
Preclinical Evaluation of Repurposed Antimalarial Artemisinins for the Treatment of Malignant Peripheral Nerve Sheath Tumors
 ,
, 
Abstract
1. Introduction
2. Results
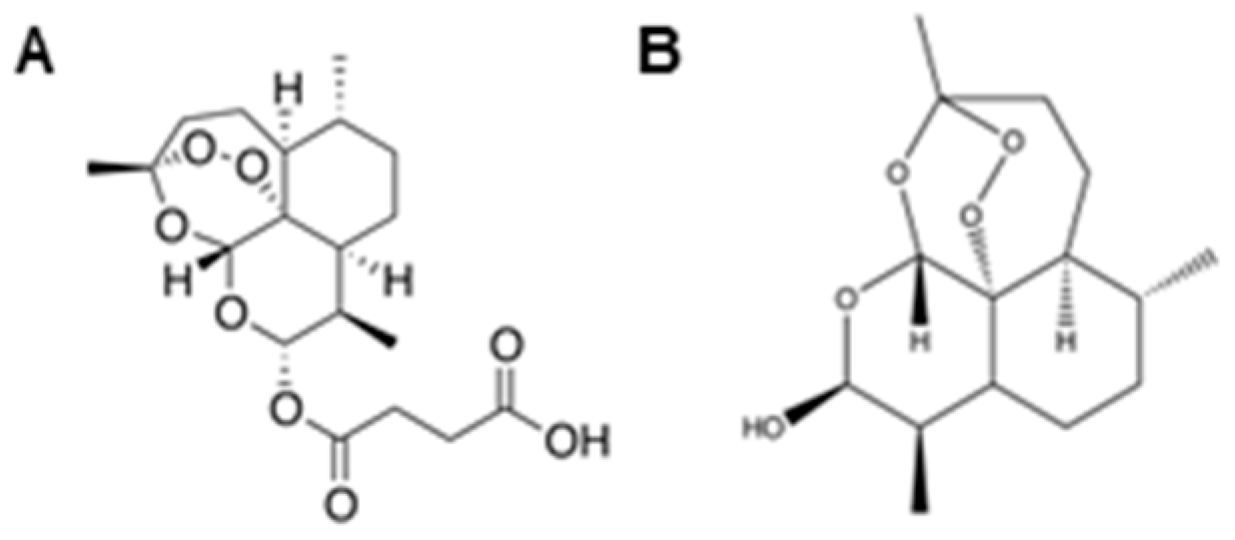
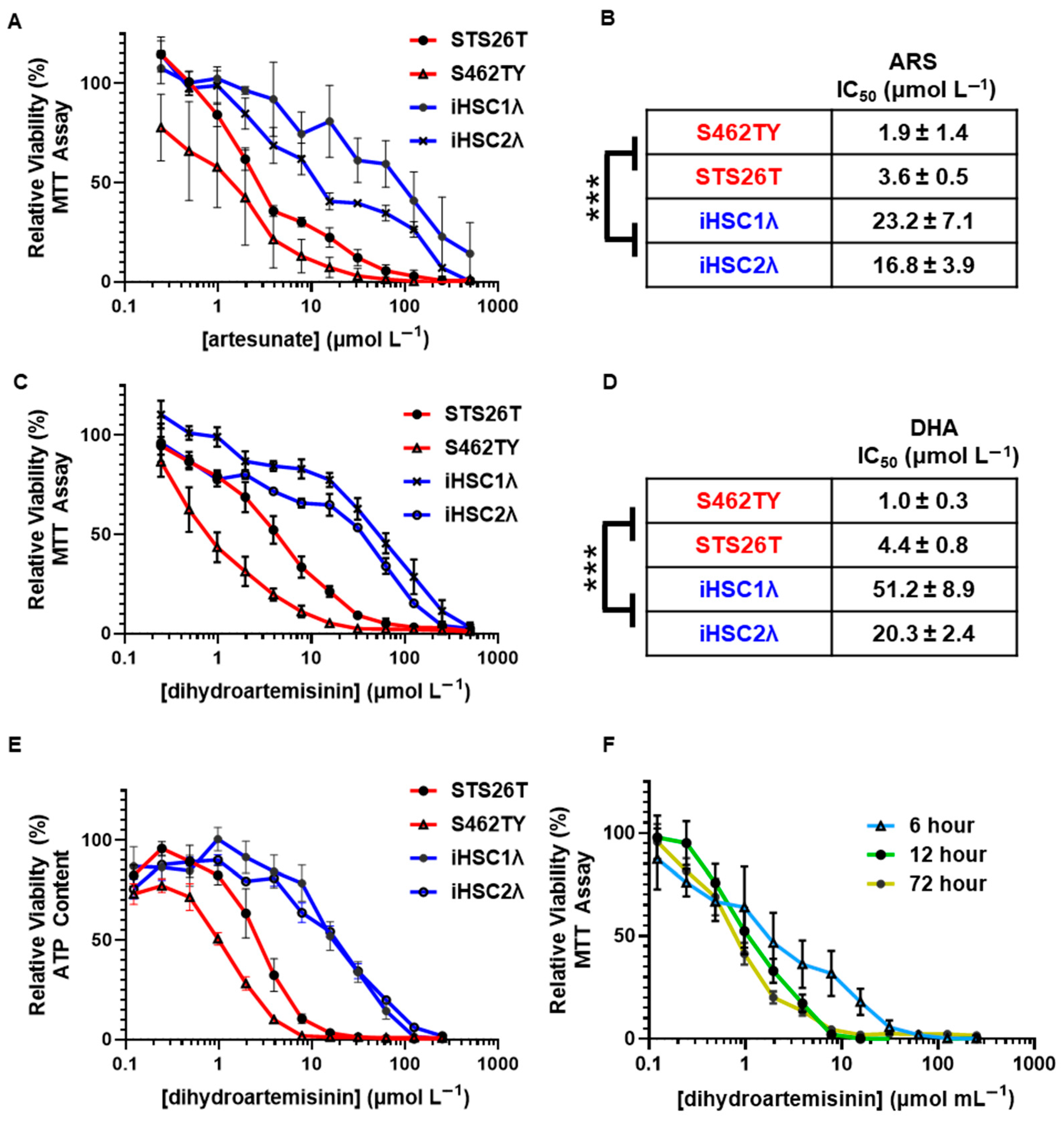
2.1. Antimalarial Artemisinins Are Selectively Cytotoxic to Malignant Peripheral Nerve Sheath Tumors
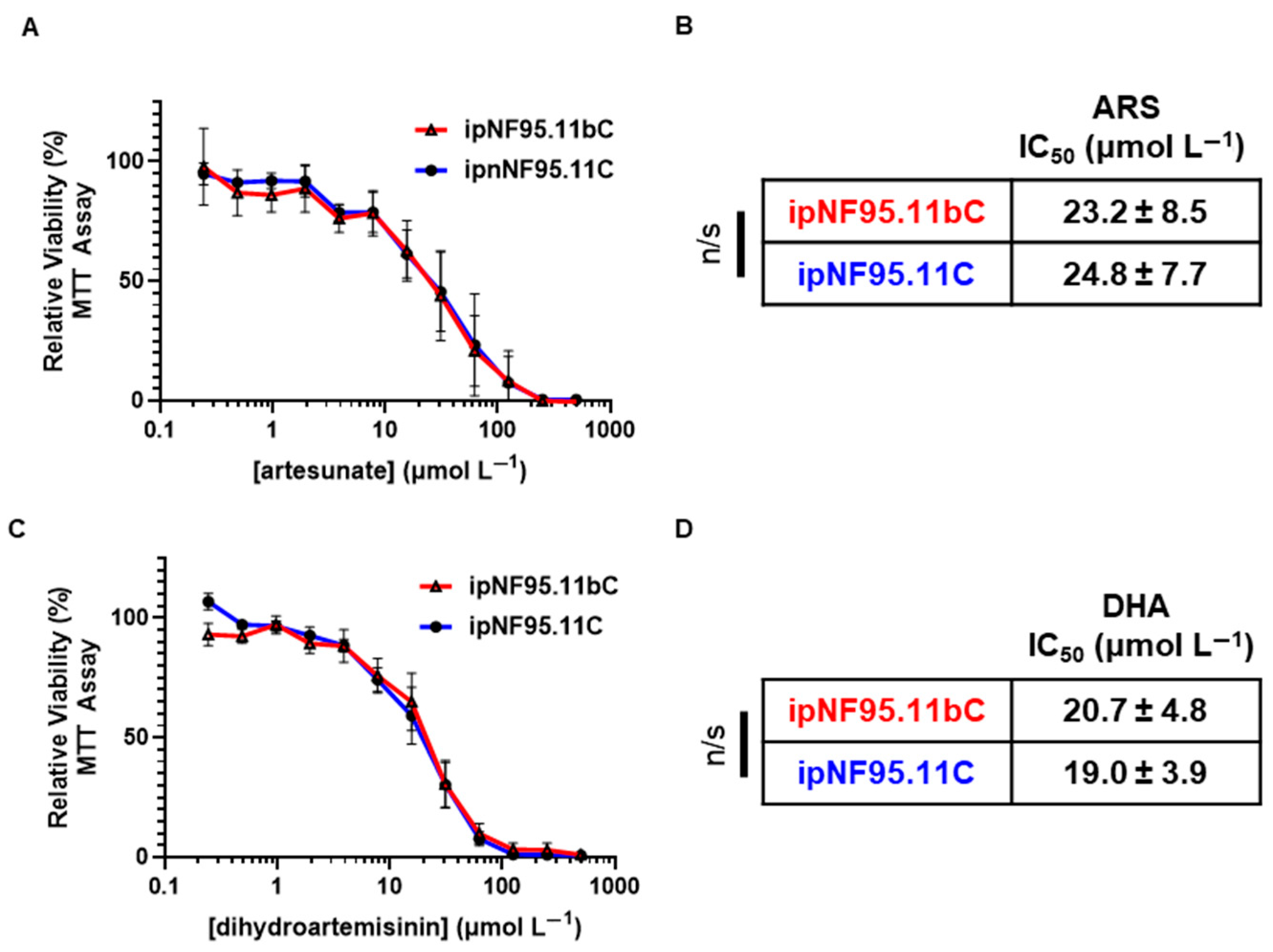
2.2. Antimalarial Artemisinins Do Not Show Selective Cytotoxicity in Plexiform Neurofibroma
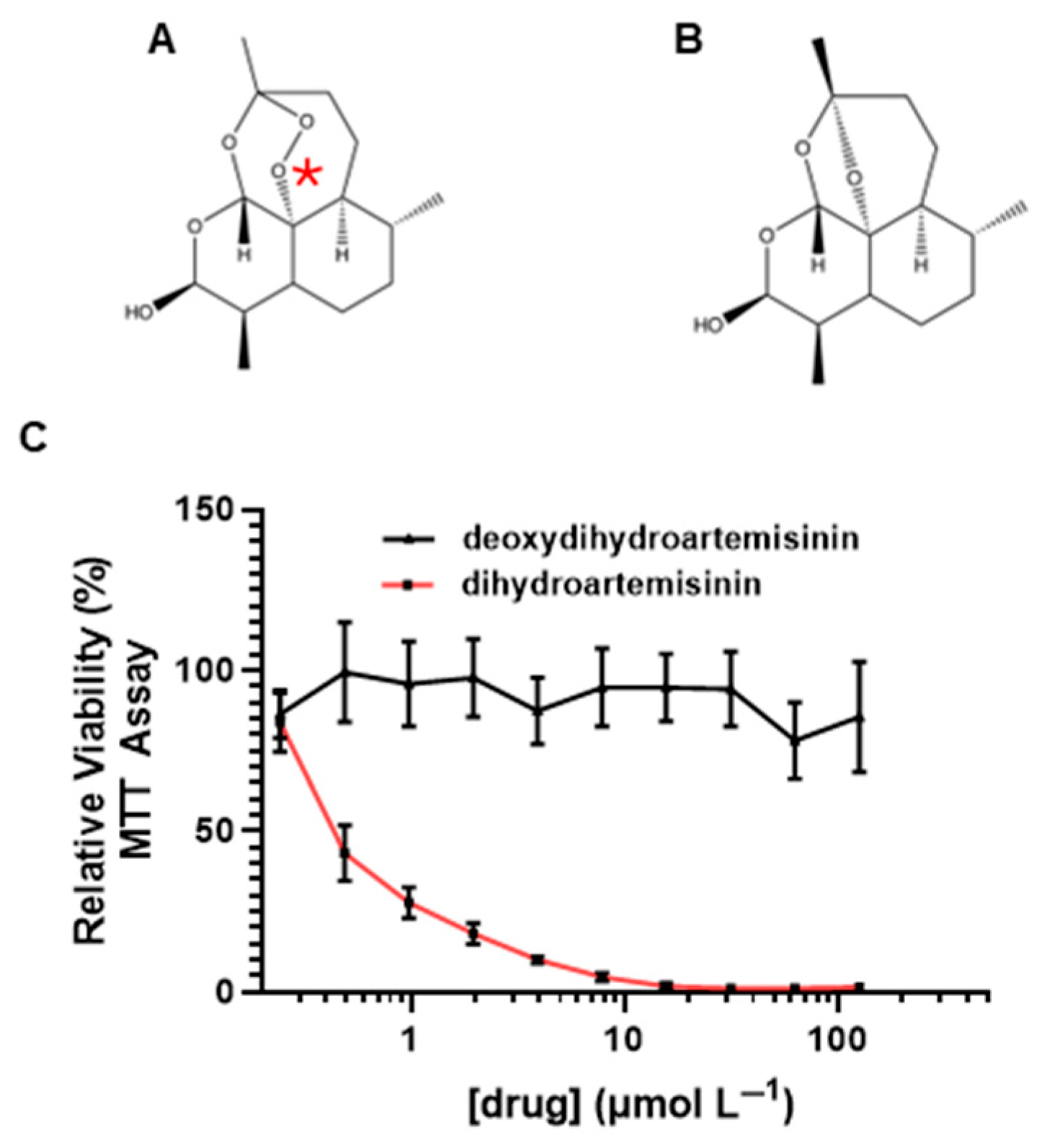
2.3. Endoperoxide Bridge Is Required for Dihydroartemisinin-Mediated Cytotoxicity in MPSNT Cells
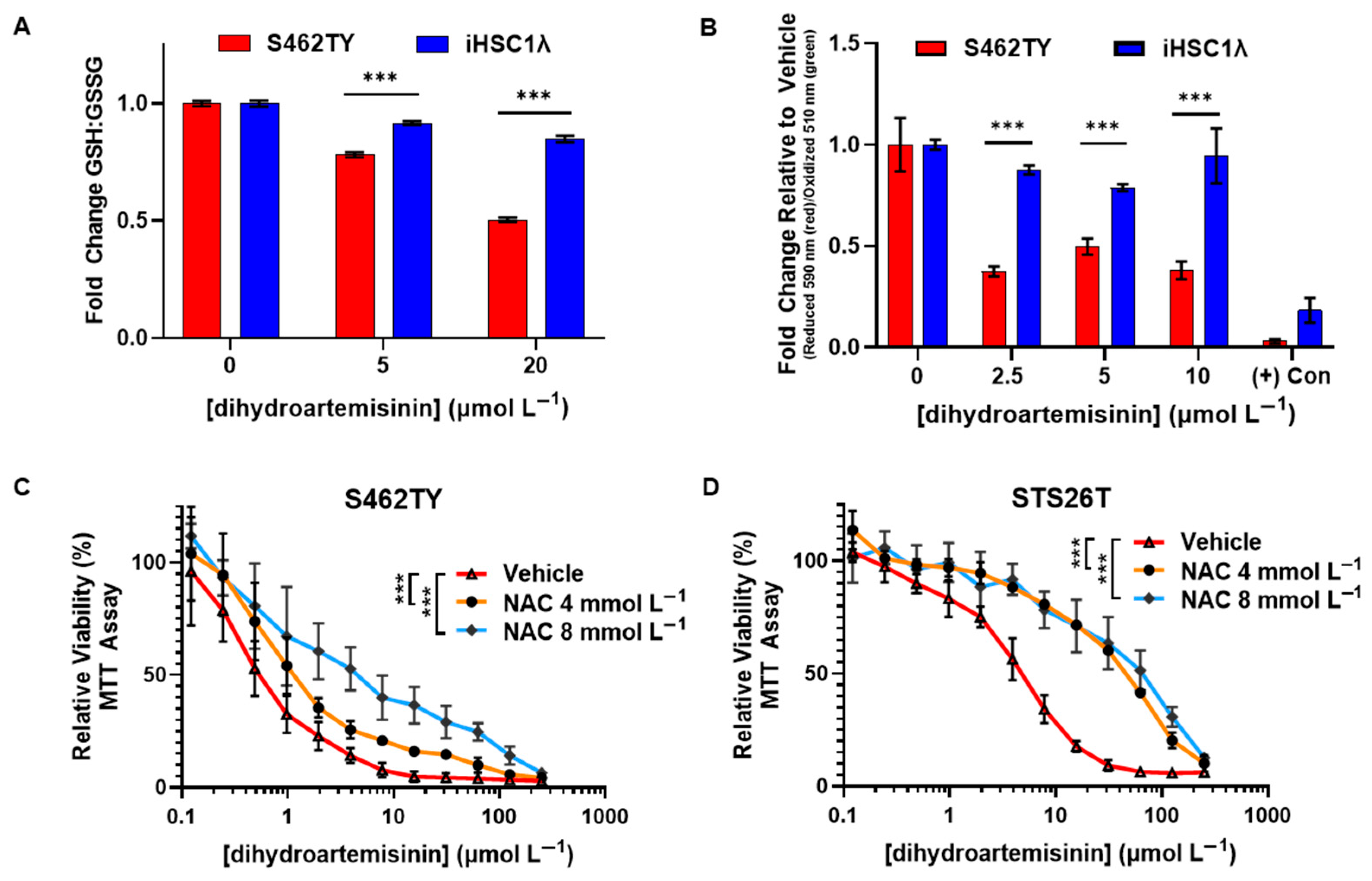
2.4. Dihydroartemisinin Disrupts the Redox Balance in MPNST and Induces Lipid Peroxidation
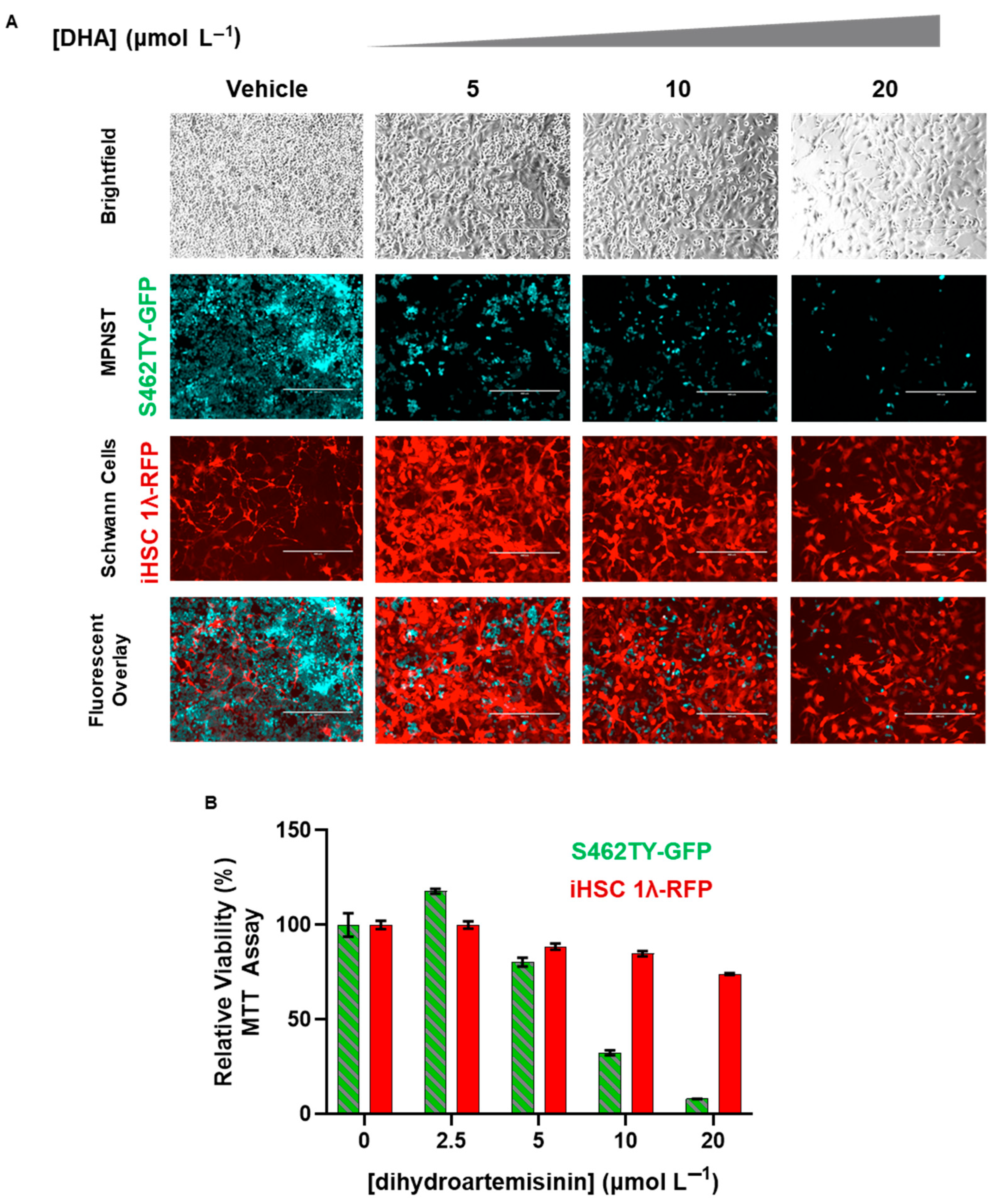
2.5. Dihydroartemisinin Selectively Removes MPNST from Co-Culture with Normal Scwhann Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Viability Assays
4.2.1. MTT Assay
4.2.2. ATP Assay
4.3. Synthesis of Deoxydihydroartemisinin
4.4. Glutathione Assay
4.5. Lipid Peroxidation Assay
4.6. Co-Culture Model System
4.7. Other Chemicals and Reagents
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ARS | artesunate |
| ATP | adenosine triphosphate |
| DHA | dihydroartemisinin |
| EMT | epithelial–mesenchymal transition |
| FDA | Food and Drug Administration |
| GSH | glutathione |
| GSSG | reduced glutathione |
| IC50 | half-maximal inhibitory concentration |
| MPNST | malignant peripheral nerve sheath tumor |
| MTT | 3-(4,5-di methyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| NF1 | neurofibromatosis type 1 |
| NAC | N-acetyl-cysteine |
| pNF | plexiform neurofibroma |
| ROS | reactive oxygen species |
| WHO | World Health Organization |
References
- Bottillo, I.; Ahlquist, T.; Brekke, H.; Danielsen, S.A.; van den Berg, E.; Mertens, F.; Lothe, R.A.; Dallapiccola, B. Germline and somatic NF1 mutations in sporadic and NF1-associated malignant peripheral nerve sheath tumours. J. Pathol. 2009, 217, 693–701. [Google Scholar] [CrossRef]
- Carroll, S.L. Molecular mechanisms promoting the pathogenesis of Schwann cell neoplasms. Acta Neuropathol. 2012, 123, 321–348. [Google Scholar] [CrossRef]
- Widemann, B.C. Current status of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Curr. Oncol. Rep. 2009, 11, 322–328. [Google Scholar] [CrossRef]
- Guo, A.; Liu, A.; Wei, L.; Song, X. Malignant peripheral nerve sheath tumors: Differentiation patterns and immunohistochemical features—A mini-review and our new findings. J. Cancer 2012, 3, 303–309. [Google Scholar] [CrossRef]
- Yamanaka, R.; Hayano, A. Radiation-Induced Malignant Peripheral Nerve Sheath Tumors: A Systematic Review. World Neurosurg. 2017, 105, 961–970 e8. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Goodsell, D.S. The molecular perspective: The ras oncogene. Oncologist 1999, 4, 263–264. [Google Scholar] [CrossRef]
- Suppiah, S.; Mansouri, S.; Mamatjan, Y.; Liu, J.C.; Bhunia, M.M.; Patil, V.; Rath, P.; Mehani, B.; Heir, P.; Bunda, S.; et al. Multiplatform molecular profiling uncovers two subgroups of malignant peripheral nerve sheath tumors with distinct therapeutic vulnerabilities. Nat. Commun. 2023, 14, 2696. [Google Scholar] [CrossRef]
- Ogrunc, M.; Di Micco, R.; Liontos, M.; Bombardelli, L.; Mione, M.; Fumagalli, M.; Gorgoulis, V.G.; d’Adda di Fagagna, F. Oncogene-induced reactive oxygen species fuel hyperproliferation and DNA damage response activation. Cell Death Differ. 2014, 21, 998–1012. [Google Scholar] [CrossRef]
- Martinez, M.; Sorzano, C.O.S.; Pascual-Montano, A.; Carazo, J.M. Gene signature associated with benign neurofibroma transformation to malignant peripheral nerve sheath tumors. PLoS ONE 2017, 12, e0178316. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali, M.G.; Shailender, G.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark. Insights 2018, 13, 1177271918755391. [Google Scholar] [CrossRef]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216, 107658. [Google Scholar] [CrossRef]
- Meshnick, S.R.; Yang, Y.Z.; Lima, V.; Kuypers, F.; Kamchonwongpaisan, S.; Yuthavong, Y. Iron-dependent free radical generation from the antimalarial agent artemisinin (qinghaosu). Antimicrob. Agents Chemother. 1993, 37, 1108–1114. [Google Scholar] [CrossRef]
- Jelinek, T. Artemisinin based combination therapy in travel medicine. Travel Med. Infect. Dis. 2013, 11, 23–28. [Google Scholar] [CrossRef]
- Cui, L.; Su, X.Z. Discovery, mechanisms of action and combination therapy of artemisinin. Expert Rev. Anti Infect. Ther. 2009, 7, 999–1013. [Google Scholar] [CrossRef]
- Augustin, Y.; Staines, H.M.; Krishna, S. Artemisinins as a novel anti-cancer therapy: Targeting a global cancer pandemic through drug repurposing. Pharmacol. Ther. 2020, 216, 107706. [Google Scholar] [CrossRef]
- Posner, G.H.; Oh, C.H.; Wang, D.; Gerena, L.; Milhous, W.K.; Meshnick, S.R.; Asawamahasadka, W. Mechanism-based design, synthesis, and in vitro antimalarial testing of new 4-methylated trioxanes structurally related to artemisinin: The importance of a carbon-centered radical for antimalarial activity. J. Med. Chem. 1994, 37, 1256–1258. [Google Scholar] [CrossRef]
- Reczek, C.R.; Birsoy, K.; Kong, H.; Martinez-Reyes, I.; Wang, T.; Gao, P.; Sabatini, D.M.; Chandel, N.S. A CRISPR screen identifies a pathway required for paraquat-induced cell death. Nat. Chem. Biol. 2017, 13, 1274–1279. [Google Scholar] [CrossRef]
- Alewine, G.; Knight, J.; Ghantae, A.; Mamrega, C.; Attiah, B.; Coover, R.A.; Fahrenholtz, C.D. Silver Nanoparticles Selectively Treat Neurofibromatosis Type 1-Associated Malignant Peripheral Nerve Sheath Tumors in a Neurofibromin-Dependent Manner. J. Pers. Med. 2022, 12, 1080. [Google Scholar] [CrossRef]
- Manz, D.H.; Blanchette, N.L.; Paul, B.T.; Torti, F.M.; Torti, S.V. Iron and cancer: Recent insights. Ann. N. Y. Acad. Sci. 2016, 1368, 149–161. [Google Scholar] [CrossRef]
- Greenshields, A.L.; Fernando, W.; Hoskin, D.W. The anti-malarial drug artesunate causes cell cycle arrest and apoptosis of triple-negative MDA-MB-468 and HER2-enriched SK-BR-3 breast cancer cells. Exp. Mol. Pathol. 2019, 107, 10–22. [Google Scholar] [CrossRef]
- Im, E.; Yeo, C.; Lee, H.J.; Lee, E.O. Dihydroartemisinin induced caspase-dependent apoptosis through inhibiting the specificity protein 1 pathway in hepatocellular carcinoma SK-Hep-1 cells. Life Sci. 2018, 192, 286–292. [Google Scholar] [CrossRef]
- White, N.J. Qinghaosu (artemisinin): The price of success. Science 2008, 320, 330–334. [Google Scholar] [CrossRef]
- von Hagens, C.; Walter-Sack, I.; Goeckenjan, M.; Osburg, J.; Storch-Hagenlocher, B.; Sertel, S.; Elsasser, M.; Remppis, B.A.; Edler, L.; Munzinger, J.; et al. Prospective open uncontrolled phase I study to define a well-tolerated dose of oral artesunate as add-on therapy in patients with metastatic breast cancer (ARTIC M33/2). Breast Cancer Res. Treat. 2017, 164, 359–369. [Google Scholar] [CrossRef]
- Newton, P.; Suputtamongkol, Y.; Teja-Isavadharm, P.; Pukrittayakamee, S.; Navaratnam, V.; Bates, I.; White, N. Antimalarial bioavailability and disposition of artesunate in acute falciparum malaria. Antimicrob. Agents Chemother. 2000, 44, 972–977. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Gutmann, D.H. Neurofibromatosis type 1: A multidisciplinary approach to care. Lancet Neurol. 2014, 13, 834–843. [Google Scholar] [CrossRef]
- Rudrapal, M.; Chetia, D. Endoperoxide antimalarials: Development, structural diversity and pharmacodynamic aspects with reference to 1,2,4-trioxane-based structural scaffold. Drug Des. Devel. Ther. 2016, 10, 3575–3590. [Google Scholar] [CrossRef]
- Araujo, N.C.; Afonso, R.; Bringela, A.; Cancela, M.L.; Cristiano, M.L.; Leite, R.B. Peroxides with antiplasmodial activity inhibit proliferation of Perkinsus olseni, the causative agent of Perkinsosis in bivalves. Parasitol. Int. 2013, 62, 575–582. [Google Scholar] [CrossRef]
- Ho, W.E.; Peh, H.Y.; Chan, T.K.; Wong, W.S. Artemisinins: Pharmacological actions beyond anti-malarial. Pharmacol. Ther. 2014, 142, 126–139. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Halasi, M.; Wang, M.; Chavan, T.S.; Gaponenko, V.; Hay, N.; Gartel, A.L. ROS inhibitor N-acetyl-L-cysteine antagonizes the activity of proteasome inhibitors. Biochem. J. 2013, 454, 201–208. [Google Scholar] [CrossRef]
- Zhang, Q.; Yi, H.; Yao, H.; Lu, L.; He, G.; Wu, M.; Zheng, C.; Li, Y.; Chen, S.; Li, L.; et al. Artemisinin Derivatives Inhibit Non-small Cell Lung Cancer Cells Through Induction of ROS-dependent Apoptosis/Ferroptosis. J. Cancer 2021, 12, 4075–4085. [Google Scholar] [CrossRef]
- Dai, X.; Zhang, X.; Chen, W.; Chen, Y.; Zhang, Q.; Mo, S.; Lu, J. Dihydroartemisinin: A Potential Natural Anticancer Drug. Int. J. Biol. Sci. 2021, 17, 603–622. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019, 28, 2752–2762. [Google Scholar] [CrossRef]
- Li, Y.; Liu, J.; Huang, J.; Wei, C.; Ge, L.; Chung, M.; Zhu, B.; Guo, Z.; Zheng, T.; Li, H.; et al. Reduced PTPRS expression promotes epithelial-mesenchymal transition of Schwann cells in NF1-related plexiform neurofibromas. Cancer Lett. 2024, 599, 217151. [Google Scholar] [CrossRef]
- Efferth, T. From ancient herb to modern drug: Artemisia annua and artemisinin for cancer therapy. Semin. Cancer Biol. 2017, 46, 65–83. [Google Scholar] [CrossRef]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Eling, N.; Reuter, L.; Hazin, J.; Hamacher-Brady, A.; Brady, N.R. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015, 2, 517–532. [Google Scholar] [CrossRef]
- Wei, T.; Liu, J. Anti-angiogenic properties of artemisinin derivatives (Review). Int. J. Mol. Med. 2017, 40, 972–978. [Google Scholar] [CrossRef]
- Vakhrusheva, O.; Erb, H.H.H.; Braunig, V.; Markowitsch, S.D.; Schupp, P.; Baer, P.C.; Slade, K.S.; Thomas, A.; Tsaur, I.; Puhr, M.; et al. Artesunate Inhibits the Growth Behavior of Docetaxel-Resistant Prostate Cancer Cells. Front. Oncol. 2022, 12, 789284. [Google Scholar] [CrossRef]
- Yin, S.; Yang, H.; Zhao, X.; Wei, S.; Tao, Y.; Liu, M.; Bo, R.; Li, J. Antimalarial agent artesunate induces G0/G1 cell cycle arrest and apoptosis via increasing intracellular ROS levels in normal liver cells. Hum. Exp. Toxicol. 2020, 39, 1681–1689. [Google Scholar] [CrossRef]
- Thio, J.; Haig, A.; Swe, E.P.P.; Nguyen, P.; Tran, K.; Kasi, M. Artemisinin-induced cholestatic liver injury and intrahepatic ductopenia. Oxf. Med. Case Rep. 2024, 2024, omae070. [Google Scholar] [CrossRef]
- Karbwang, J.; Na-Bangchang, K.; Congpuong, K.; Molunto, P.; Thanavibul, A. Pharmacokinetics and bioavailability of oral and intramuscular artemether. Eur. J. Clin. Pharmacol. 1997, 52, 307–310. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Y.; Liu, G.; Xu, M. New clinical application prospects of artemisinin and its derivatives: A scoping review. Infect. Dis. Poverty 2023, 12, 115. [Google Scholar] [CrossRef]
- Batty, K.T.; Thu, L.T.; Davis, T.M.; Ilett, K.F.; Mai, T.X.; Hung, N.C.; Tien, N.P.; Powell, S.M.; Thien, H.V.; Binh, T.Q.; et al. A pharmacokinetic and pharmacodynamic study of intravenous vs oral artesunate in uncomplicated falciparum malaria. Br. J. Clin. Pharmacol. 1998, 45, 123–129. [Google Scholar] [CrossRef]
- Angus, B.J.; Thaiaporn, I.; Chanthapadith, K.; Suputtamongkol, Y.; White, N.J. Oral artesunate dose-response relationship in acute falciparum malaria. Antimicrob. Agents Chemother. 2002, 46, 778–782. [Google Scholar] [CrossRef]
- Abanyie, F.; Acharya, S.D.; Leavy, I.; Bowe, M.; Tan, K.R. Safety and Effectiveness of Intravenous Artesunate for Treatment of Severe Malaria in the United States-April 2019 Through December 2020. Clin. Infect. Dis. 2021, 73, 1965–1972. [Google Scholar] [CrossRef]
- Abanyie, F.; Ng, J.; Tan, K.R. Post-artesunate Delayed Hemolysis in Patients With Severe Malaria in the United States-April 2019 Through July 2021. Clin. Infect. Dis. 2023, 76, e857–e863. [Google Scholar] [CrossRef]
- Attiah, B.; Alewine, G.; Easter, M.K.; Coover, R.A.; Fahrenholtz, C.D. Silver Nanoparticles Selectively Treat Neurofibromatosis Type 1-Associated Plexiform Neurofibroma Cells at Doses That Do Not Affect Patient-Matched Schwann Cells. Pharmaceutics 2024, 16, 371. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Subtype Classification |
|---|---|
| S462TY | NF1-associated MPNST |
| STS26T | Sporadic MPNST |
| iHSC1λ | Schwann cell |
| iHSC2λ | Schwann cell |
| ipNF95.11bC 1 | NF1-associated plexiform neurofibroma |
| ipnNF95.11C 1 | NF1-associated Schwann cell |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duensing, H.M.; Dixon, J.M.; Hunter, O.R.; Graves, N.C.; Smith, N.C.; Tomes, A.J.; Fahrenholtz, C.D. Preclinical Evaluation of Repurposed Antimalarial Artemisinins for the Treatment of Malignant Peripheral Nerve Sheath Tumors. Int. J. Mol. Sci. 2025, 26, 6628. https://doi.org/10.3390/ijms26146628
Duensing HM, Dixon JM, Hunter OR, Graves NC, Smith NC, Tomes AJ, Fahrenholtz CD. Preclinical Evaluation of Repurposed Antimalarial Artemisinins for the Treatment of Malignant Peripheral Nerve Sheath Tumors. International Journal of Molecular Sciences. 2025; 26(14):6628. https://doi.org/10.3390/ijms26146628
Chicago/Turabian StyleDuensing, Heather M., Jalen M. Dixon, Owen R. Hunter, Nicolina C. Graves, Nickalus C. Smith, Andersen J. Tomes, and Cale D. Fahrenholtz. 2025. "Preclinical Evaluation of Repurposed Antimalarial Artemisinins for the Treatment of Malignant Peripheral Nerve Sheath Tumors" International Journal of Molecular Sciences 26, no. 14: 6628. https://doi.org/10.3390/ijms26146628
APA StyleDuensing, H. M., Dixon, J. M., Hunter, O. R., Graves, N. C., Smith, N. C., Tomes, A. J., & Fahrenholtz, C. D. (2025). Preclinical Evaluation of Repurposed Antimalarial Artemisinins for the Treatment of Malignant Peripheral Nerve Sheath Tumors. International Journal of Molecular Sciences, 26(14), 6628. https://doi.org/10.3390/ijms26146628