
Gene Therapies in Dermatological Diseases: A Breakthrough in Treatment

 , ,
, , 
Abstract
1. Introduction
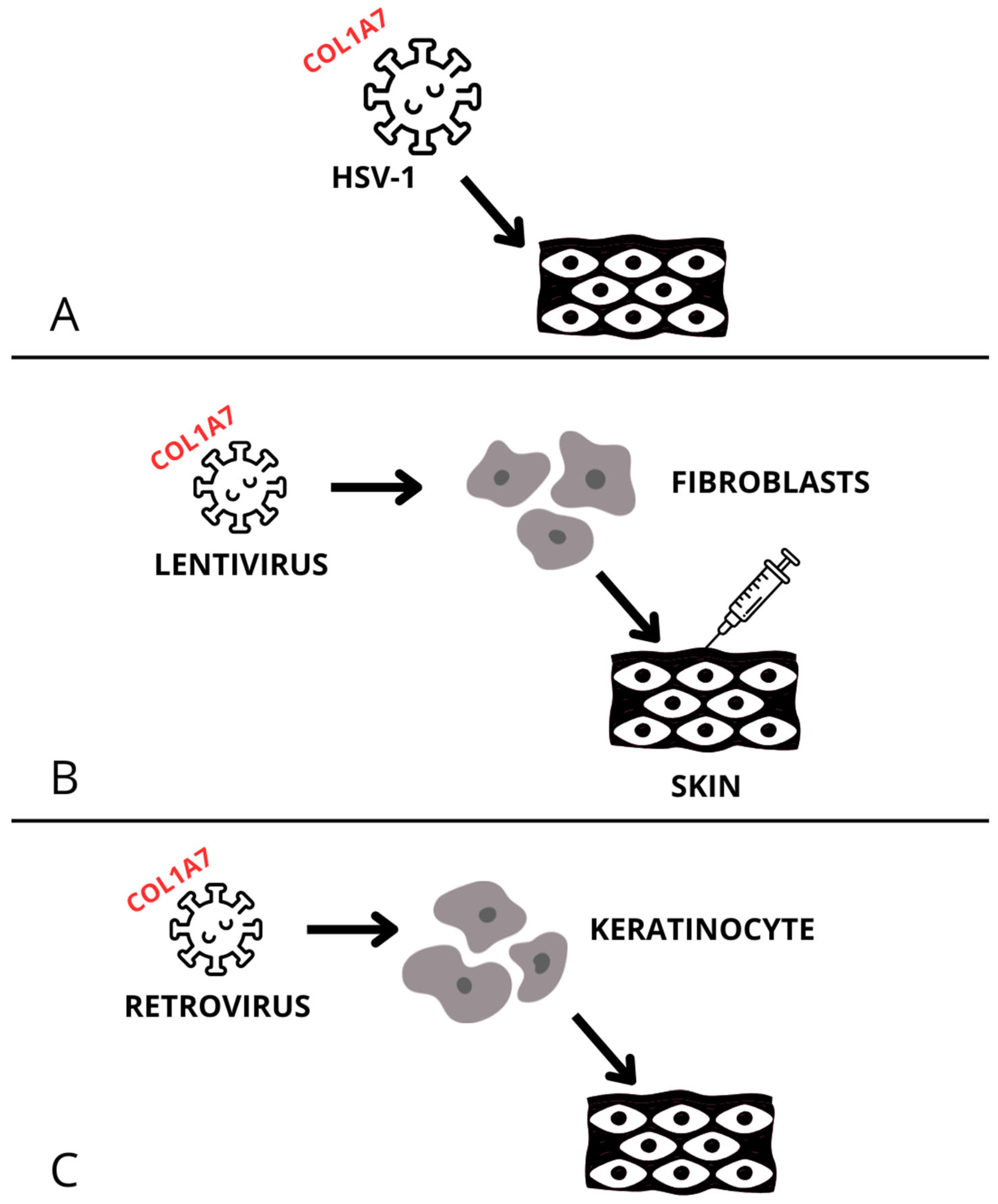
2. Recessive Dystrophic Epidermolysis Bullosa (RDEB)
3. Melanoma
3.1. Small Interfering RNA (siRNA)-Based Treatment
3.2. CAR-T
3.3. Oncolytic Viruses
4. Psoriasis
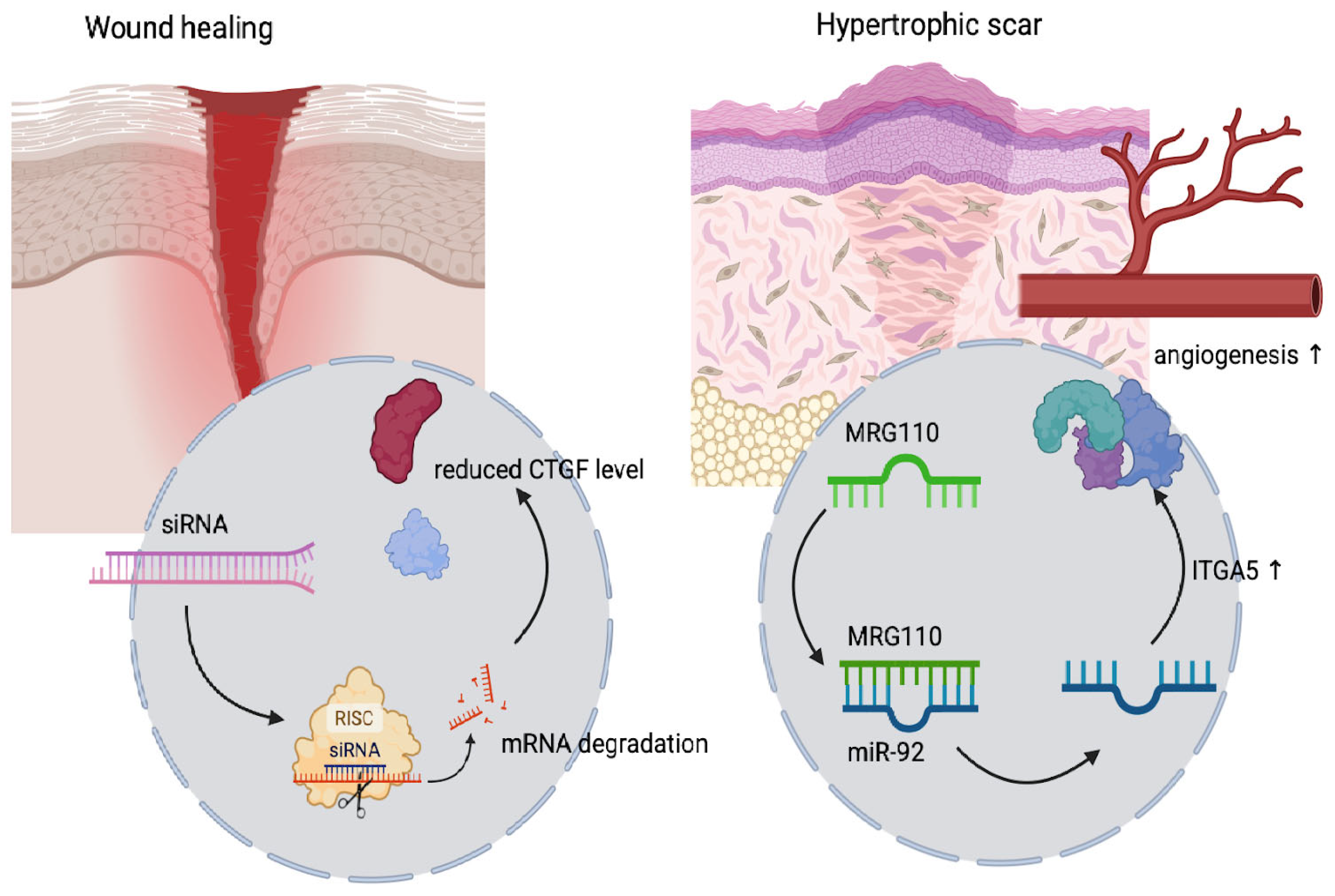
5. Wound Healing
6. Ichthyosis
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| B-VEC | Beremagene Geperpavec |
| RDEB | Recessive Dystrophic Epidermolysis Bullosa |
| FDA | Food and Drug Administration |
| siRNA | Small Interfering RNA |
| CAR-T | Chimeric Antigen Receptor T-cell |
| T-VEC | Talimogene Laherparepvec |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| AAVs | Adeno-Associated Viruses |
| AFs | Anchoring Fibrils |
| DDEB | Dominant Dystrophic Epidermolysis Bullosa |
| HSV-1 | Herpes Simplex Virus Type 1 |
| C7 | Type VII Collagen |
| mRNA | Messenger RNA |
| RISC | RNA-Induced Silencing Complex |
| CDC2 | Cell Division Cycle 2 |
| WEE1 | WEE1 G2 Checkpoint Kinase |
| RRCPP | RGD-R8 Cell-Penetrating Peptide |
| LMW PEI | Low Molecular Weight Polyethylenimine |
| R8 | Octaarginine Cell-Penetrating Peptide |
| PEG | Polyethylene Glycol |
| RGD | Arginine-Glycine-Aspartic Acid |
| B16F10 | Mouse Melanoma Cell Line |
| C26 | Mouse Colon Cancer Cell Line |
| 293T | Human Embryonic Kidney Cell Line |
| 4T1 | Mouse Breast Cancer Cell Line |
| RRCPP/siWee1 | RRCPP Complex with siRNA Targeting WEE1 |
| NS | Normal SalineRRCPP/siNC—RRCPP with Non-Coding siRNA |
| PD-1 | Programmed Cell Death Protein 1 |
| mTOR | Mammalian Target of Rapamycin |
| INF-γ | Interferon Gamma |
| WT1 | Wilms Tumor 1 |
| SPACE-EGF | Self-assembling Peptide Amphiphile Conjugated with EGF |
| c-Myc | Cellular Myelocytomatosis Oncogene |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| MHC | Major Histocompatibility Complex |
| CD3ζ | CD3 Zeta Chain |
| TME | Tumor Microenvironment |
| VEGFR-2 | Vascular Endothelial Growth Factor Receptor 2 |
| CD-16 | Fc Gamma Receptor III |
| CD-70 | Cluster of Differentiation 70 |
| HER-2 | Human Epidermal Growth Factor Receptor 2 |
| B7-H3 | B7 Homolog 3 |
| IL-2 | Interleukin 2 |
| TRP1 | Tyrosinase-Related Protein 1 |
| ICP34.5 | Infected Cell Protein 34.5 |
| ICP47 | Infected Cell Protein 47 |
| GM-CSF | Granulocyte-Macrophage Colony-Stimulating Factor |
| K17 | Keratin 17 |
| NFAT2 | Nuclear Factor of Activated T-cells 2 |
| TRAF3IP2 | TRAF3 Interacting Protein 2AKR1B10 |
| AKR1B10 | Aldo-Keto Reductase Family 1 Member B10 |
| POMP | Proteasome Maturation Protein |
| FGFR2 | Fibroblast Growth Factor Receptor 2 |
| FGFR2r | Mutated/Regulatory Variant of FGFR2 |
| TRAF3IP2 | TRAF3 Interacting Protein 2 |
| WTAP | Wilms Tumor 1 Associated Protein |
| EGR1 | Early Growth Response 1 |
| PLK2 | Polo-Like Kinase 2 |
| GRHL2 | Grainyhead-Like Transcription Factor 2 |
| SGPL1 | Sphingosine-1-Phosphate Lyase 1 |
| CTSB | Cathepsin B |
| miR-125b | MicroRNA-125b |
| miR-31 | MicroRNA-31 |
| miR-210 | MicroRNA-210 |
| DEFB4 | Beta-Defensin 4 |
| TSLP | Thymic Stromal Lymphopoietin |
| KRT17 | Keratin 17 |
| TNFα | Tumor Necrosis Factor Alpha |
| DIPEA | Diisopropylethylamine |
| CK2 | Casein Kinase 2 |
| IL-17A | Interleukin 17A |
| NFKBIZ | Nuclear Factor Kappa B Inhibitor Zeta |
| SOCS1 | Suppressor of Cytokine Signaling 1 |
| PP6 | Protein Phosphatase 6 |
| NF-κB | Nuclear factor kappa B |
| PCSK9 | Proprotein Convertase Subtilisin/Kexin Type 9 |
| IL-6 | Interleukin 6 |
| IL-21 | Interleukin 21 |
| IL-23 | Interleukin 23 |
| LNPs | Lipid Nanoparticles |
| IL-1β | Interleukin 1 β |
| IL-6 siRNA | Interleukin 6 small interfering RNA |
| C8B2-si-STAT3 | siRNA Construct Targeting STAT3 |
| AQP1 | Aquaporin 1 |
| HIF-1α | Hypoxia-Inducible Factor 1 Alpha |
| KRT16 | Keratin 16 |
| VEGF | Vascular Endothelial Growth Factor |
| ERK | Extracellular Signal-Regulated Kinase |
| PI3K/mTOR | Phosphoinositide 3-Kinase/Mammalian Target of Rapamycin |
| DFUs | Diabetic Foot Ulcers |
| VLUs | Venous Leg Ulcers |
| PUs | Pressure Ulcers |
| GF | Growth Factor |
| PDGF | Platelet-Derived Growth Factor |
| FGF | Fibroblast Growth Factor |
| EGF | Epidermal Growth Factor |
| CTGF | Connective Tissue Growth Factor |
| MRG-110 | Antagomir Targeting miR-92a |
| ITGA5 | Integrin Alpha 5 |
| miR-92a | MicroRNA-92a |
| miR-192 | MicroRNA-192 |
| ASOs | Antisense Oligonucleotides |
| BMT101,LEMS401, RXI-109 | Therapeutic siRNA or ASO |
| Productscp-asiRNA | Cell-Penetrating Antisense Small Interfering RNA |
| RNAi | RNA Interference |
| COX-2 | Cyclooxygenase-2 |
| MRG-110 | miRagen compound 110 |
| TGF-β/Smad | SMAD-dependent signaling mediated by TGF-β |
| PRP | Platelet-Rich Plasma |
| ARCI | Autosomal recessive congenital ichthyosis |
| LI | Lamellar ichthyosis |
| HI | Harlequin ichthyosis |
| CIE | Congenital ichthyosiform erythroderma |
| TGM1 | Transglutaminase 1 |
| ABEs | Adenine base editors |
| SgRNSs | Single gene RNAs |
| ABCA12 | ATP binding cassette subfamily A member 12 |
| TALENs | Transcription activator-like effector nucleases |
| KID | Keratitis-ichthyosis-deafness |
References
- Bal, J.; Bartnik, E.; Biedrzycka, M.; Bocian, E.; Bosek, L.; Brojer, E.; Charzewska, A.; Czartoryska, B.; Daca-Roszak, P.; Daniel, W.A.; et al. Genetyka Medyczna i Molekularna; Wydawnictwo Naukowe PWN SA: Warszawa, Poland, 2023. [Google Scholar]
- Ingusci, S.; Goins, W.F.; Cohen, J.B.; Miyagawa, Y.; Knipe, D.M.; Glorioso, J.C. Next-generation replication-defective HSV vectors for delivery of large DNA payloads. Mol. Ther. 2025, 33, 2205–2216. [Google Scholar] [CrossRef] [PubMed]
- Drewa, G.; Ferenc, T. Genetyka Medyczna. Podręcznik dla Studentów; Edra Urban and Partner: Wrocław, Poland, 2018. [Google Scholar]
- Gurevich, I.; Agarwal, P.; Zhang, P.; Dolorito, J.A.; Oliver, S.; Liu, H.; Reitze, N.; Sarma, N.; Bagci, I.S.; Sridhar, K.; et al. In vivo topical gene therapy for recessive dystrophic epidermolysis bullosa: A phase 1 and 2 trial. Nat. Med. 2022, 28, 780–788. [Google Scholar] [PubMed]
- Lwin, S.M.; Syed, F.; Di, W.L.; Kadiyirire, T.; Liu, L.; Guy, A.; Petrova, A.; Abdul-Wahab, A.; Reid, F.; Phillips, R.; et al. Safety and early efficacy outcomes for lentiviral fibroblast gene therapy in recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4, e126243. [Google Scholar]
- Gonzalez, J.C.; Park, K.W.; Evans, D.B.; Sharma, R.; Sahaym, O.; Gopalakrishnan, S.; Dar, A.I.; Valdez, T.A.; Sharma, A. Nano Approaches to Nucleic Acid Delivery: Barriers, Solutions, and Current Landscape. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2025, 17, e70010. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zeng, L.; Gowda, B.H.J.; Ahmed, M.G.; Abourehab, M.A.S.; Chen, Z.S.; Zhang, C.; Li, J.; Kesharwani, P. Advancements in nanoparticle-based treatment approaches for skin cancer therapy. Mol. Cancer 2023, 22, 10. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Slominski, A.T.; Slominski, R.M.; Raman, C.; Chen, J.Y.; Athar, M.; Elmets, C. Neuroendocrine signaling in the skin with a special focus on the epidermal neuropeptides. Am. J. Physiol. Cell Physiol. 2022, 323, C1757–C1776. [Google Scholar] [CrossRef]
- Slominski, R.M.; Chen, J.Y.; Raman, C.; Slominski, A.T. Photo-neuro-immuno-endocrinology: How the ultraviolet radiation regulates the body, brain, and immune system. Proc. Natl. Acad. Sci. USA 2024, 121, e2308374121. [Google Scholar] [CrossRef]
- Has, C.; Bauer, J.W.; Bodemer, C.; Bolling, M.C.; Bruckner-Tuderman, L.; Diem, A.; Fine, J.D.; Heagerty, A.; Hovnanian, A.; Marinkovich, M.P.; et al. Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br. J. Dermatol. 2020, 183, 614–627. [Google Scholar]
- Wallace, M.; Dasgupta, R.; Cravero, K.; Yang, H.; South, A.P.; Nikbakht, N. The Utility of Collagen VII Topical Gene Therapy in Treatment of Surgical Defect after Excision of Recessive Dystrophic Epidermolysis Bullosa associated Squamous Cell Carcinoma. Br. J. Dermatol. 2025, ljaf128, ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Osborn, M.J.; Panda, S.; Reineke, T.M.; Tolar, J.; Nyström, A. Progress in skin gene therapy: From the inside and out. Mol. Ther. 2025, 33, 2065–2081. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Li, M.; Intong-Wheeler, L.R.A.; Tran, K.; Marucci, D.; Murrell, D.F. Epidemiology and Outcome of Squamous Cell Carcinoma in Epidermolysis Bullosa in Australia and New Zealand. Acta Derm. Venereol. 2018, 98, 70–76. [Google Scholar] [PubMed]
- Jeon, I.K.; On, H.R.; Kim, S.C. Quality of Life and Economic Burden in Recessive Dystrophic Epidermolysis Bullosa. Ann. Dermatol. 2016, 28, 6–14. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.; Wang, X.; Li, Y.; Qiu, B.; Bushmalyova, A.; He, Z.; Wang, W.; Lara-Sáez, I. CRISPR-Cas9-based non-viral gene editing therapy for topical treatment of recessive dystrophic epidermolysis bullosa. Mol. Ther.-Methods Clin. Dev. 2023, 31, 101134. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guide, S.V.; Gonzalez, M.E.; Bağcı, I.S.; Agostini, B.; Chen, H.; Feeney, G.; Steimer, M.; Kapadia, B.; Sridhar, K.; Quesada Sanchez, L.; et al. Trial of Beremagene Geperpavec (B-VEC) for Dystrophic Epidermolysis Bullosa. N. Engl. J. Med. 2022, 387, 2211–2219. [Google Scholar]
- Available online: https://www.fda.gov/media/168356/download (accessed on 19 May 2023).
- Available online: https://ir.krystalbio.com/news-releases/news-release-details/krystal-biotech-announces-european-commission-approval-vyjuvekr (accessed on 28 April 2025).
- Siprashvili, Z.; Nguyen, N.T.; Gorell, E.S.; Loutit, K.; Khuu, P.; Furukawa, L.K.; Lorenz, H.P.; Leung, T.H.; Keene, D.R.; Rieger, K.E.; et al. Safety and Wound Outcomes Following Genetically Corrected Autologous Epidermal Grafts in Patients With Recessive Dystrophic Epidermolysis Bullosa. JAMA 2016, 316, 1808–1817. [Google Scholar]
- Eichstadt, S.; Barriga, M.; Ponakala, A.; Teng, C.; Nguyen, N.T.; Siprashvili, Z.; Nazaroff, J.; Gorell, E.S.; Chiou, A.S.; Taylor, L.; et al. Phase 1/2a clinical trial of gene-corrected autologous cell therapy for recessive dystrophic epidermolysis bullosa. JCI Insight 2019, 4, e130554. [Google Scholar] [PubMed]
- So, J.Y.; Nazaroff, J.; Iwummadu, C.V.; Harris, N.; Gorell, E.S.; Fulchand, S.; Bailey, I.; McCarthy, D.; Siprashvili, Z.; Marinkovich, M.P.; et al. Long-term safety and efficacy of gene-corrected autologous keratinocyte grafts for recessive dystrophic epidermolysis bullosa. Orphanet J. Rare Dis. 2022, 17. [Google Scholar]
- Available online: https://www.fda.gov/media/186513/download (accessed on 28 April 2025).
- Eddy, K.; Chen, S. Overcoming Immune Evasion in Melanoma. Int. J. Mol. Sci. 2020, 21, 8984. [Google Scholar]
- Goyal, R.; Chopra, H.; Singh, I.; Dua, K.; Gautam, R.K. Insights on prospects of nano-siRNA based approaches in treatment of Cancer. Front. Pharmacol. 2022, 13, 985670. [Google Scholar] [CrossRef]
- Isazadeh, H.; Oruji, F.; Shabani, S.; Behroozi, J.; Nasiri, H.; Isazadeh, A.; Akbari, M. Advances in siRNA delivery approaches in cancer therapy: Challenges and opportunities. Mol. Biol. Rep. 2023, 50, 9529–9543. [Google Scholar] [CrossRef]
- Zhang, X.; Cai, A.; Gao, Y.; Zhang, Y.; Duan, X.; Men, K. Treatment of Melanoma by Nano-conjugate-Delivered Wee1 siRNA. Mol. Pharm. 2021, 18, 3387–3400. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Shi, X.; Song, H.; Zhang, C.; Wang, X.; Huang, P.; Dong, A.; Zhang, Y.; Kong, D.; Wang, W. Polymer-lipid hybrid nanovesicle-enabled combination of immunogenic chemotherapy and RNAi-mediated PD-L1 knockdown elicits antitumor immunity against melanoma. Biomaterials 2021, 268, 120579. [Google Scholar] [CrossRef]
- Li, C.; Han, X. Melanoma Cancer Immunotherapy Using PD-L1 siRNA and Imatinib Promotes Cancer-Immunity Cycle. Pharm. Res. 2020, 37, 109. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Gonzalez, M.R.; Vazquez-Garza, E.; Garcia-Rivas, G.; Rodriguez-Aguayo, C.; Chavez-Reyes, A. Therapeutic Effects of WT1 Silencing via Respiratory Administration of Neutral DOPC Liposomal-siRNA in a Lung Metastasis Melanoma Murine Model. Non-Coding RNA 2023, 9, 21. [Google Scholar] [CrossRef]
- Ruan, R.; Chen, M.; Sun, S.; Wei, P.; Zou, L.; Liu, J.; Gao, D.; Wen, L.; Ding, W. Topical and Targeted Delivery of siRNAs to Melanoma Cells Using a Fusion Peptide Carrier. Sci. Rep. 2016, 6, 29159. [Google Scholar] [CrossRef]
- Wells, D.A. A Review of CAR T-Cell Therapies Approved for the Treatment of Relapsed and Refractory B-Cell Lymphomas. J. Hematol. Oncol. Pharm. 2022, 12, 30–42. [Google Scholar]
- Dabas, P.; Danda, A. Revolutionizing cancer treatment: A comprehensive review of CAR-T cell therapy. Med. Oncol. 2023, 40, 275. [Google Scholar] [CrossRef]
- Bahmanyar, M.; Vakil, M.K.; Al-Awsi, G.R.L.; Kouhpayeh, S.A.; Mansoori, H.; Mansoori, Y.; Salahi, A.; Nikfar, G.; Tavassoli, A.; Behmard, E.; et al. Opportunities and obstacles for the melanoma immunotherapy using T cell and chimeric antigen receptor T (CAR-T) applications: A literature review. Mol. Biol. Rep. 2022, 49, 10627–10633. [Google Scholar] [CrossRef]
- Soltantoyeh, T.; Akbari, B.; Karimi, A.; Chalbatani, G.M.; Ghahri-Saremi, N.; Hadjati, J.; Hamblin, M.R.; Mirzaei, H.R. Chimeric Antigen Receptor (CAR) T Cell Therapy for Metastatic Melanoma: Challenges and Road Ahead. Cells 2021, 10, 1450. [Google Scholar] [CrossRef]
- Rosa, C.M.; Vancheswaran, A.; Ariyan, C.E. T-cell immunotherapy for melanoma. Surg. Oncol. 2024, 57, 102160. [Google Scholar]
- Jilani, S.; Saco, J.D.; Mugarza, E.; Pujol-Morcillo, A.; Chokry, J.; Ng, C.; Abril-Rodriguez, G.; Berger-Manerio, D.; Pant, A.; Hu, J.; et al. CAR-T cell therapy targeting surface expression of TYRP1 to treat cutaneous and rare melanoma subtypes. Nat. Commun. 2024, 15, 1244. [Google Scholar] [CrossRef] [PubMed]
- Papaioannou, I.; Owen, J.S.; Yáñez-Muñoz, R.J. Clinical applications of gene therapy for rare diseases: A review. Int. J. Exp. Pathol. 2023, 104, 154–176. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.; Yan, S.C.; Segherlou, Z.H.; Hosseini-Siyanaki, M.-R.; Poe, J.; Perez-Vega, C.; Chiocca, E.A.; Lucke-Wold, B. Oncolytic Viral Therapy: A Review and Promising Future Directions. J. Neurosurg. 2024, 140, 319–327. [Google Scholar] [CrossRef]
- Haitz, K.; Khosravi, H.; Lin, J.Y.; Menge, T.; Nambudiri, V.E. Review of talimogene laherparepvec: A first-in-class oncolytic viral treatment of advanced melanoma. J. Am. Acad. Dermatol. 2020, 83, 189–196. [Google Scholar] [CrossRef]
- Dummer, R.; Hoeller, C.; Gruter, I.P.; Michielin, O. Combining talimogene laherparepvec with immunotherapies in melanoma and other solid tumors. Cancer Immunol. Immunother. 2017, 66, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Kalsi, S.; Galenkamp, A.L.; Singh, R.; Khosla, A.A.; McGranaghan, P.; Cintolo-Gonzalez, J. Talimogene laherparepvec (T-VEC) and Emerging Intralesional Immunotherapies for Metastatic Melanoma: A Review. Curr. Oncol. Rep. 2024, 26, 1651–1663. [Google Scholar] [CrossRef]
- Armstrong, A.W.; Read, C. Pathophysiology, clinical presentation, and treatment of psoriasis: A review. JAMA 2020, 323, 1945–1960. [Google Scholar] [CrossRef]
- Ni, X.; Lai, Y. Keratinocyte: A trigger or an executor of psoriasis? J. Leukoc. Biol. 2020, 108, 485–491. [Google Scholar] [CrossRef]
- Lee, H.J.; Kim, M. Challenges and Future Trends in the Treatment of Psoriasis. Int. J. Mol. Sci. 2023, 24, 13313. [Google Scholar]
- Zhou, X.; Chen, Y.; Cui, L.; Shi, Y.; Guo, C. Advances in the pathogenesis of psoriasis: From keratinocyte perspective. Cell Death Dis. 2022, 13, 81. [Google Scholar] [CrossRef]
- Xiao, C.Y.; Zhu, Z.L.; Zhang, C.; Fu, M.; Qiao, H.J.; Wang, G.; Dang, E.L. Small interfering RNA targeting of keratin 17 reduces inflammation in imiquimod-induced psoriasis-like dermatitis. Chin. Med. J. 2020, 133, 2910–2918. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, F.; Zhao, J.; Wei, K.; Jiang, P.; Shi, Y.; Chang, C.; Zheng, Y.; Shan, Y.; Li, Y.; He, B.; et al. Targeted siRNA Therapy for Psoriasis: Translating Preclinical Potential into Clinical Treatments. Immunotargets Ther. 2024, 13, 259–271. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kong, Y.; Wu, R.; Zhang, S.; Zhao, M.; Wu, H.; Lu, Q.; Fu, S.; Su, Y. Wilms’ tumor 1-associating protein contributes to psoriasis by promoting keratinocytes proliferation via regulating cyclinA2 and CDK2. Int. Immunopharmacol. 2020, 88, 106918. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.; Su, Y.; Wu, R.; Zhang, P.; Feng, C. Overexpression of Wilms tumor 1 promotes IL-1β expression by upregulating histone acetylation in keratinocytes. Int. Immunopharmacol. 2021, 96, 107793. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Zhang, B.; Xie, H.; Huang, C.; Wu, Q. GRHL2 regulates keratinocyte EMT-MET dynamics and scar formation during cutaneous wound healing. Cell Death Dis. 2024, 15, 748. [Google Scholar] [CrossRef]
- Desmet, E.; Van Gele, M.; Grine, L.; Remaut, K.; Lambert, J. Towards the development of a RNAi-based topical treatment for psoriasis: Proof-of-concept in a 3D psoriasis skin model. Exp. Dermatol. 2018, 27, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Martinez Junior, A.M.; Ruiz, T.F.R.; Vilamaior, P.S.L.; Tiera, V.A.O.; Taboga, S.R.; Tiera, M.J. Topical delivery of siRNA to psoriatic skin model using high molecular weight chitosan derivatives: In vitro and in vivo studies. Drug Deliv. Transl. Res. 2025. ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Mandal, A.; Kumbhojkar, N.; Reilly, C.; Dharamdasani, V.; Ukidve, A.; Ingber, D.E.; Mitragotri, S. Treatment of psoriasis with NFKBIZ siRNA using topical ionic liquid formulations. Sci Adv. 2020, 6, eabb6049. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lee, W.R.; Chou, W.L.; Lin, Z.C.; Sung, C.T.; Lin, C.Y.; Fang, J.Y. Laser-assisted nanocarrier delivery to achieve cutaneous siRNA targeting for attenuating psoriasiform dermatitis. J. Control. Release 2022, 347, 590–606. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.C.; Hung, C.F.; Aljuffali, I.A.; Lin, M.H.; Fang, J.Y. RNA-Based Antipsoriatic Gene Therapy: An Updated Review Focusing on Evidence from Animal Models. Drug Des. Dev. Ther. 2024, 18, 1277–1296. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhou, J.; Bian, H.; Wu, N. Protein inhibitor of activated STAT3 (PIAS3) attenuates psoriasis and associated inflammation. J. Dermatol. 2023, 50, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Ndeupen, S.; Qin, Z.; Jacobsen, S.; Bouteau, A.; Estanbouli, H.; Igyártó, B.Z. The mRNA-LNP platform’s lipid nanoparticle component used in preclinical vaccine studies is highly inflammatory. iScience 2021, 24, 103479. [Google Scholar] [CrossRef]
- Zeng, A.; Liu, Y.; Wang, P.; Cao, Y.; Guo, W. Using siRNA-Based Anti-Inflammatory Lipid Nanoparticles for Gene Regulation in Psoriasis. Int. J. Nanomed. 2025, 20, 4519–4533. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ahn, I.; Kang, C.S.; Han, J. Where should siRNAs go: Applicable organs for siRNA drugs. Exp. Mol. Med. 2023, 55, 1283–1292. [Google Scholar] [CrossRef]
- Karar, J.; Maity, A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, J.; Fan, H.; Wang, T.; Lin, L.; Cai, T. Silencing KRT16 inhibits keratinocyte proliferation and VEGF secretion in psoriasis via inhibition of ERK signaling pathway. Kaohsiung J. Med. Sci. 2019, 35, 284–296. [Google Scholar] [CrossRef]
- Yi, R.C.; Akbik, M.; Smith, L.R.; Klionsky, Y.; Feldman, S.R. Therapeutic Advancements in Psoriasis and Psoriatic Arthritis. J. Clin. Med. 2025, 14, 1312. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Las Heras, K.; Igartua, M.; Santos-Vizcaino, E.; Hernandez, R.M. Chronic wounds: Current status, available strategies and emerging therapeutic solutions. J. Control. Release 2020, 328, 532–550. [Google Scholar] [CrossRef]
- Cwajda-Białasik, J.; Mościcka, P.; Szewczyk, M. Selected methods of treatment of chronic wounds. Pielęgniarstwo Chir. I Angiol./Surg. Vasc. Nurs. 2019, 13, 1–11. [Google Scholar]
- Mullin, J.A.; Rahmani, E.; Kiick, K.L.; Sullivan, M.O. Growth factors and growth factor gene therapies for treating chronic wounds. Bioeng. Transl. Med. 2024, 9, e10642. [Google Scholar] [CrossRef]
- Shaabani, E.; Sharifiaghdam, M.; Faridi-Majidi, R.; De Smedt, S.C.; Braeckmans, K.; Fraire, J.C. Gene therapy to enhance angiogenesis in chronic wounds. Mol. Ther.-Nucleic Acids 2022, 29, 871–899. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Kieser, R.E.; Khan, S.; Bejar, N.; Kiss, D.L. The Dawning of a New Enterprise: RNA Therapeutics for the Skin. J. Dermatol. Skin Sci. 2023, 5, 4–13. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Karimzadeh, F.; Soltani Fard, E.; Nadi, A.; Malekzadeh, R.; Elahian, F.; Mirzaei, S.A. Advances in skin gene therapy: Utilizing innovative dressing scaffolds for wound healing, a comprehensive review. J. Mater. Chem. B. 2024, 12, 6033–6062. [Google Scholar] [CrossRef] [PubMed]
- Karam, F.; Sayadi, M.; Dadi, S.; Sarab, G.A. Overexpression of miR-192 in fibroblasts accelerates wound healing in diabetic rats: Research article. Eur. J. Med. Res. 2025, 30, 239. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koutsoukos, S.A.; Bilousova, G. Highlights of Gene and Cell Therapy for Epidermolysis Bullosa and Ichthyosis. Dermatol. Ther. 2024, 14, 2379–2392. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joosten, M.D.W.; Clabbers, J.M.K.; Jonca, N.; Mazereeuw-Hautier, J.; Gostyński, A.H. New developments in the molecular treatment of ichthyosis: Review of the literature. Orphanet J. Rare Dis. 2022, 17, 1–15. [Google Scholar]
- Sercia, L.; Romano, O.; Marini, G.; Enzo, E.; Forcato, M.; De Rosa, L.; De Luca, M. A cellular disease model toward gene therapy of TGM1-dependent lamellar ichthyosis. Mol. Ther. Methods Clin. Dev. 2024, 32, 101311. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chulpanova, D.S.; Shaimardanova, A.A.; Ponomarev, A.S.; Elsheikh, S.; Rizvanov, A.A.; Solovyeva, V.V. Current Strategies for the Gene Therapy of Autosomal Recessive Congenital Ichthyosis and Other Types of Inherited Ichthyosis. Int. J. Mol. Sci. 2022, 23, 2506. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peña-Corona, S.I.; Gutiérrez-Ruiz, S.C.; Echeverria, M.L.D.C.; Cortés, H.; González-Del Carmen, M.; Leyva-Gómez, G. Advances in the treatment of autosomal recessive congenital ichthyosis, a look towards the repositioning of drugs. Front. Pharmacol. 2023, 14, 1274248. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]

{kind=link}
{kind=link}
| RNA Name | Target Gene/Pathway | Role in Psoriasis |
|---|---|---|
| miR-155 | SOCS1 | Promotes inflammation via Th17 pathway |
| miR-340 | STAT3 | Inhibits keratinocyte hyperproliferation |
| miR-21 | Multiple (e.g., TGF-β, immune regulators) | Enhances inflammation and epidermal thickening |
| miR-31 | PP6 | Promotes keratinocyte proliferation; drives inflammation via NF-κB and IL-1β/Th17 axis |
| IL-6 siRNA | IL-6 | Reduces inflammatory cytokine production |
| K17 siRNA | Keratin 17 | Lowers keratinocyte activation and inflammation |
| Pcsk9 siRNA | PCSK9 | Regulates lipid metabolism and inflammatory signaling |
| TNF-α siRNA | TNF-α | Decreases TNF-α expression; reduces skin inflammation |
| Disease | Summary of Gene Therapy Treatments | References |
| RDEB (Recessive Dystrophic Epidermolysis Bullosa) |
| [4,15,16,17,18] |
| [19,20,21,22] | |
| [5,14] | |
| Melanoma |
| [24,25,26,27,28,29] |
| [31,32,33,34,35,36] | |
| [37,38,39,40,41] | |
| Psoriasis |
| [46] |
| [42,43,44,45] | |
| [48,49] | |
| [59] | |
| Wound Healing |
| [63] |
| [64,69] | |
| [70] | |
| [68] | |
| Ichthyosis |
| [73] |
| [73] | |
| [72,74] | |
| [72] | |
| [75] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lisińska, W.; Cegiełka, P.; Zalewska, Z.; Bien, N.; Sobolewska-Sztychny, D.; Narbutt, J.; Lesiak, A. Gene Therapies in Dermatological Diseases: A Breakthrough in Treatment. Int. J. Mol. Sci. 2025, 26, 6592. https://doi.org/10.3390/ijms26146592
Lisińska W, Cegiełka P, Zalewska Z, Bien N, Sobolewska-Sztychny D, Narbutt J, Lesiak A. Gene Therapies in Dermatological Diseases: A Breakthrough in Treatment. International Journal of Molecular Sciences. 2025; 26(14):6592. https://doi.org/10.3390/ijms26146592
Chicago/Turabian StyleLisińska, Wiktoria, Patryk Cegiełka, Zuzanna Zalewska, Natalia Bien, Dorota Sobolewska-Sztychny, Joanna Narbutt, and Aleksandra Lesiak. 2025. "Gene Therapies in Dermatological Diseases: A Breakthrough in Treatment" International Journal of Molecular Sciences 26, no. 14: 6592. https://doi.org/10.3390/ijms26146592
APA StyleLisińska, W., Cegiełka, P., Zalewska, Z., Bien, N., Sobolewska-Sztychny, D., Narbutt, J., & Lesiak, A. (2025). Gene Therapies in Dermatological Diseases: A Breakthrough in Treatment. International Journal of Molecular Sciences, 26(14), 6592. https://doi.org/10.3390/ijms26146592