
Machine Learning Framework for Ovarian Cancer Diagnostics Using Plasma Lipidomics and Metabolomics

 , ,
, , 
Abstract
1. Introduction
2. Results
2.1. Clinical Characteristics of Study Participants
2.2. Plasma Lipidome/Metabolome Data
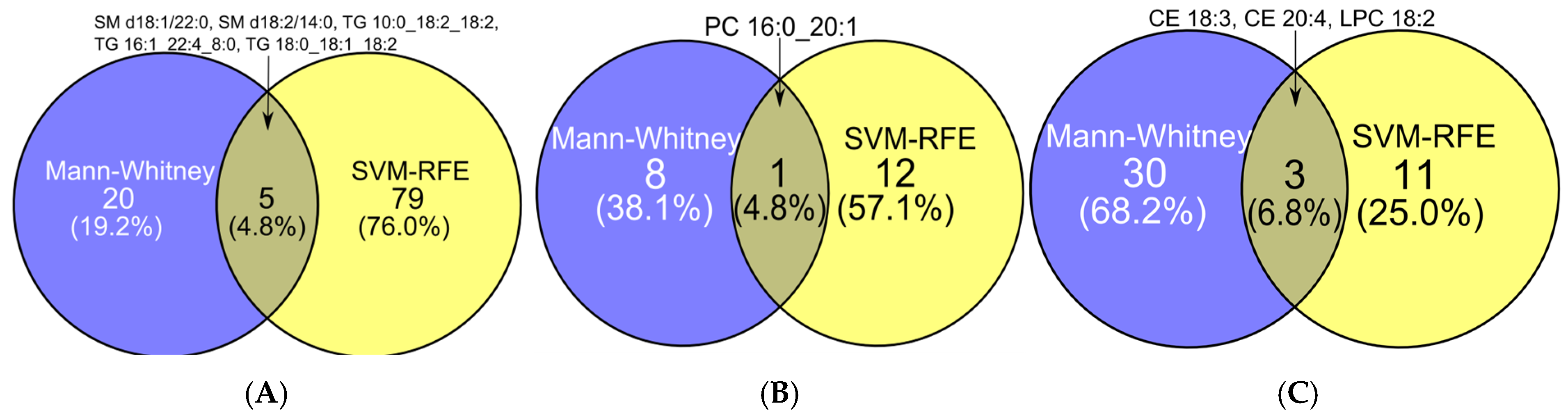
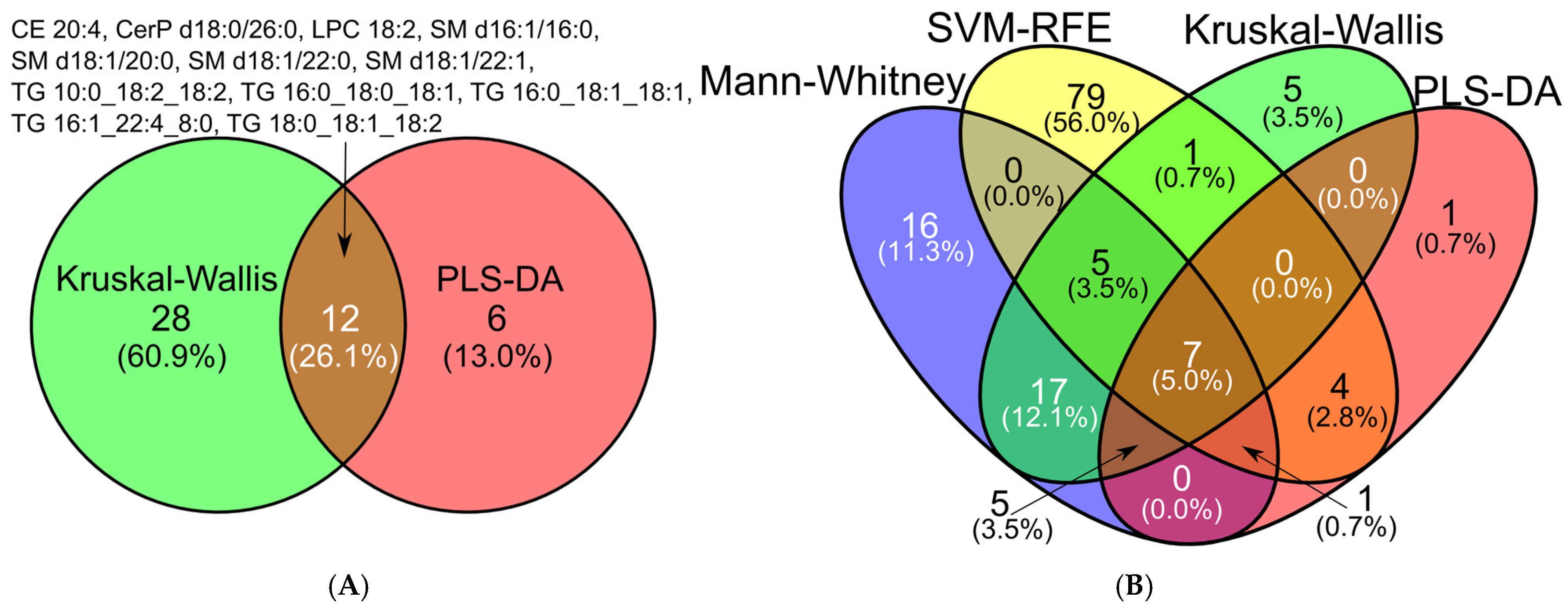
2.3. Feature Selection
2.3.1. Comparative Performance and Stability of Feature Selection Methods in Binary Classification
2.3.2. Comparative Performance and Stability of Feature Selection Methods in Multiclass Analysis
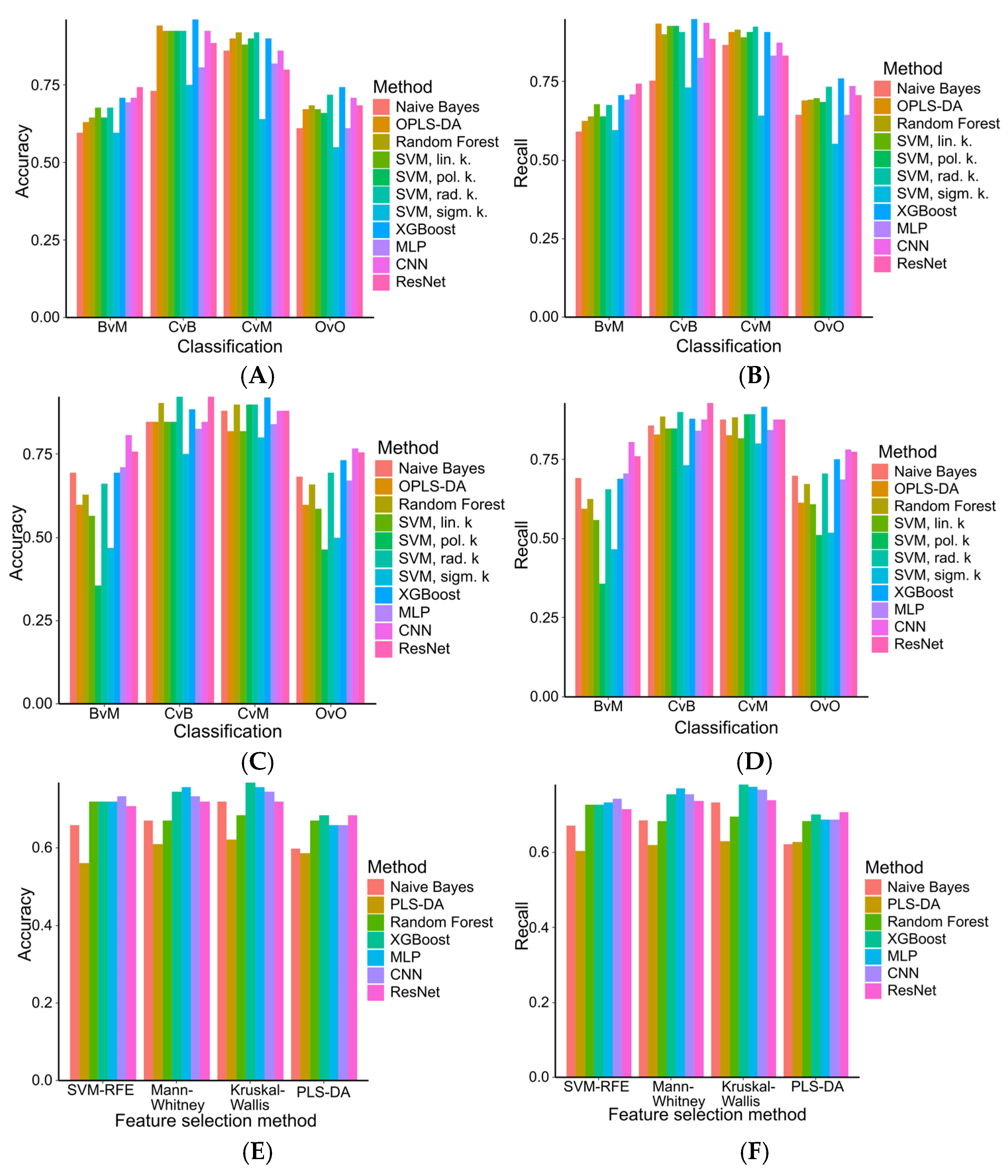
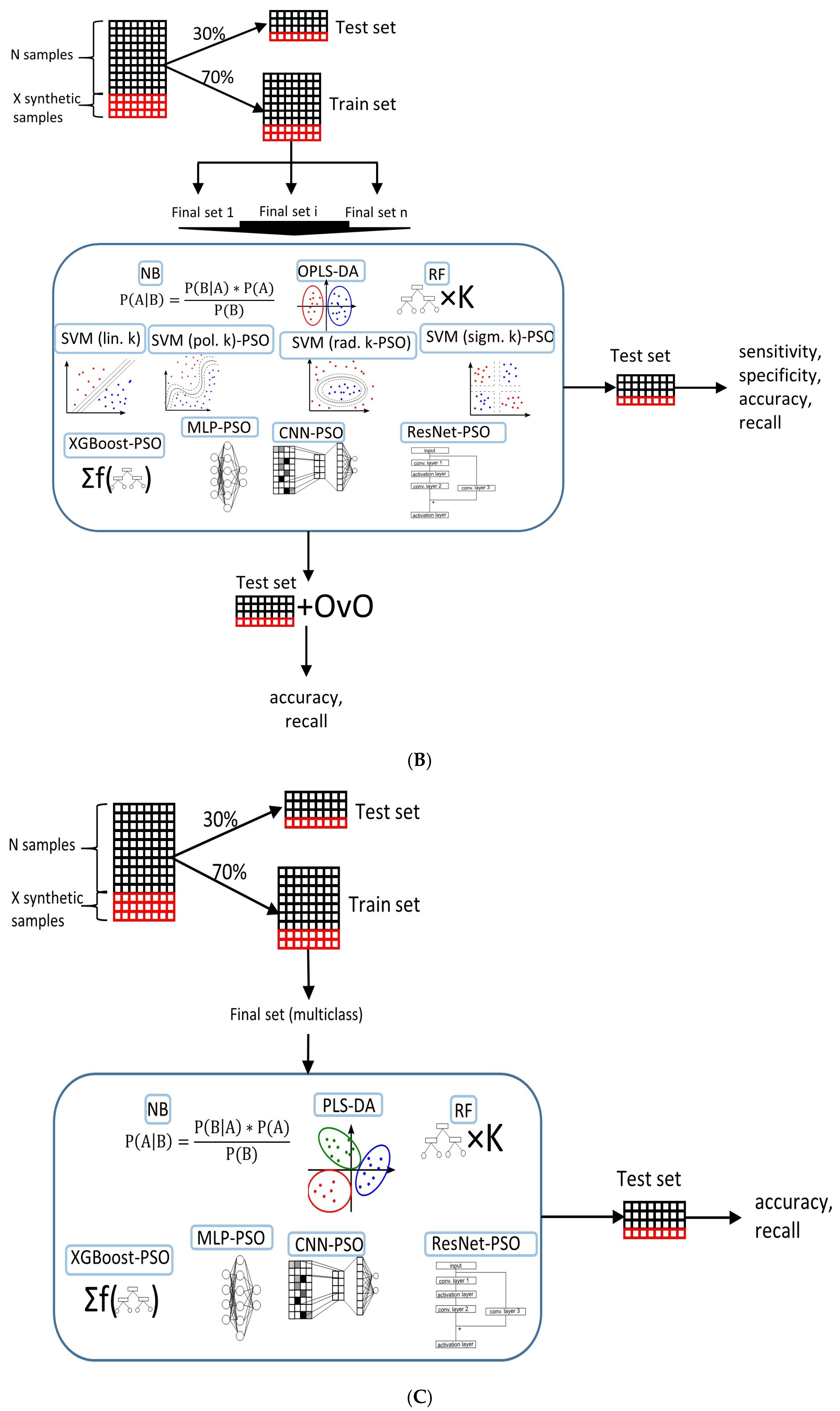
2.4. Machine Learning Models in Ovarian Tumor Classification
3. Discussion
4. Materials and Methods
4.1. Study Design
4.2. Lipidomic Analysis of Blood Plasma Samples (HPLC-MS)
4.3. Metabolomic Analysis by NMR Spectroscopy
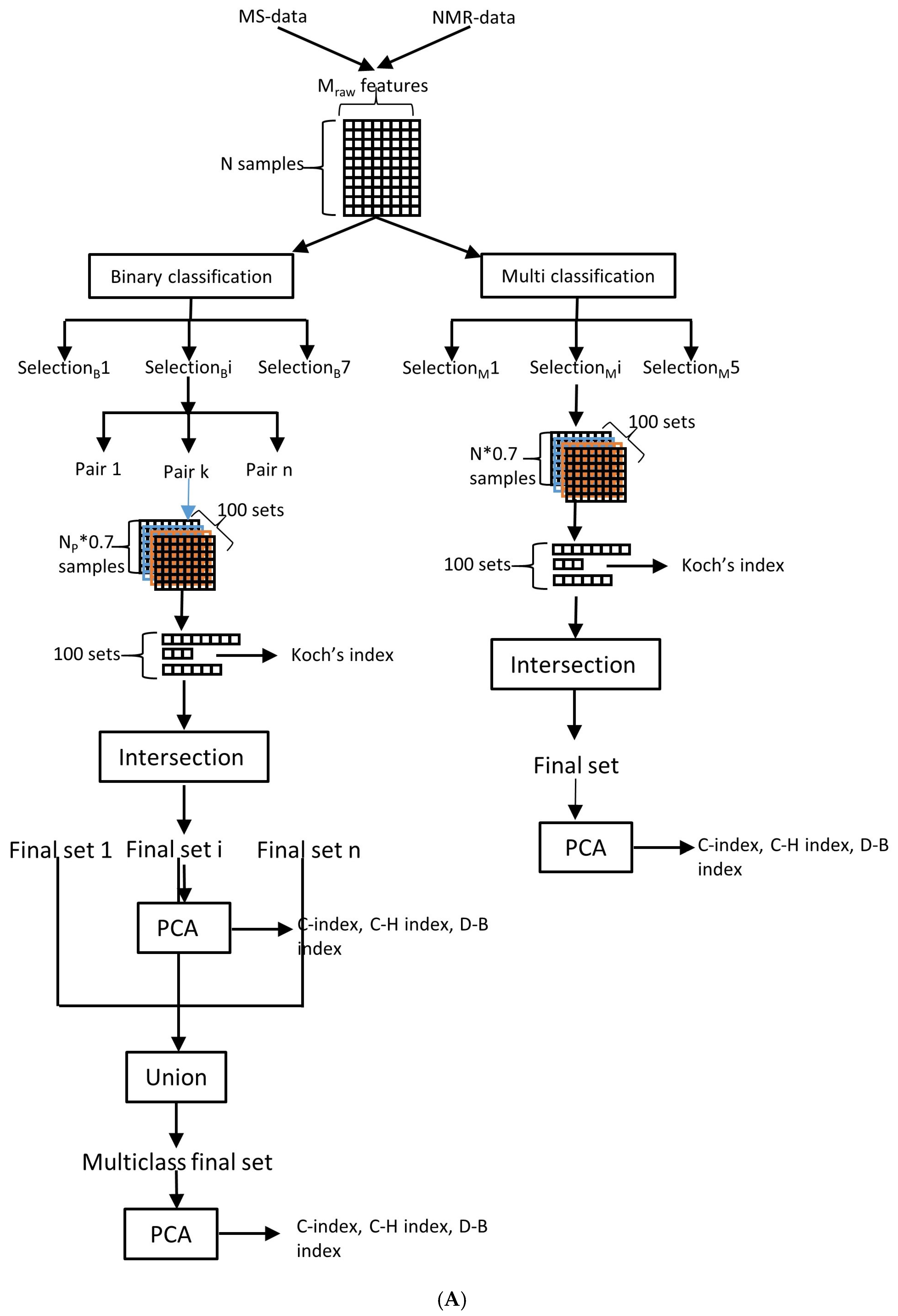
4.4. Feature Selection and Stability Analysis
4.5. Classification Model Selection
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANN | Artificial neural network. |
| CE | Cholesterol esters |
| Cer(P) | Ceramide (phosphate) |
| CNN | Convolutional Neural Network |
| DG | Diacylglycerols |
| FIGO | International Federation of Gynecology and Obstetrics |
| LASSO | Least Absolute Shrinkage and Selection operator |
| MLP | Multilayer Perceptron |
| NB | Naive Bayes |
| OPLS-DA | Orthogonal Projection on Latent Structures-Discriminant Analysis |
| PLS-DA | Partial Least Squares Discriminant Analysis |
| PC (P-/O-) | (plasmenyl-/plasmanyl) Phosphatidylcholine |
| PE (P-/O-) | (plasmenyl-/plasmanyl) Phosphatidylethanolamine |
| PSO | Particle Swarm Optimization |
| ResNet | Residual Convolutional Neural Network |
| RFE | Recursive Feature Elimination |
| RF | Random Forest |
| SM | Sphingomyelins |
| SVM | Support Vector Machine |
| TG | Triacylglycerol |
| XGBoost | Extreme Gradient Boosting |
| VIP | Variable Importance projection |
References
- International Agency for Research on Cancer. Global Cancer Statistics; International Agency for Research on Cancer: Lyon, France, 2022. [Google Scholar]
- Liest, A.L.; Omran, A.S.; Mikiver, R.; Rosenberg, P.; Uppugunduri, S. RMI and ROMA are equally effective in discriminating between benign and malignant gynecological tumors: A prospective population-based study. Acta Obstet. Gynecol. Scand. 2019, 98, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.T.; Webber, E.M.; Sawaya, G.F. Screening for Ovarian Cancer: An Updated Evidence Review for the U.S. Preventive Services Task Force; Agency for Healthcare Research and Quality (US): Rockville, MD, USA, 2018. [Google Scholar]
- Matsas, A.; Stefanoudakis, D.; Troupis, T.; Kontzoglou, K.; Eleftheriades, M.; Christopoulos, P.; Panoskaltsis, T.; Stamoula, E.; Iliopoulos, D.C. Tumor Markers and Their Diagnostic Significance in Ovarian Cancer. Life 2023, 13, 1689. [Google Scholar] [CrossRef]
- Gupta, R.K.; Dholariya, S.; Radadiya, M.; Agarwal, P. NGAL/MMP-9 as a Biomarker for Epithelial Ovarian Cancer: A Case–Control Diagnostic Accuracy Study Rohit. Saudi J. Med. Med. Sci. 2022, 10, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Pawlik, W.; Pawlik, J.; Kozłowski, M.; Łuczkowska, K.; Kwiatkowski, S.; Kwiatkowska, E.; Machaliński, B.; Cymbaluk-Płoska, A. The clinical importance of il-6, il-8, and tnf-α in patients with ovarian carcinoma and benign cystic lesions. Diagnostics 2021, 11, 1625. [Google Scholar] [CrossRef]
- Sakares, W.; Wongkhattiya, W.; Vichayachaipat, P.; Chaiwut, C.; Yodsurang, V.; Nutthachote, P. Accuracy of CCL20 expression level as a liquid biopsy-based diagnostic biomarker for ovarian carcinoma. Front. Oncol. 2022, 12, 1038835. [Google Scholar] [CrossRef] [PubMed]
- De Silva, S.; Alli-Shaik, A.; Gunaratne, J. Machine Learning-Enhanced Extraction of Biomarkers for High-Grade Serous Ovarian Cancer from Proteomics Data. Sci. Data 2024, 11, 685. [Google Scholar] [CrossRef]
- Ning, L.; Lang, J.; Wu, L. Plasma circN4BP2L2 is a promising novel diagnostic biomarker for epithelial ovarian cancer. BMC Cancer 2022, 22, 6. [Google Scholar] [CrossRef]
- Rong, J.; Sun, G.; Zhu, J.; Zhu, Y.; Chen, Z. Combination of plasma-based lipidomics and machine learning provides a useful diagnostic tool for ovarian cancer. J. Pharm. Biomed. Anal. 2025, 253, 116559. [Google Scholar] [CrossRef]
- Long, F.; Pu, X.Y.; Wang, X.; Ma, D.X.; Gao, S.H.; Shi, J.; Zhong, X.C.; Ran, R.; Wang, L.L.; Chen, Z.; et al. A metabolic fingerprint of ovarian cancer: A novel diagnostic strategy employing plasma EV-based metabolomics and machine learning algorithms. J. Ovarian Res. 2025, 18, 26. [Google Scholar] [CrossRef]
- Chagovets, V.; Starodubtseva, N.; Tokareva, A.; Novoselova, A.; Patysheva, M.; Larionova, I.; Prostakishina, E.; Rakina, M.; Kazakova, A.; Topolnitskiy, E.; et al. Specific changes in amino acid profiles in monocytes of patients with breast, lung, colorectal and ovarian cancers. Front. Immunol. 2023, 14, 1332043. [Google Scholar] [CrossRef]
- Iurova, M.V.; Chagovets, V.V.; Pavlovich, S.V.; Starodubtseva, N.L.; Khabas, G.N.; Chingin, K.S.; Tokareva, A.O.; Sukhikh, G.T.; Frankevich, V.E. Lipid Alterations in Early-Stage High-Grade Serous Ovarian Cancer. Front. Mol. Biosci. 2022, 9, 770983. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhao, L.; Jiang, Y.; Li, L.; Guo, M.; Mu, Y.; Zhu, H. Integrated analysis of plasma and urine reveals unique metabolomic profiles in idiopathic inflammatory myopathies subtypes. J. Cachexia Sarcopenia Muscle 2022, 13, 2456–2472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.D.; Xue, C.; Kolachalama, V.B.; Donald, W.A. Interpretable Machine Learning on Metabolomics Data Reveals Biomarkers for Parkinson’s Disease. ACS Cent. Sci. 2023, 9, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Yan, Q.; He, D.; Walker, D.I.; Uppal, K.; Wang, X.; Orimoloye, H.T.; Jones, D.P.; Ritz, B.R.; Heck, J.E. The neonatal blood spot metabolome in retinoblastoma. EJC Paediatr. Oncol. 2023, 2, 100123. [Google Scholar] [CrossRef]
- Pyragius, C.E.; Fuller, M.; Ricciardelli, C.; Oehler, M.K. Aberrant lipid metabolism: An emerging diagnostic and therapeutic target in ovarian cancer. Int. J. Mol. Sci. 2013, 14, 7742–7756. [Google Scholar] [CrossRef]
- Ahmed-Salim, Y.; Galazis, N.; Bracewell-Milnes, T.; Phelps, D.L.; Jones, B.P.; Chan, M.; Munoz-Gonzales, M.D.; Matsuzono, T.; Smith, J.R.; Yazbek, J.; et al. The application of metabolomics in ovarian cancer management: A systematic review. Int. J. Gynecol. Cancer 2021, 31, 754–774. [Google Scholar] [CrossRef]
- Fan, L.; Zhang, W.; Yin, M.; Zhang, T.; Wu, X.; Zhang, H.; Sun, M.; Li, Z.; Hou, Y.; Zhou, X.; et al. Identification of metabolic biomarkers to diagnose epithelial ovarian cancer using a UPLC/QTOF/MS platform. Acta Oncol. 2012, 51, 473–479. [Google Scholar] [CrossRef]
- Wörheide, M.A.; Krumsiek, J.; Kastenmüller, G.; Arnold, M. Multi-omics integration in biomedical research—A metabolomics-centric review. Anal. Chim. 2021, 1141, 144–162. [Google Scholar] [CrossRef]
- Papoutsoglou, G.; Tarazona, S.; Lopes, M.B.; Klammsteiner, T.; Ibrahimi, E.; Eckenberger, J.; Novielli, P.; Tonda, A.; Simeon, A.; Shigdel, R.; et al. Machine learning approaches in microbiome research: Challenges and best practices. Front. Microbiol. 2023, 14, 1261889. [Google Scholar] [CrossRef]
- Brix, F.; Demetrowitsch, T.; Jensen-Kroll, J.; Zacharias, H.U.; Szymczak, S.; Laudes, M.; Schreiber, S.; Schwarz, K. Evaluating the Effect of Data Merging and Postacquisition Normalization on Statistical Analysis of Untargeted High-Resolution Mass Spectrometry Based Urinary Metabolomics Data. Anal. Chem. 2024, 96, 33–40. [Google Scholar] [CrossRef]
- Chua, A.E.; Pfeifer, L.D.; Sekera, E.R.; Hummon, A.B.; Desaire, H. Workflow for Evaluating Normalization Tools for Omics Data Using Supervised and Unsupervised Machine Learning. J. Am. Soc. Mass Spectrom. 2023, 34, 2775–2784. [Google Scholar] [CrossRef] [PubMed]
- Tokareva, A.; Starodubtseva, N.; Frankevich, V.; Silachev, D. Minimizing Cohort Discrepancies: A Comparative Analysis of Data Normalization Approaches in Biomarker Research. Computation 2024, 12, 137. [Google Scholar] [CrossRef]
- Tokareva, A.O.; Chagovets, V.V.; Kononikhin, A.S.; Starodubtseva, N.L.; Nikolaev, E.N.; Frankevich, V.E. Comparison of the effectiveness of variable selection method for creating a diagnostic panel of biomarkers for mass spectrometric lipidome analysis. J. Mass Spectrom. 2021, 56, e4702. [Google Scholar] [CrossRef] [PubMed]
- Abd-Elnaby, M.; Alfonse, M.; Roushdy, M. Classification of breast cancer using microarray gene expression data: A survey. J. Biomed. Inform. 2021, 117, 103764. [Google Scholar] [CrossRef]
- Pudjihartono, N.; Fadason, T.; Kempa-Liehr, A.W.; O’Sullivan, J.M. A Review of Feature Selection Methods for Machine Learning-Based Disease Risk Prediction. Front. Bioinform. 2022, 2, 927312. [Google Scholar] [CrossRef]
- Wu, Z.; Chen, H.; Ke, S.; Mo, L.; Qiu, M.; Zhu, G.; Zhu, W.; Liu, L. Identifying potential biomarkers of idiopathic pulmonary fibrosis through machine learning analysis. Sci. Rep. 2023, 13, 16559. [Google Scholar] [CrossRef]
- Tian, Y.; Tao, K.; Li, S.; Chen, X.; Wang, R.; Zhang, M.; Zhai, Z. Identification of m6A-Related Biomarkers in Systemic Lupus Erythematosus: A Bioinformation-Based Analysis. J. Inflamm. Res. 2024, 17, 507–526. [Google Scholar] [CrossRef]
- Zhu, T.; Ma, Y.; Wang, J.; Xiong, W.; Mao, R.; Cui, B.; Min, Z.; Song, Y.; Chen, Z. Serum Metabolomics Reveals Metabolomic Profile and Potential Biomarkers in Asthma. Allergy Asthma Immunol. Res. 2024, 16, 235–252. [Google Scholar] [CrossRef]
- Chardin, D.; Humbert, O.; Bailleux, C.; Burel-Vandenbos, F.; Rigau, V.; Pourcher, T.; Barlaud, M. Primal-dual for classification with rejection (PD-CR): A novel method for classification and feature selection—An application in metabolomics studies. BMC Bioinform. 2021, 22, 594. [Google Scholar] [CrossRef]
- Zhou, D.; Zhu, W.; Sun, T.; Wang, Y.; Chi, Y.; Chen, T.; Lin, J. iMAP: A Web Server for Metabolomics Data Integrative Analysis. Front. Chem. 2021, 9, 659656. [Google Scholar] [CrossRef]
- Alamro, H.; Thafar, M.A.; Albaradei, S.; Gojobori, T.; Essack, M.; Gao, X. Exploiting machine learning models to identify novel Alzheimer’s disease biomarkers and potential targets. Sci. Rep. 2023, 13, 4979. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Greenwood, P.; Klein, M.S. Deep learning for rapid identification of microbes using metabolomics profiles. Metabolites 2021, 11, 863. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhou, Z.; Dong, J.; Fu, Y.; Li, Y.; Luan, Z.; Peng, X. Predicting breast cancer 5-year survival using machine learning: A systematic review. PLoS ONE 2021, 16, e0250370. [Google Scholar] [CrossRef]
- Chagovets, V.V.; Vasil’ev, V.G.; Iurova, M.V.; Khabas, G.N.; Pavlovich, S.V.; Starodubtseva, N.L.; Mayboroda, O.A. Metabolic “footprints” of the circulating cancer mucins: CA125 in the high-grade ovarian cancer. Bull. Russ. State Med. Univ. 2021, 6, 10–16. [Google Scholar] [CrossRef]
- Pal, M.; Foody, G.M. Feature selection for classification of hyperspectral data by SVM. IEEE Trans. Geosci. Remote Sens. 2010, 48, 2297–2307. [Google Scholar] [CrossRef]
- Gonzalez-Covarrubias, V. Lipidomics in longevity and healthy aging. Biogerontology 2013, 14, 663–672. [Google Scholar] [CrossRef]
- Naudin, S.; Sampson, J.N.; Moore, S.C.; Albanes, D.; Freedman, N.D.; Weinstein, S.J.; Stolzenberg-Solomon, R. Lipidomics and pancreatic cancer risk in two prospective studies. Eur. J. Epidemiol. 2023, 38, 783–793. [Google Scholar] [CrossRef]
- Li, F.; Qin, X.; Chen, H.; Qiu, L.; Guo, Y.; Liu, H.; Chen, G.; Song, G.; Wang, X.; Li, F.; et al. Lipid profiling for early diagnosis and progression of colorectal cancer using direct-infusion electrospray ionization Fourier transform ion cyclotron resonance mass spectrometry. Rapid Commun. Mass Spectrom. 2013, 27, 24–34. [Google Scholar] [CrossRef]
- Trabert, B.; Hathaway, C.A.; Rice, M.S.; Rimm, E.B.; Sluss, P.M.; Terry, K.L.; Zeleznik, O.A.; Tworoger, S.S. Ovarian Cancer Risk in Relation to Blood Cholesterol and Triglycerides. Cancer Epidemiol. Biomark. Prev. A Publ. Am. Assoc. Cancer Res. Cosponsored Am. Soc. Prev. Oncol. 2021, 30, 2044–2051. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, D.; Luo, Y.; Guo, S.; Wang, T.; Liu, J.; Liu, Y.; Li, Z. Tissue and serum lipidome shows altered lipid composition with diagnostic potential in mycosis fungoides. Oncotarget 2017, 8, 48041–48050. [Google Scholar] [CrossRef]
- Cotte, A.K.; Cottet, V.; Aires, V.; Mouillot, T.; Rizk, M.; Vinault, S.; Binquet, C.; De Barros, J.P.P.; Hillon, P.; Delmas, D. Phospholipid profiles and hepatocellular carcinoma risk and prognosis in cirrhotic patients. Oncotarget 2019, 10, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- López, N.C.; García-Ordás, M.T.; Vitelli-Storelli, F.; Fernández-Navarro, P.; Palazuelos, C.; Alaiz-Rodríguez, R. Evaluation of feature selection techniques for breast cancer risk prediction. Int. J. Environ. Res. Public Health 2021, 18, 10670. [Google Scholar] [CrossRef] [PubMed]
- Okser, S.; Pahikkala, T.; Aittokallio, T. Genetic variants and their interactions in disease risk prediction—Machine learning and network perspectives. BioData Min. 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, M.C.; Grisci, B.I.; Dorn, M. Analysis and comparison of feature selection methods towards performance and stability. Expert Syst. Appl. 2024, 249, 123667. [Google Scholar] [CrossRef]
- He, Z.; Yu, W. Stable feature selection for biomarker discovery. Comput. Biol. Chem. 2010, 34, 215–225. [Google Scholar] [CrossRef]
- Mohamed, M.; Abdullah, A.; Zaki, A.M.; Rizk, F.H.; Eid, M.M.; El-Kenway, E.M. Advances and Challenges in Feature Selection Methods: A Comprehensive Review. J. Artif. Intell. Metaheuristics 2024, 7, 67–77. [Google Scholar] [CrossRef]
- Talal, A.A. Abdullah; Mohd Soperi Mohd Zahid; Waleed Ali A Review of Interpretable ML in Healthcare: Taxonomy, Applications, Challenges, and Future Directions. Symmetry 2021, 13, 2439. [Google Scholar]
- Harrison, C.J.; Sidey-Gibbons, C.J. Machine learning in medicine: A practical introduction to natural language processing. BMC Med. Res. Methodol. 2021, 21, 158. [Google Scholar] [CrossRef]
- Ban, D.; Housley, S.N.; Matyunina, L.V.; McDonald, L.D.E.; Bae-Jump, V.L.; Benigno, B.B.; Skolnick, J.; McDonald, J.F. A personalized probabilistic approach to ovarian cancer diagnostics. Gynecol. Oncol. 2024, 182, 168–175. [Google Scholar] [CrossRef]
- Wu, Z.; Zhu, M.; Kang, Y.; Leung, E.L.H.; Lei, T.; Shen, C.; Jiang, D.; Wang, Z.; Cao, D.; Hou, T. Do we need different machine learning algorithms for QSAR modeling? A comprehensive assessment of 16 machine learning algorithms on 14 QSAR data sets. Brief. Bioinform. 2021, 22, bbaa321. [Google Scholar] [CrossRef]
- Farzaneh, F.; Salimnezhad, M.; Hosseini, M.S.; Ganjoei, T.A.; Arab, M.; Talayeh, M. D-dimer, Fibrinogen and Tumor Marker Levels in Patients with benign and Malignant Ovarian Tumorsneovascularization. Asian Pac. J. Cancer Prev. 2023, 24, 4263–4268. [Google Scholar] [CrossRef] [PubMed]
- Hasenburg, A.; Eichkorn, D.; Vosshagen, F.; Obermayr, E.; Geroldinger, A.; Zeillinger, R.; Bossart, M. Biomarker-based early detection of epithelial ovarian cancer based on a five-protein signature in patient’s plasma—A prospective trial. BMC Cancer 2021, 21, 1037. [Google Scholar] [CrossRef] [PubMed]
- Shan, D.; Cheng, S.; Ma, Y.; Peng, H. Serum levels of tumor markers and their clinical significance in epithelial ovarian cancer. J. Cent. South Univ. 2023, 48, 1039–1049. [Google Scholar] [CrossRef]
- Periyasamy, A.; Gopisetty, G.; Subramanium, M.J.; Velusamy, S.; Rajkumar, T. Identification and validation of differential plasma proteins levels in epithelial ovarian cancer. J. Proteom. 2020, 226, 103893. [Google Scholar] [CrossRef]
- Nazarizadeh, A.; Banirostam, T.; Biglari, T.; Kalantarhormozi, M.; Chichagi, F.; Behnoush, A.H.; Habibi, M.A.; Shahidi, R. Integrated neural network and evolutionary algorithm approach for liver fibrosis staging: Can artificial intelligence reduce patient costs? JGH Open 2024, 8, e13075. [Google Scholar] [CrossRef]
- Qaderi, K.; Sharifipour, F.; Dabir, M.; Shams, R.; Behmanesh, A. Artificial intelligence (AI) approaches to male infertility in IVF: A mapping review. Eur. J. Med. Res. 2025, 30, 246. [Google Scholar] [CrossRef]
- Nahar, A.; Paul, S.; Saikia, M.J. A systematic review on machine learning approaches in cerebral palsy research. PeerJ 2024, 12, e18270. [Google Scholar] [CrossRef]
- Smiley, A.; Villarreal-Zegarra, D.; Reategui-Rivera, C.M.; Escobar-Agreda, S.; Finkelstein, J. Methodological and reporting quality of machine learning studies on cancer diagnosis, treatment, and prognosis. Front. Oncol. 2025, 15, 1555247. [Google Scholar] [CrossRef]
- Gómez-Pascual, A.; Naccache, T.; Xu, J.; Hooshmand, K.; Wretlind, A.; Gabrielli, M.; Lombardo, M.T.; Shi, L.; Buckley, N.J.; Tijms, B.M.; et al. Paired plasma lipidomics and proteomics analysis in the conversion from mild cognitive impairment to Alzheimer’s disease. Comput. Biol. Med. 2024, 176, 108588. [Google Scholar] [CrossRef]
- Wang, K.; Theeke, L.A.; Liao, C.; Wang, N.; Lu, Y.; Xiao, D.; Xu, C. Deep learning analysis of UPLC-MS/MS-based metabolomics data to predict Alzheimer’s disease. J. Neurol. Sci. 2023, 453, 120812. [Google Scholar] [CrossRef]
- Zhang, T.H.; Hasib, M.M.; Chiu, Y.C.; Han, Z.F.; Jin, Y.F.; Flores, M.; Chen, Y.; Huang, Y. Transformer for Gene Expression Modeling (T-GEM): An Interpretable Deep Learning Model for Gene Expression-Based Phenotype Predictions. Cancers 2022, 14, 4763. [Google Scholar] [CrossRef] [PubMed]
- Kalkan, H.; Akkaya, U.M.; Inal-Gültekin, G.; Sanchez-Perez, A.M. Prediction of Alzheimer’s Disease by a Novel Image-Based Representation of Gene Expression. Genes 2022, 13, 1406. [Google Scholar] [CrossRef] [PubMed]
- El-Melegy, M.; Mamdouh, A.; Ali, S.; Badawy, M.; El-Ghar, M.A.; Alghamdi, N.S.; El-Baz, A. Prostate Cancer Diagnosis via Visual Representation of Tabular Data and Deep Transfer Learning. Bioengineering 2024, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Karim, A.; Su, Z.; West, P.K.; Keon, M.; The NYGC ALS Consortium; Shamsani, J.; Brennan, S.; Wong, T.; Milicevic, O.; Teunisse, G.; et al. Molecular Classification and Interpretation of Amyotrophic Lateral Sclerosis Using Deep Convolution Neural Networks and Shapley Values. Genes 2021, 12, 1754. [Google Scholar] [CrossRef]
- Lecun, Y.; Bengio, Y.; Hinton, G. Deep learning. Nature 2015, 521, 436–444. [Google Scholar] [CrossRef]
- Ng, W.; Minasny, B.; de Sousa Mendes, W.; Demattê, J.A.M. The influence of training sample size on the accuracy of deep learning models for the prediction of soil properties with near-infrared spectroscopy data. SOIL 2020, 6, 565–578. [Google Scholar] [CrossRef]
- Yilmaz, E.O.; Kavzoglu, T. Analysis of the effect of training sample size on the performance of 2D CNN models. Intercont. Geoinf. Days 2021, 2, 241–244. [Google Scholar]
- Kim, D.; Seo, S.B.; Yoo, N.H.; Shin, G. A Study on Sample Size Sensitivity of Factory Manufacturing Dataset for CNN-Based Defective Product Classification. Computation 2022, 10, 142. [Google Scholar] [CrossRef]
- Alenizy, H.A.; Berri, J. Transforming tabular data into images via enhanced spatial relationships for CNN processing. Sci. Rep. 2025, 15, 17004. [Google Scholar] [CrossRef]
- Elmannai, H.; El-Rashidy, N.; Mashal, I.; Alohali, M.A.; Farag, S.; El-Sappagh, S.; Saleh, H. Polycystic Ovary Syndrome Detection Machine Learning Model Based on Optimized Feature Selection and Explainable Artificial Intelligence. Diagnostics 2023, 13, 1056. [Google Scholar] [CrossRef]
- Sah, S.; Bifarin, O.O.; Moore, S.G.; Gaul, D.A.; Chung, H.; Kwon, S.Y.; Cho, H.; Cho, C.H.; Kim, J.H.; Kim, J.; et al. Serum Lipidome Profiling Reveals a Distinct Signature of Ovarian Cancer in Korean Women. Cancer Epidemiol. Biomark. Prev. 2024, 33, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Tanioka, S.; Ishida, F.; Nakano, F.; Kawakita, F.; Kanamaru, H.; Nakatsuka, Y.; Nishikawa, H.; Suzuki, H. Machine Learning Analysis of Matricellular Proteins and Clinical Variables for Early Prediction of Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Mol. Neurobiol. 2019, 56, 7128–7135. [Google Scholar] [CrossRef] [PubMed]
- Belsti, Y.; Moran, L.; Du, L.; Mousa, A.; De Silva, K.; Enticott, J.; Teede, H. Comparison of machine learning and conventional logistic regression-based prediction models for gestational diabetes in an ethnically diverse population; the Monash GDM Machine learning model. Int. J. Med. Inform. 2023, 179, 105228. [Google Scholar] [CrossRef] [PubMed]
- Bunkhumpornpat, C.; Sinapiromsaran, K.; Lursinsap, C. Safe-level-SMOTE: Safe-level-synthetic minority over-sampling technique for handling the class imbalanced problem. Lect. Notes Comput. Sci. (Incl. Subser. Lect. Notes Artif. Intell. Lect. Notes Bioinform.) 2009, 5476 LNAI, 475–482. [Google Scholar] [CrossRef]
- Kivrak, M.; Avci, U.; Uzun, H.; Ardic, C. The Impact of the SMOTE Method on Machine Learning and Ensemble Learning Performance Results in Addressing Class Imbalance in Data Used for Predicting Total Testosterone Deficiency in Type 2 Diabetes Patients. Diagnostics 2024, 14, 2634. [Google Scholar] [CrossRef]
- Ramezankhani, A.; Pournik, O.; Shahrabi, J.; Azizi, F.; Hadaegh, F.; Khalili, D. The impact of oversampling with SMOTE on the performance of 3 classifiers in prediction of type 2 diabetes. Med. Decis. Mak. 2016, 36, 137–144. [Google Scholar] [CrossRef]
- Hassanzadeh, R.; Farhadian, M.; Rafieemehr, H. Hospital mortality prediction in traumatic injuries patients: Comparing different SMOTE-based machine learning algorithms. BMC Med. Res. Methodol. 2023, 23, 101. [Google Scholar] [CrossRef]
- Mohseni-Takalloo, S.; Mohseni, H.; Mozaffari-Khosravi, H.; Mirzaei, M.; Hosseinzadeh, M. The effect of data balancing approaches on the prediction of metabolic syndrome using non-invasive parameters based on random forest. BMC Bioinform. 2024, 25, 18. [Google Scholar] [CrossRef]
- Blagus, R.; Lusa, L. SMOTE for high-dimensional class-imbalanced data. BMC Bioinform. 2013, 14, 106. [Google Scholar] [CrossRef]
- Welvaars, K.; Oosterhoff, J.H.F.; van den Bekerom, M.P.J.; Doornberg, J.N.; van Haarst, E.P.; van der Zee, J.A.; van Andel, G.A.; Lagerveld, B.W.; Hovius, M.C.; Kauer, P.C.; et al. Implications of resampling data to address the class imbalance problem (IRCIP): An evaluation of impact on performance between classification algorithms in medical data. JAMIA Open 2023, 6, ooad033. [Google Scholar] [CrossRef]
- Starodubtseva, N.L.; Tokareva, A.O.; Rodionov, V.V.; Brzhozovskiy, A.G.; Bugrova, A.E.; Chagovets, V.V.; Kometova, V.V.; Kukaev, E.N.; Soares, N.C.; Kovalev, G.I.; et al. Integrating Proteomics and Lipidomics for Evaluating the Risk of Breast Cancer Progression: A Pilot Study. Biomedicines 2023, 11, 1786. [Google Scholar] [CrossRef] [PubMed]
- Tonoyan, N.M.; Chagovets, V.V.; Starodubtseva, N.L.; Tokareva, A.O.; Chingin, K.; Kozachenko, I.F.; Adamyan, L.V.; Frankevich, V.E. Alterations in lipid profile upon uterine fibroids and its recurrence. Sci. Rep. 2021, 11, 11447. [Google Scholar] [CrossRef] [PubMed]
- Koelmel, J.P.; Kroeger, N.M.; Ulmer, C.Z.; Bowden, J.A.; Patterson, R.E.; Cochran, J.A.; Beecher, C.W.W.; Garrett, T.J.; Yost, R.A. LipidMatch: An automated workflow for rule-based lipid identification using untargeted high-resolution tandem mass spectrometry data. BMC Bioinform. 2017, 18, 331. [Google Scholar] [CrossRef] [PubMed]
- Tokareva, A.O.; Chagovets, V.V.; Kononikhin, A.S.; Starodubtseva, N.L.; Nikolaev, E.N.; Frankevich, V.E. Normalization methods for reducing interbatch effect without quality control samples in liquid chromatography-mass spectrometry-based studies. Anal. Bioanal. Chem. 2021, 413, 3479–3486. [Google Scholar] [CrossRef]
- Sud, M.; Fahy, E.; Cotter, D.; Brown, A.; Dennis, E.A.; Glass, C.K.; Merrill, A.H.; Murphy, R.C.; Raetz, C.R.H.; Russell, D.W.; et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007, 35, 527–532. [Google Scholar] [CrossRef]
- Galindo-Prieto, B.; Eriksson, L.; Trygg, J. Variable influence on projection (VIP) for orthogonal projections to latent structures (OPLS). J. Chemom. 2014, 28, 623–632. [Google Scholar] [CrossRef]
- Menze, B.H.; Kelm, B.M.; Masuch, R.; Himmelreich, U.; Bachert, P.; Petrich, W.; Hamprecht, F.A. A comparison of random forest and its Gini importance with standard chemometric methods for the feature selection and classification of spectral data. BMC Bioinform. 2009, 10, 213. [Google Scholar] [CrossRef]
- Guyon, I.; Weston, J.; Barhill, S. Gene selection for cancer classification using Support Vector Machines. Mach. Learn. 2002, 46, 389–422. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection Via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Kursa, M.B.; Rudnicki, W.R. Feature selection with the boruta package. J. Stat. Softw. 2010, 36, 1–13. [Google Scholar] [CrossRef]
- Koch, L.F. Index of Biotal Dispersity. Ecology 1957, 38, 145–148. [Google Scholar] [CrossRef]
- Hubert, L.J.; Levin, J.R. A general statistical framework for assessing categorical clustering in free recall. Psychol. Bull. 1976, 83, 1072–1080. [Google Scholar] [CrossRef]
- Davies, D.L.; Bouldin, D.W. A Cluster Separation Measure. IEEE Trans. Pattern Anal. Mach. Intell. 1979, PAMI-1, 224–227. [Google Scholar] [CrossRef]
- Caliñski, T.; Harabasz, J. A Dendrite Method Foe Cluster Analysis. Commun. Stat. 1974, 3, 1–27. [Google Scholar] [CrossRef]
- Sharma, A.; Vans, E.; Shigemizu, D.; Boroevich, K.A.; Tsunoda, T. DeepInsight: A methodology to transform a non-image data to an image for convolution neural network architecture. Sci. Rep. 2019, 9, 11399. [Google Scholar] [CrossRef]
- Clerc, M.; Kennedy, J. The Particle Swarm—Explosion, Stability, and Convergence in a Multidimensional Complex Space. Mutat. Res. DNAging 2002, 6, 58–73. [Google Scholar] [CrossRef]
- Thévenot, E.A.; Roux, A.; Xu, Y.; Ezan, E.; Junot, C. Analysis of the Human Adult Urinary Metabolome Variations with Age, Body Mass Index, and Gender by Implementing a Comprehensive Workflow for Univariate and OPLS Statistical Analyses. J. Proteome Res. 2015, 14, 3322–3335. [Google Scholar] [CrossRef]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Meyer, D. Support Vector Machines. The Interface to libsvm in package. R News 2024, 8, e107. [Google Scholar]
- Friedman, J.; Hastie, T.; Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 2010, 33, 1–22. [Google Scholar] [CrossRef]
- Chen, T.; Guestrin, C. XGBoost: A scalable tree boosting system. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 13–17 August 2016; Volume 13–17, pp. 785–794. [Google Scholar] [CrossRef]
- Dudek, A.; Walesiak, M. The Choice of Variable Normalization Method in Cluster Analysis. Education Excellence and Innovation Management: A 2025 Vision to Sustain Economic Development during Global Challenges. In Proceedings of the 35th International Business Information Management Association Conference (IBIMA), Seville, Spain, 1–2 April 2020; pp. 325–340. [Google Scholar]
- Siriseriwan, W. A Collection of Oversampling Techniques for Class Imbalance Problem Based on SMOTE 2024. Available online: https://reddertar.r-universe.dev/smotefamily (accessed on 7 July 2025).
- Wild, F. Latent Semantic Analysis 2022. Available online: https://cran.r-project.org/web/packages/lsa/index.html (accessed on 7 July 2025).
- Donaldson, J. T-Distributed Stochastic Neighbor Embedding for R (t-SNE) 2022. Available online: https://CRAN.R-project.org/package=tsne (accessed on 7 July 2025).
- Barber, C.B. Convex Hull in Arbitrary Dimension. 2018. Available online: https://cran.r-project.org/src/contrib/Archive/cxhull/ (accessed on 7 July 2025).
- Kuhn, M. Building predictive models in R using the caret package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef]
- Kalinowski, T.; Falbe, D.; Allaire, J.; Chollet, F.; RStudio; Google; Tang, Y.; Van Der Bijl, W.; Studer, M.; Keydana, S. R Interface to “Keras”. 2024. Available online: https://keras3.posit.co/index.html (accessed on 7 July 2025).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variable | OC (n = 103) | Benign Tumor (n = 107) | Control Group (n = 19) | p Value (Kruskal–Wallis H Test) |
|---|---|---|---|---|
| Age, years, Median (Q1;Q3) | 51.0 (39.0;60.0) | 38.0 (34.0;45.0) | 39.5 (34.0;60.3) | <0.001 |
| BMI (kg/m2), Median (Q1;Q3) | 25.0 (22.0;27.8) | 23.5 (21.0;27.0) | 22.5 (20.8;25.3) | 0.27 |
| Benign ovarian tumors, n (%) | - | cystadenoma—30 (28%) endometrioid cyst—56 (52%) mature teratoma—21(20%) | - | - |
| Borderline tumors, n (%) | 28 (27%) | - | - | - |
| Low-grade OC, n (%) | 16 (16%) | - | - | - |
| FIGO stage (high-grade OC), n (%) | IA—5( 4.9%) IIB—5 (4.9%) IIC—1 (1.0%) IIIA—4 (3.9%) IIIC—40 (39%) IVA—4 (3.9%) | - | - |
| Method | Benign vs. Malignant | Control vs. Benign | Control vs. Malignant | Mean (Score) |
|---|---|---|---|---|
| Mann–Whitney | 0.41 | 0.31 | 0.47 | 0.40 (5) |
| Welch | 0.35 | 0.27 | 0.35 | 0.32 (4) |
| OPLS-DA | 0.50 | 0.51 | 0.57 | 0.53 (6) |
| RF | 0.19 | 0.20 | 0.16 | 0.18 (3) |
| SVM-RFE | 0.94 | 0.59 | 0.71 | 0.75 (7) |
| LASSO | 0.20 | 0.14 | 0.06 | 0.14 (1) |
| Boruta | 0.17 | 0.14 | 0.12 | 0.14 (2) |
| Metric | Method | Benign vs. Malignant | Control vs. Benign | Control vs. Malignant | Combined Feature Set | Mean (Score) |
|---|---|---|---|---|---|---|
| Hubert–Levin’s C index | Mann–Whitney | 0.46 | 0.42 | 0.45 | 0.44 | 0.44 (7) |
| Welch | 0.46 | 0.42 | 0.46 | 0.44 | 0.44 (6) | |
| OPLS-DA | 0.45 | 0.46 | 0.47 | 0.47 | 0.46 (4) | |
| RF | 100.00 | 100.00 | 100.00 | 0.47 | 75.12 (1) | |
| SVM-RFE | 0.47 | 0.44 | 0.46 | 0.45 | 0.45 (5) | |
| LASSO | 0.46 | 100.00 | 100.00 | 0.46 | 50.23 (2) | |
| Boruta | 0.44 | 100.00 | 100.00 | 0.44 | 50.22 (3) | |
| Davies–Bouldin’s index | Mann–Whitney | 3.35 | 4.09 | 3.87 | 3.95 | 3.81 (7) |
| Welch | 3.33 | 4.37 | 4.46 | 4.49 | 4.16 (6) | |
| OPLS-DA | 4.76 | 16.44 | 15.23 | 13.39 | 12.45 (4) | |
| RF | 100.00 | 100.00 | 100.00 | 7.55 | 76.89 (1) | |
| SVM-RFE | 10.75 | 7.43 | 7.66 | 8.77 | 8.65 (5) | |
| LASSO | 2.80 | 100.00 | 100.00 | 2.80 | 51.40 (3) | |
| Boruta | 2.72 | 100.00 | 100.00 | 2.72 | 51.36 (2) | |
| Calinski–Harabasz pseudo-F statistic | Mann–Whitney | −12.79 | 47.35 | −4.53 | 1.53 | 7.89 (4) |
| Welch | −13.42 | 45.04 | 1.10 | 7.99 | 10.18 (5) | |
| OPLS-DA | 4.25 | 30.06 | 24.15 | 23.39 | 20.46 (7) | |
| RF | −100.00 | −100.00 | −100.00 | 15.92 | −71.02 (1) | |
| SVM-RFE | 11.66 | 11.86 | 20.36 | 10.89 | 13.69 (6) | |
| LASSO | −14.29 | −100.00 | −100.00 | −14.29 | −57.14 (2) | |
| Boruta | 8.82 | −100.00 | −100.00 | 8.82 | −45.59 (3) |
| Method | Koch’s Index (Score) | Hubert–Levin’s C Index (Score) | Davies–Bouldin’s Index (Score) | Calinski–Harabasz Pseudo-F Statistic (Score) | Sum Score (Rank) |
|---|---|---|---|---|---|
| Kruskall–Wallis | 0.47 (5) | 0.44 (4) | 3.95 (5) | 1.37 (4) | 18 (1) |
| PLS-DA | 0.46 (4) | 0.42 (5) | 7.69 (4) | 15.19 (5) | 18 (1) |
| RF | 0.18 (1) | 100.00 (3) | 100.00 (3) | −100.00 (3) | 10 (2) |
| LASSO | 0.21 (3) | 100.00 (2) | 100.00 (2) | −100.00 (2) | 9 (3) |
| Boruta | 0.18 (2) | 100.00 (1) | 100.00 (1) | −100.00 (1) | 5 (4) |
| Model, Feature Selection Method | Predicted Outcome | Clinical Group | ||
|---|---|---|---|---|
| Control (n = 20) | Benign (n = 32) | Malignant (n = 30) | ||
| XGBoost, Kruskal–Wallis set | control | 18 (90%) | 3 | 2 |
| benign | 2 | 27 (84%) | 10 | |
| malignant | 0 | 2 | 18 (60%) | |
| OvO CNN, Mann–Whitney set | control | 17 (85%) | 5 | 3 |
| benign | 0 | 20 (63%) | 1 | |
| malignant | 3 | 7 | 26 (87%) | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokareva, A.; Iurova, M.; Starodubtseva, N.; Chagovets, V.; Novoselova, A.; Kukaev, E.; Frankevich, V.; Sukhikh, G. Machine Learning Framework for Ovarian Cancer Diagnostics Using Plasma Lipidomics and Metabolomics. Int. J. Mol. Sci. 2025, 26, 6630. https://doi.org/10.3390/ijms26146630
Tokareva A, Iurova M, Starodubtseva N, Chagovets V, Novoselova A, Kukaev E, Frankevich V, Sukhikh G. Machine Learning Framework for Ovarian Cancer Diagnostics Using Plasma Lipidomics and Metabolomics. International Journal of Molecular Sciences. 2025; 26(14):6630. https://doi.org/10.3390/ijms26146630
Chicago/Turabian StyleTokareva, Alisa, Mariia Iurova, Natalia Starodubtseva, Vitaliy Chagovets, Anastasia Novoselova, Evgenii Kukaev, Vladimir Frankevich, and Gennady Sukhikh. 2025. "Machine Learning Framework for Ovarian Cancer Diagnostics Using Plasma Lipidomics and Metabolomics" International Journal of Molecular Sciences 26, no. 14: 6630. https://doi.org/10.3390/ijms26146630
APA StyleTokareva, A., Iurova, M., Starodubtseva, N., Chagovets, V., Novoselova, A., Kukaev, E., Frankevich, V., & Sukhikh, G. (2025). Machine Learning Framework for Ovarian Cancer Diagnostics Using Plasma Lipidomics and Metabolomics. International Journal of Molecular Sciences, 26(14), 6630. https://doi.org/10.3390/ijms26146630