19 patients as a biomarker of severity of the infection. BMC Infect. Dis. 2025, 25, 778. [Google Scholar] [CrossRef]
- Balnis, J.; Madrid, A.; Drake, L.A.; Vancavage, R.; Tiwari, A.; Patel, V.J.; Ramos, R.B.; Schwarz, J.J.; Yucel, R.; Singer, H.A.; et al. Blood DNA methylation in post-acute sequelae of COVID-19 (PASC): A prospective cohort study. EBioMedicine 2024, 106, 105251. [Google Scholar] [CrossRef]
- Granvik, C.; Persson, I.-L.; Barros, G.W.F.; Ahlm, C.; Forsell, M.N.E.; Tevell, S.; Sundh, J.; Blomberg, A.; Lind, A.; Cajander, S.; et al. Long-Term Physical Capacity Following COVID-19: A Prospective, Three-Year Study. medRxiv 2025. [Google Scholar] [CrossRef]
- Pagliaro, P.; Penna, C. ACE/ACE2 Ratio: A Key Also in 2019 Coronavirus Disease (Covid-19)? Front. Med. 2020, 7, 335. [Google Scholar] [CrossRef]
- Carruthers, B.M.; Jain, A.K.; De Meirleir, K.L.; Peterson, D.L.; Klimas, N.G.; Lerner, A.M.; Bested, A.C.; Flor-Henry, P.; Joshi, P.; Powles, A.C.P.; et al. Myalgic Encephalomyelitis/Chronic Fatigue Syndrome. J. Chronic Fatigue Syndr. 2003, 11, 7–115. [Google Scholar] [CrossRef]
- WHO. A Clinical Case Definition of Post COVID-19 Condition by a Delphi Consensus. 2021. Available online: https://www.who.int/publications/i/item/WHO-2019-nCoV-Post_COVID-19_condition-Clinical_case_definition-2021.1 (accessed on 6 October 2021).
- Peppercorn, K.; Edgar, C.; Al Momani, S.; Rodger, E.J.; Tate, W.P.; Chatterjee, A. Application of DNA Methylome Analysis to Patients with ME/CFS. Methods Mol. Biol. 2025, 2920, 141–160. [Google Scholar] [CrossRef]
- Ludgate, J.L.; Wright, J.; Stockwell, P.A.; Morison, I.M.; Eccles, M.R.; Chatterjee, A. A streamlined method for analysing genome-wide DNA methylation patterns from low amounts of FFPE DNA. BMC Med. Genomics 2017, 10, 54. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Stockwell, P.A.; Almomani, S.; Eccles, M.R.; Chatterjee, A. Protocol for generating high-quality genome-scale DNA methylation sequencing data from human cancer biospecimens. STAR Protoc. 2023, 4, 102714. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, P.A.; Chatterjee, A.; Rodger, E.J.; Morison, I.M. DMAP: Differential methylation analysis package for RRBS and WGBS data. Bioinformatics 2014, 30, 1814–1822. [Google Scholar] [CrossRef]
- Chatterjee, A.; Stockwell, P.A.; Rodger, E.J.; Morison, I.M. Comparison of alignment software for genome-wide bisulphite sequence data. Nucleic. Acids. Res. 2012, 40, e79. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chr | Start | End | Location | GeneID | %Difference LC v. HC | %Difference ME v. HC | %Difference LC v. ME |
|---|---|---|---|---|---|---|---|
| 18 | 34854648 | 34854693 | intron | CELF4 | −13.9 | +15.1 | −29.0 |
| 1 | 90074598 | 90074699 | Intergenic | −36.4 | −15.7 | −20.7 | |
| 16 | 4527540 | 4527641 | Promoter | NMRAL1 | −32.0 | −12.5 | −19.5 |
| 5 | 39185995 | 39186145 | intron | FYB | −33.3 | −14.5 | −18.8 |
| 2 | 240241154 | 240241261 | Intergenic | +13.4 | +32.2 | −18.8 | |
| 7 | 158766236 | 158766379 | Intergenic | +18.4 | +35.4 | −17.0 | |
| 8 | 61778005 | 61778136 | exon | CHD7 | −29.2 | −14.5 | −14.7 |
| 17 | 76129099 | 76129221 | intron | TMC8 | −24.7 | −13.3 | −11.4 |
| 6 | 30854000 | 30854160 | intron | DDR1 | −22.4 | −11.0 | −11.4 |
| 17 | 75429645 | 75429795 | Intergenic | −21.9 | −10.6 | −11.3 | |
| 1 | 59090260 | 59090360 | Intergenic | +12.4 | +22.8 | −10.6 | |
| 2 | 121955379 | 121955533 | Intergenic | +22.3 | +12.2 | +10.1 | |
| 16 | 3137553 | 3137716 | Intergenic | ZNF205 | +26.1 | +14.7 | +11.4 |
| 19 | 1047184 | 1047347 | exon | ABCA7 | +24.6 | +11.8 | +12.8 |
| 7 | 158250978 | 158251159 | Intergenic | +28.1 | +15.3 | +12.8 | |
| 14 | 55587537 | 55587752 | Promoter | LGALS3 | +28.5 | +14.8 | +13.7 |
| 15 | 75336231 | 75336352 | intron | PPCDC | +26.5 | +10.8 | +15.7 |
| 8 | 58055165 | 58055309 | Intergenic | +27.6 | +11.7 | +15.9 | |
| 21 | 46714776 | 46714890 | Intergenic | +31.5 | +13.8 | +17.7 | |
| 8 | 58055310 | 58055463 | Intergenic | +34.2 | +15.7 | +18.5 | |
| 1 | 19110747 | 19110909 | Intergenic | −16.7 | −35.4 | +18.7 | |
| 17 | 76661321 | 76661487 | Intergenic | +11.8 | −11.3 | +23.1 | |
| 10 | 118025165 | 118025303 | Intergenic | +13.1 | −13.1 | +26.2 | |
| 6 | 36969405 | 36969621 | Promoter | FGD2 | +21.7 | −11.1 | +32.8 |
| 20 | 3732943 | 3733092 | exon | HSPA12B | +18.3 | −15.6 | +33.9 |
| 3 | 126945870 | 126946029 | Intergenic | +11.4 | −25.9 | +37.3 |
| A. Promoters | ||||||
|---|---|---|---|---|---|---|
| Chromosome | Start | End | Associated Genes | DM * (%) (LC vs. HC) | DM * (%) (HC vs. ME) | DM * (%) (LC vs. ME) |
| 19 | 13841885 | 13841989 | CCDC130 | +14.2 | +11.8 | +2.4 |
| 14 | 77495636 | 77495807 | IRF2BPL | −12.2 | −10.2 | −2.0 |
| 17 | 33776642 | 33776791 | SLFN13 | +18.1 | +11.7 | +6.4 |
| 16 | 84076941 | 84077080 | SLC38A8 | +12.7 | +13.0 | −0.3 |
| 19 | 36249868 | 36250044 | HSPB6 | +11.1 | +10.3 | +0.8 |
| 16 | 4527540 | 4527641 | NMRAL1 | −32.0 | −12.5 | −19.5 |
| 14 | 55587537 | 55587752 | LGALS3 | +28.5 | +14.8 | +13.7 |
| 20 | 57581333 | 57581441 | CTSZ | +21.2 | +18.2 | +2.8 |
| 20 | 35170171 | 35170286 | MYL9 | +13.2 | +11.6 | +1.6 |
| 12 | 2027243 | 2027352 | CACNA2D4 | +17.7 | +17.6 | +0.1 |
| 6 | 36969405 | 36969621 | FGD2 | +21.7 | −11.1 | +32.8 |
| 6 | 31939186 | 31939321 | DOM3Z | +21.4 | +15.6 | +5.8 |
| B. Exons | ||||||
| 1 | 245851466 | 245851609 | KIF26B | −19.0 | −18.7 | −0.3 |
| 8 | 61778005 | 61778136 | CHD7 | −29.2 | −14.5 | −14.7 |
| 1 | 226821736 | 226821914 | ITPKB | −12.1 | −13.1 | +1.0 |
| 17 | 40463432 | 40463555 | STAT5A | +16.2 | +11.4 | +4.8 |
| 20 | 3732943 | 3733092 | HSPA12B | +18.3 | −15.6 | +33.9 |
| 19 | 1047184 | 1047347 | ABCA7 | +24.6 | +11.8 | +12.8 |
| Patient | Age | Sex | Patient | Age | Sex | Control | Age | Sex |
|---|---|---|---|---|---|---|---|---|
| ME030 | 40 | F | LC01 | 43 | F | HC18 | 46 | F |
| ME028 | 19 | F | LC02 | 27 | F | HC39 | 26 | F |
| ME027 | 65 | F | LC03 | 65 | F | HC10 | 59 | F |
| ME029 | 40 | M | LC04 | 42 | M | HC37 | 40 | M |
| ME007 | 27 | F | LC05 | 36 | F | HC38 | 31 | F |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peppercorn, K.; Sharma, S.; Edgar, C.D.; Stockwell, P.A.; Rodger, E.J.; Chatterjee, A.; Tate, W.P. Comparing DNA Methylation Landscapes in Peripheral Blood from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID Patients. Int. J. Mol. Sci. 2025, 26, 6631. https://doi.org/10.3390/ijms26146631
Peppercorn K, Sharma S, Edgar CD, Stockwell PA, Rodger EJ, Chatterjee A, Tate WP. Comparing DNA Methylation Landscapes in Peripheral Blood from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID Patients. International Journal of Molecular Sciences. 2025; 26(14):6631. https://doi.org/10.3390/ijms26146631
Chicago/Turabian StylePeppercorn, Katie, Sayan Sharma, Christina D. Edgar, Peter A. Stockwell, Euan J. Rodger, Aniruddha Chatterjee, and Warren P. Tate. 2025. "Comparing DNA Methylation Landscapes in Peripheral Blood from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID Patients" International Journal of Molecular Sciences 26, no. 14: 6631. https://doi.org/10.3390/ijms26146631
APA StylePeppercorn, K., Sharma, S., Edgar, C. D., Stockwell, P. A., Rodger, E. J., Chatterjee, A., & Tate, W. P. (2025). Comparing DNA Methylation Landscapes in Peripheral Blood from Myalgic Encephalomyelitis/Chronic Fatigue Syndrome and Long COVID Patients. International Journal of Molecular Sciences, 26(14), 6631. https://doi.org/10.3390/ijms26146631