
Neuroinflammation Based Neurodegenerative In Vitro Model of SH-SY5Y Cells—Differential Effects on Oxidative Stress and Insulin Resistance Relevant to Alzheimer’s Pathology
 , , and
, , and
Abstract
1. Introduction
2. Results
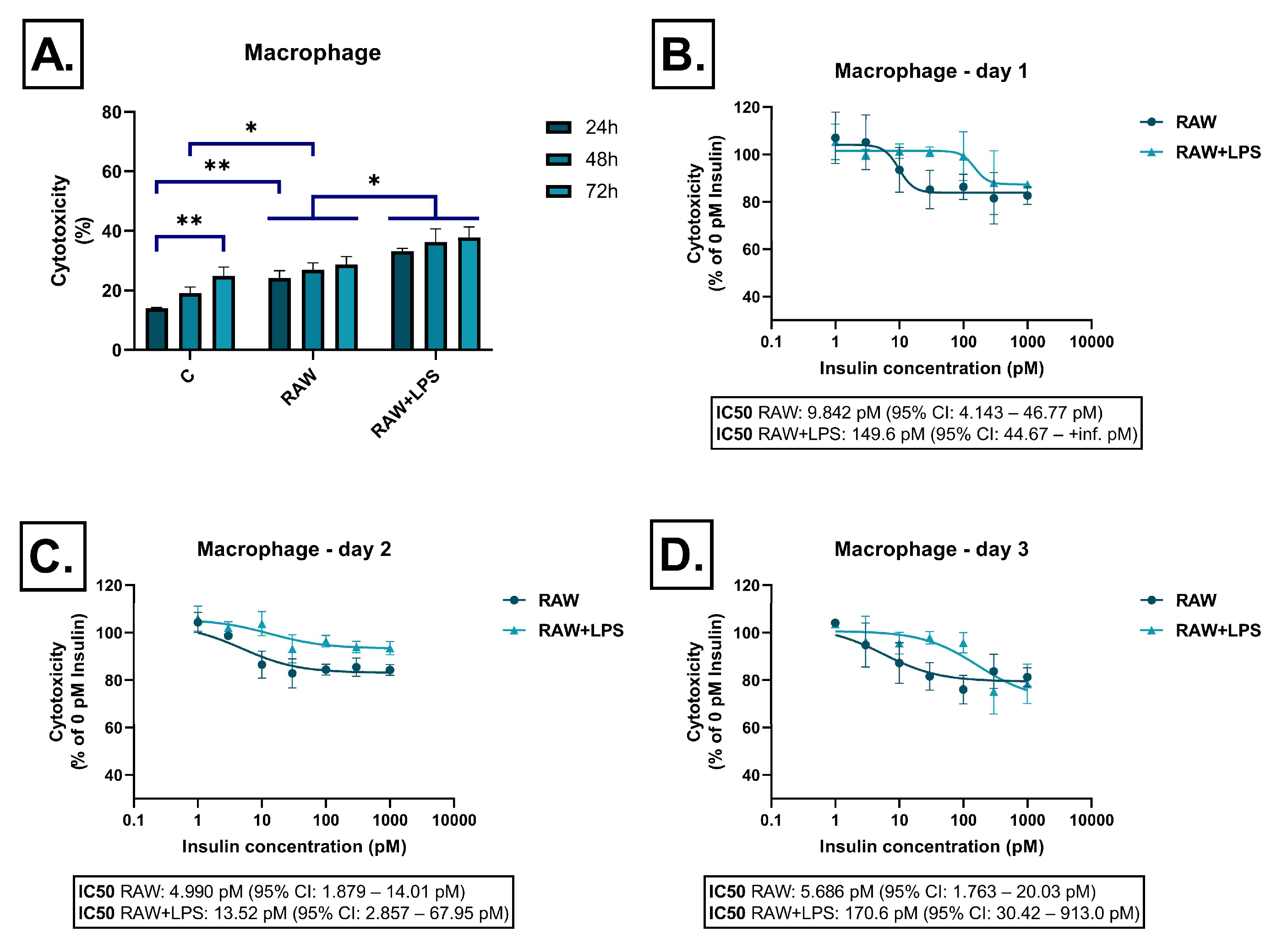
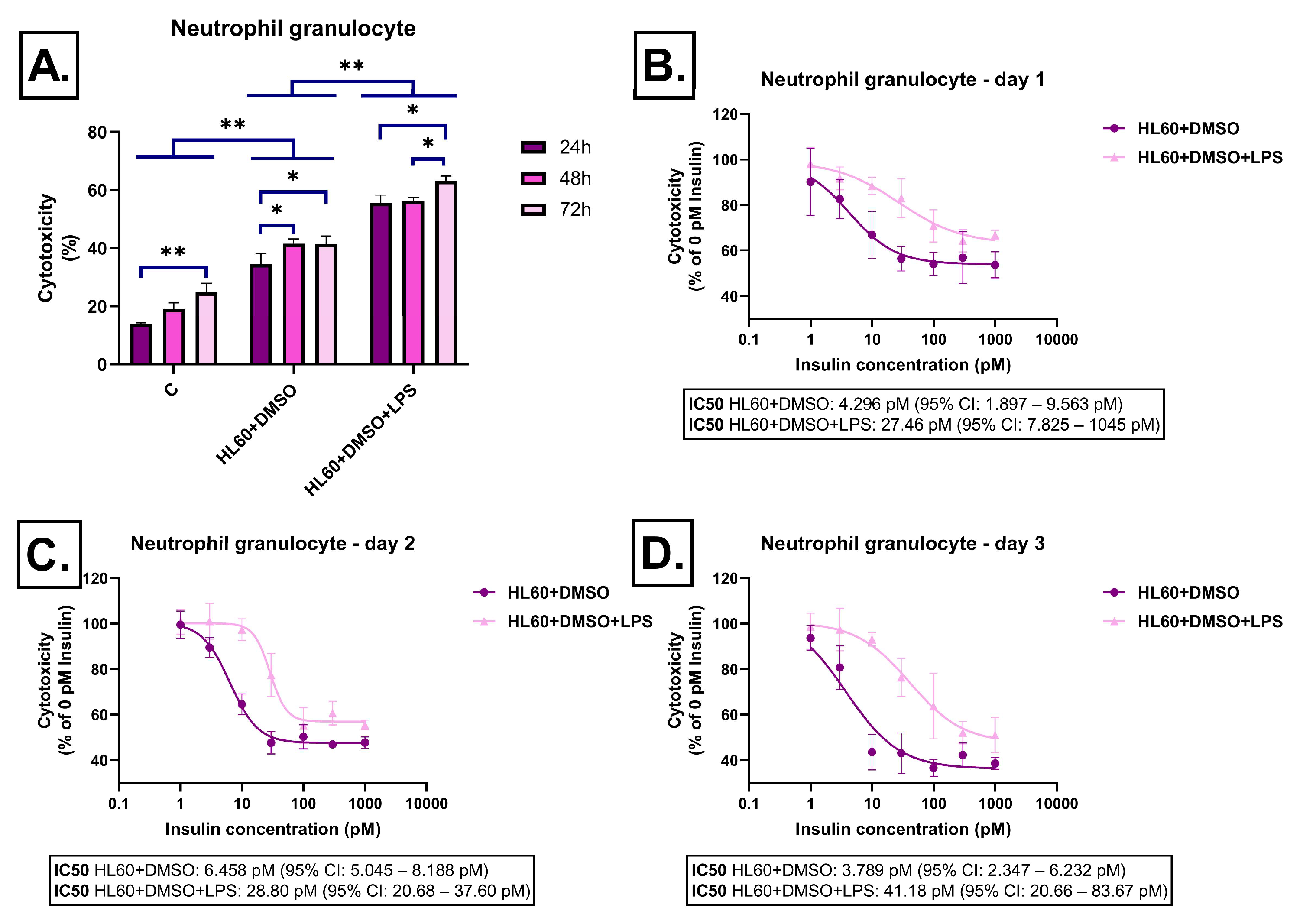
2.1. Effect of Immune Cell-Derived Conditioned Media on Cell Viability
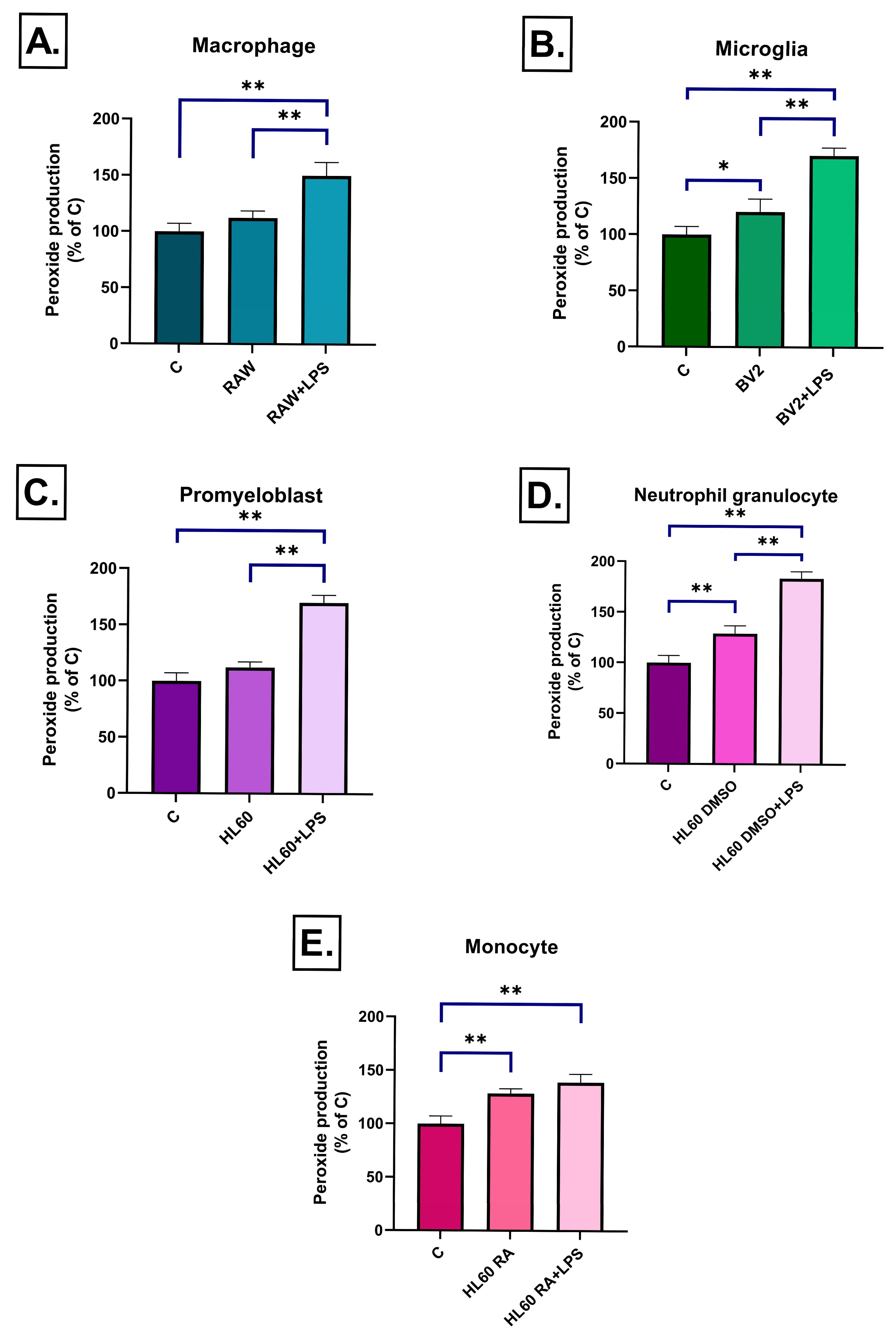
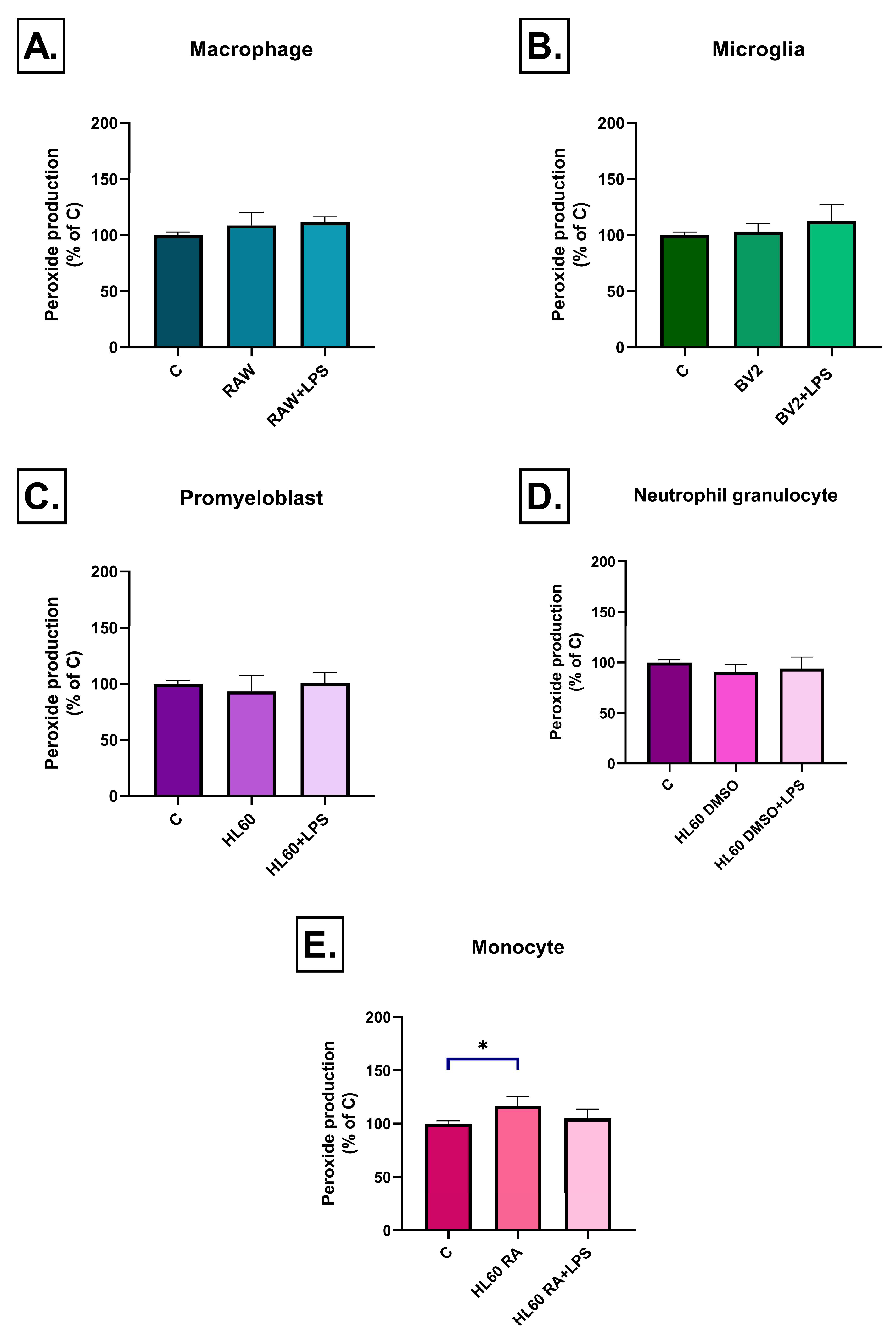
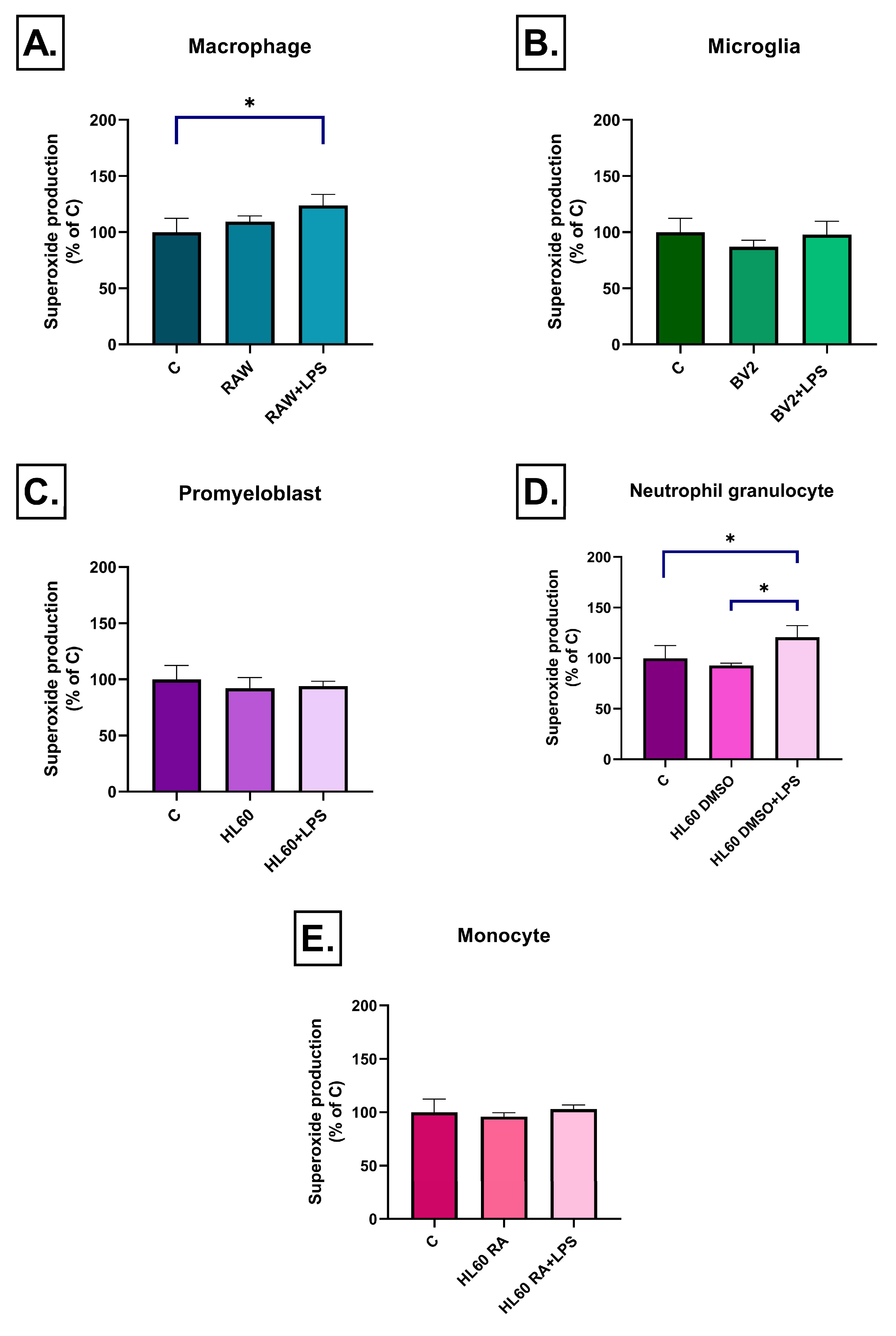
2.2. Effect of Conditioned Media on ROS Production
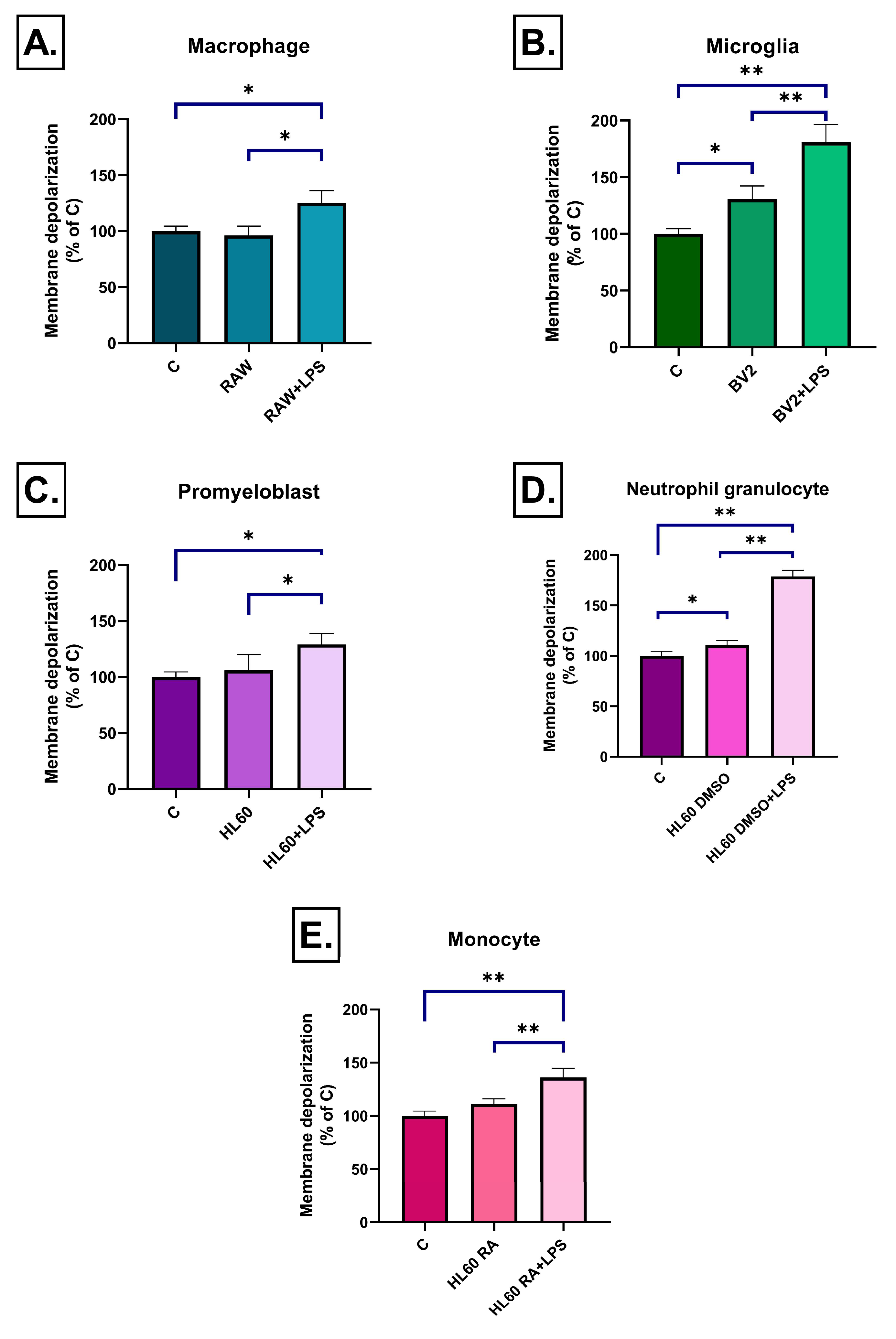
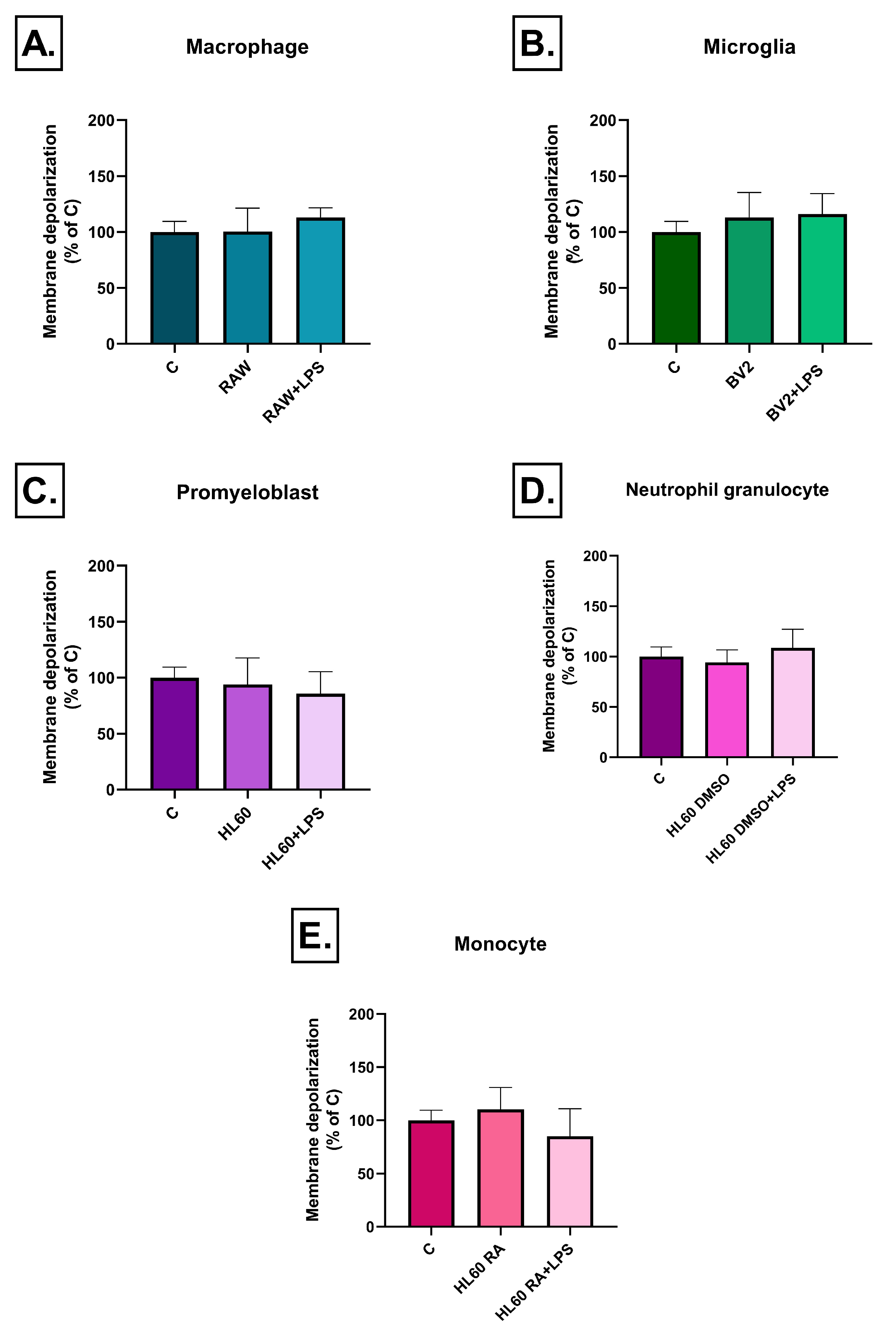
2.3. Effect of Inflammatory Conditioned Media on Mitochondrial Membrane Potential
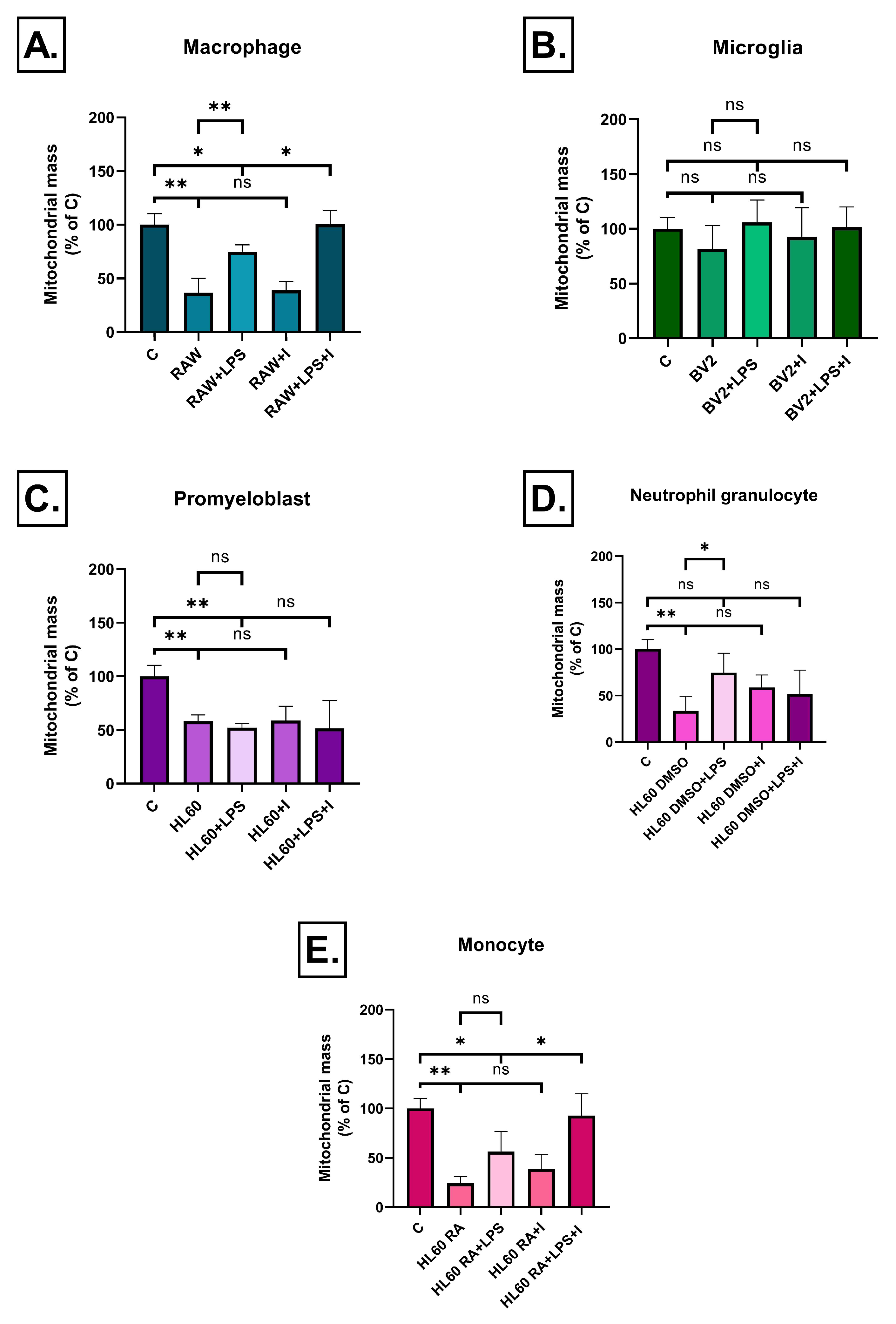
2.4. Effect of Inflammatory Conditioned Media on Mitochondrial Mass
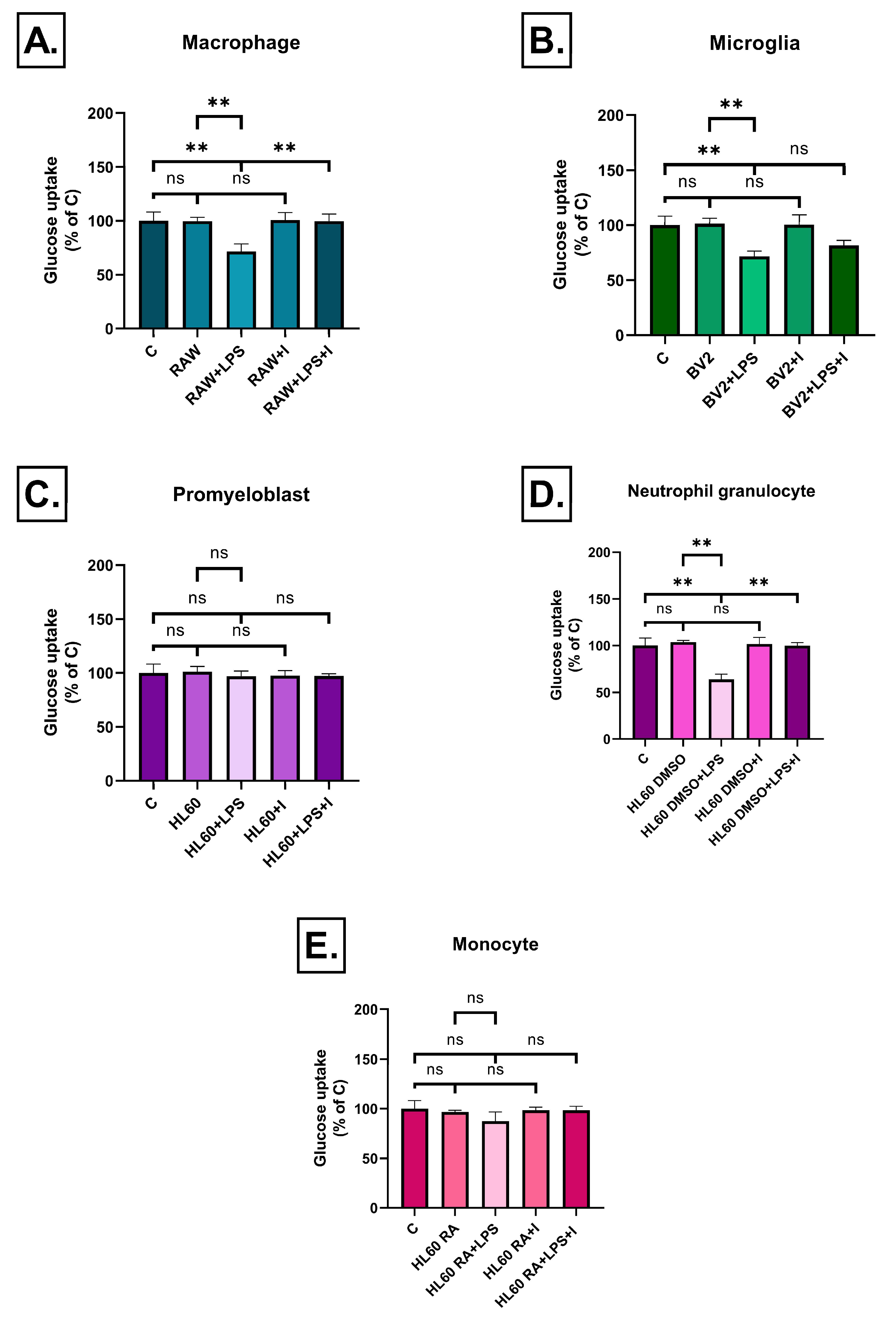
2.5. Effect of Inflammatory Conditioned Media on Glucose Uptake
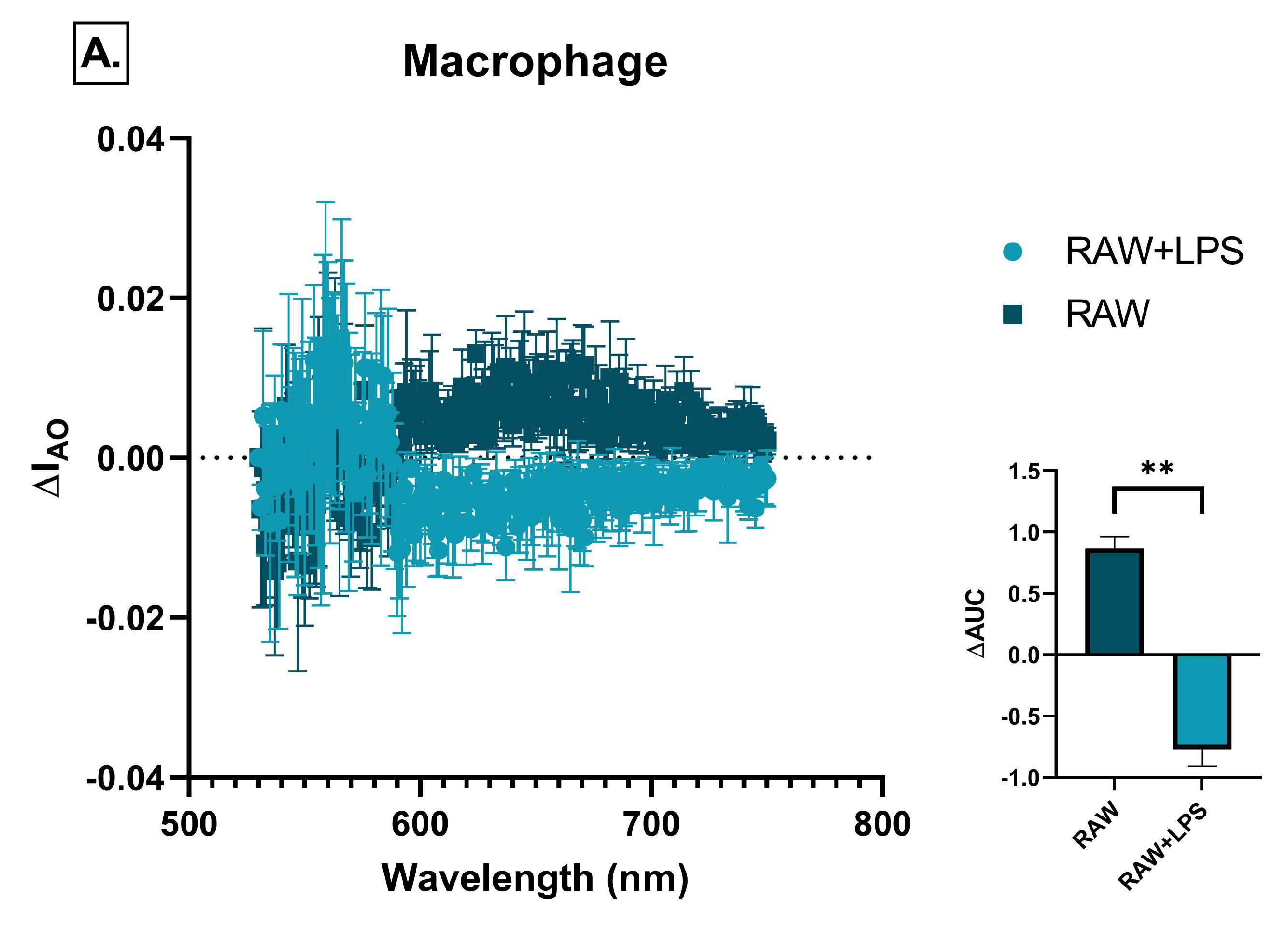
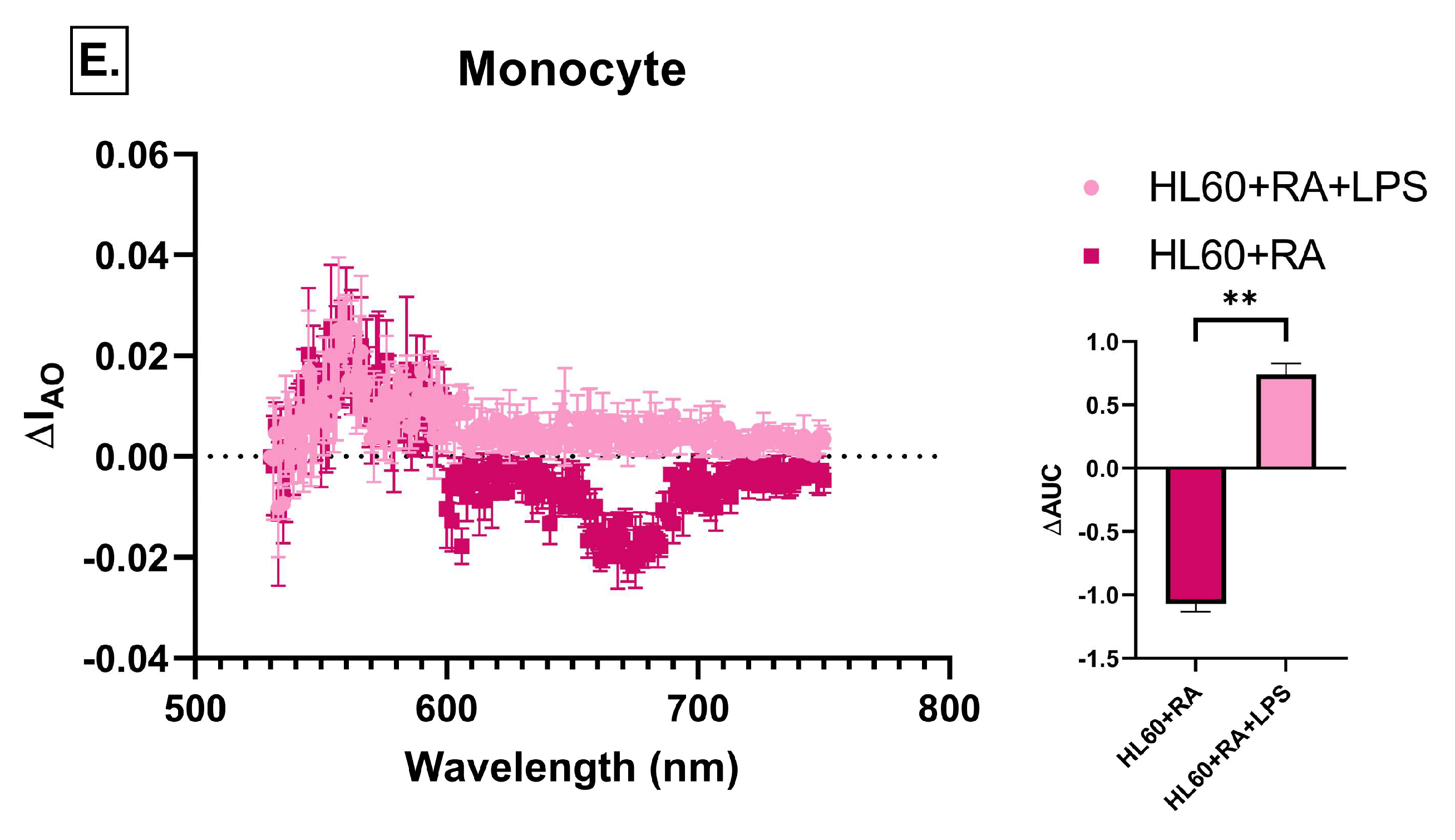
2.6. Effect of Immune Cell-Conditioned Media on Autophagic Activity
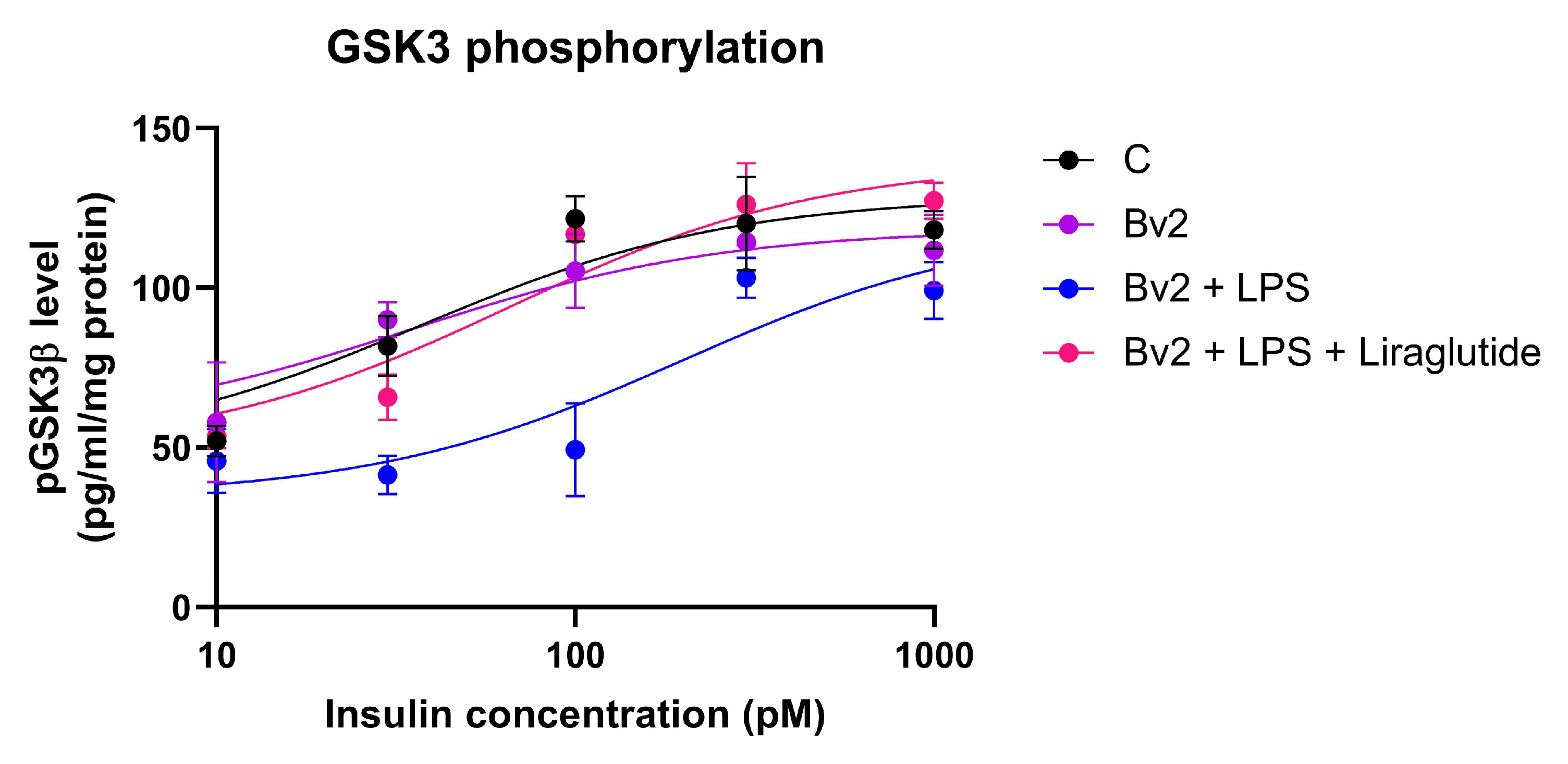
2.7. Effect of Immune Cell-Conditioned Media on Insulin Signaling
2.8. Effect of Immune Cell-Conditioned Media on Protein Aggregation
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Cultures and Treatments
- (1)
- Neutrophil-like differentiation using 1.25% DMSO [46]
- (2)
- Monocyte-like differentiation using 100 nM RA. In both cases, differentiation was initiated on day 0 and continued for five days, with medium replacement and differentiation agent supplementation on day 3. Undifferentiated HL60 cells were also used in the experiments.
4.3. Lactate Dehydrogenase (LDH) Release Cytotoxicity Assay
4.4. Assessment of ROS Levels and Mitochondrial Membrane Potential
4.5. Measurement of Mitochondrial Content
4.6. 2-NBDG Glucose Uptake Assay
4.7. Detection of Autophagic Vacuoles by Acridine Orange
4.8. ELISA for Phospho-GSK-3 Alpha/Beta (S21/S9)
4.9. Thioflavin S Protein Aggregation Staining
4.10. Statistical Evaluation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-NBDG | 2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose (fluorescent glucose analog) |
| AD | Alzheimer’s Disease |
| AO | Acridine Orange (fluorescent dye for acidic organelles/autophagy detection) |
| APP | Amyloid Precursor Protein |
| ATP | Adenosine Triphosphate |
| Aβ | Amyloid-Beta Peptide |
| C | Control |
| DAPI | 4′,6-Diamidino-2-Phenylindole (nuclear stain) |
| DCFDA | 2′,7′-Dichlorofluorescin Diacetate (indicator of hydrogen peroxide) |
| DMEM/F12 | Dulbecco’s Modified Eagle Medium/Nutrient Mixture F-12 |
| DMSO | Dimethyl Sulfoxide |
| ELISA | Enzyme-Linked Immunosorbent Assay |
| ER | Endoplasmic Reticulum |
| FBS | Fetal Bovine Serum |
| GLP-1 | Glucagon-Like Peptide-1 |
| GSK-3β | Glycogen Synthase Kinase-3 Beta |
| HE | Hydroethidine |
| IL-1β | Interleukin-1 Beta |
| IL-6 | Interleukin-6 |
| iPSC | Induced Pluripotent Stem Cell |
| JC-1 | 5,5′,6,6′-Tetrachloro-1,1′,3,3′-Tetraethylbenzimidazolylcarbocyanine Iodide |
| LDH | Lactate Dehydrogenase |
| LPS | Lipopolysaccharide |
| NFT | Neurofibrillary Tangle |
| NLRP3 | NOD-, LRR-, and Pyrin Domain-Containing Protein 3 |
| NO | Nitric Oxide |
| pGSK-3β | Phosphorylated Glycogen Synthase Kinase-3 Beta |
| PARP | Poly (ADP-Ribose) Polymerase |
| PBS | Phosphate-Buffered Saline |
| PI3K | Phosphoinositide 3-Kinase |
| RA | Retinoic Acid |
| ROS | Reactive Oxygen Species |
| SD | Standard Deviation |
| SH-SY5Y | Human Neuroblastoma Cell Line |
| Thioflavin S | Fluorescent dye for beta-sheet-rich protein aggregates |
| TNF-α | Tumor Necrosis Factor-Alpha |
References
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement. 2018, 4, 575–590. [Google Scholar] [CrossRef]
- Parbo, P.; Ismail, R.; Sommerauer, M.; Stokholm, M.G.; Hansen, A.K.; Hansen, K.V.; Amidi, A.; Schaldemose, J.L.; Gottrup, H.; Braendgaard, H.; et al. Does inflammation precede tau aggregation in early Alzheimer’s disease? A PET study. Neurobiol. Dis. 2018, 117, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Di Benedetto, S. Neuroimmune crosstalk in chronic neuroinflammation: Microglial interactions and immune modulation. Front. Cell. Neurosci. 2025, 19, 1575022. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Microglial phagocytosis of live neurons. Nat. Rev. Neurosci. 2014, 15, 209–216. [Google Scholar] [CrossRef]
- Simpson, D.S.A.; Oliver, P.L. ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease. Antioxidants 2020, 9, 743. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-beta in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Liu, Y.; Dai, Y.; Li, Q.; Chen, C.; Chen, H.; Song, Y.; Hua, F.; Zhang, Z. Beta-amyloid activates NLRP3 inflammasome via TLR4 in mouse microglia. Neurosci. Lett. 2020, 736, 135279. [Google Scholar] [CrossRef]
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2021, 17, 157–172. [Google Scholar] [CrossRef]
- Sultana, R.; Butterfield, D.A. Role of oxidative stress in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2010, 19, 341–353. [Google Scholar] [CrossRef] [PubMed]
- Bhat, A.H.; Dar, K.B.; Anees, S.; Zargar, M.A.; Masood, A.; Sofi, M.A.; Ganie, S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015, 74, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef]
- Liu, J.; Li, L. Targeting Autophagy for the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Front. Mol. Neurosci. 2019, 12, 203. [Google Scholar] [CrossRef]
- Pickford, F.; Masliah, E.; Britschgi, M.; Lucin, K.; Narasimhan, R.; Jaeger, P.A.; Small, S.; Spencer, B.; Rockenstein, E.; Levine, B.; et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J. Clin. Investig. 2008, 118, 2190–2199. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhang, Y.; Zhang, J.; Zhang, X.; Yang, G. Molecular Mechanism of Autophagy: Its Role in the Therapy of Alzheimer’s Disease. Curr. Neuropharmacol. 2020, 18, 720–739. [Google Scholar] [CrossRef]
- Bagamery, F.; Varga, K.; Kecsmar, K.; Vincze, I.; Szoko, E.; Tabi, T. Lack of insulin resistance in response to streptozotocin treatment in neuronal SH-SY5Y cell line. J. Neural Transm. 2020, 127, 71–80. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef]
- Kothari, V.; Luo, Y.; Tornabene, T.; O’Neill, A.M.; Greene, M.W.; Geetha, T.; Babu, J.R. High fat diet induces brain insulin resistance and cognitive impairment in mice. Biochim. Biophys. Acta Mol. Basis. Dis. 2017, 1863, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Varga, K.; Paszternak, A.; Kovacs, V.; Guczogi, A.; Sikur, N.; Patakfalvi, D.; Bagamery, F.; Szoko, E.; Tabi, T. Differential Cytoprotective Effect of Resveratrol and Its Derivatives: Focus on Antioxidant and Autophagy-Inducing Effects. Int. J. Mol. Sci. 2024, 25, 11274. [Google Scholar] [CrossRef]
- Hotamisligil, G.S.; Peraldi, P.; Budavari, A.; Ellis, R.; White, M.F.; Spiegelman, B.M. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science 1996, 271, 665–668. [Google Scholar] [CrossRef]
- Mullins, R.J.; Diehl, T.C.; Chia, C.W.; Kapogiannis, D. Insulin Resistance as a Link between Amyloid-Beta and Tau Pathologies in Alzheimer’s Disease. Front. Aging Neurosci. 2017, 9, 118. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef]
- Baker, L.D.; Cross, D.J.; Minoshima, S.; Belongia, D.; Watson, G.S.; Craft, S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 2011, 68, 51–57. [Google Scholar] [CrossRef]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef]
- de la Monte, S.M. Type 3 diabetes is sporadic Alzheimer׳s disease: Mini-review. Eur. Neuropsychopharmacol. 2014, 24, 1954–1960. [Google Scholar] [CrossRef]
- Park, J.; Wetzel, I.; Marriott, I.; Dreau, D.; D’Avanzo, C.; Kim, D.Y.; Tanzi, R.E.; Cho, H. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nat. Neurosci. 2018, 21, 941–951. [Google Scholar] [CrossRef]
- Boje, K.M.; Arora, P.K. Microglial-produced nitric oxide and reactive nitrogen oxides mediate neuronal cell death. Brain Res. 1992, 587, 250–256. [Google Scholar] [CrossRef]
- Combs, C.K.; Karlo, J.C.; Kao, S.C.; Landreth, G.E. beta-Amyloid stimulation of microglia and monocytes results in TNFalpha-dependent expression of inducible nitric oxide synthase and neuronal apoptosis. J. Neurosci. 2001, 21, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.; Hwang, I.; Park, S.; Hong, S.; Hwang, B.; Cho, Y.; Son, J.; Yu, J.W. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ. 2019, 26, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Chien, C.H.; Lee, M.J.; Liou, H.C.; Liou, H.H.; Fu, W.M. Microglia-Derived Cytokines/Chemokines Are Involved in the Enhancement of LPS-Induced Loss of Nigrostriatal Dopaminergic Neurons in DJ-1 Knockout Mice. PLoS ONE 2016, 11, e0151569. [Google Scholar] [CrossRef]
- Chung, L.Y.; Lin, Y.T.; Liu, C.; Tai, Y.C.; Lin, H.Y.; Lin, C.H.; Chen, C.C. Neuroinflammation Upregulated Neuronal Toll-Like Receptors 2 and 4 to Drive Synucleinopathy in Neurodegeneration. Front. Pharmacol. 2022, 13, 845930. [Google Scholar] [CrossRef]
- Sutinen, E.M.; Korolainen, M.A.; Hayrinen, J.; Alafuzoff, I.; Petratos, S.; Salminen, A.; Soininen, H.; Pirttila, T.; Ojala, J.O. Interleukin-18 alters protein expressions of neurodegenerative diseases-linked proteins in human SH-SY5Y neuron-like cells. Front. Cell. Neurosci. 2014, 8, 214. [Google Scholar] [CrossRef]
- Han, S.; Gao, H.; Chen, S.; Wang, Q.; Li, X.; Du, L.J.; Li, J.; Luo, Y.Y.; Li, J.X.; Zhao, L.C.; et al. Procyanidin A1 Alleviates Inflammatory Response induced by LPS through NF-kappaB, MAPK, and Nrf2/HO-1 Pathways in RAW264.7 cells. Sci. Rep. 2019, 9, 15087. [Google Scholar] [CrossRef]
- Facchin, B.M.; Dos Reis, G.O.; Vieira, G.N.; Mohr, E.T.B.; da Rosa, J.S.; Kretzer, I.F.; Demarchi, I.G.; Dalmarco, E.M. Inflammatory biomarkers on an LPS-induced RAW 264.7 cell model: A systematic review and meta-analysis. Inflamm. Res. 2022, 71, 741–758. [Google Scholar] [CrossRef]
- Zenaro, E.; Pietronigro, E.; Della Bianca, V.; Piacentino, G.; Marongiu, L.; Budui, S.; Turano, E.; Rossi, B.; Angiari, S.; Dusi, S.; et al. Neutrophils promote Alzheimer’s disease-like pathology and cognitive decline via LFA-1 integrin. Nat. Med. 2015, 21, 880–886. [Google Scholar] [CrossRef]
- Langsdorf, E.F.; Mao, X.; Chang, S.L. A role for reactive oxygen species in endotoxin-induced elevation of MOR expression in the nervous and immune systems. J. Neuroimmunol. 2011, 236, 57–64. [Google Scholar] [CrossRef]
- Jatczak-Pawlik, I.; Ewiak-Paszynska, A.; Domowicz, M.; Jurewicz, A.; Stasiolek, M. Intracellular Accumulation and Secretion of YKL-40 (CHI3L1) in the Course of DMSO-Induced HL-60 Cell Differentiation. Pharmaceuticals 2024, 17, 443. [Google Scholar] [CrossRef]
- Greuel, B.K.; Da Silva, D.E.; Robert-Gostlin, V.N.; Klegeris, A. Natural Compounds Oridonin and Shikonin Exhibit Potentially Beneficial Regulatory Effects on Select Functions of Microglia. Brain Sci. 2024, 14, 328. [Google Scholar] [CrossRef]
- Klegeris, A.; McGeer, P.L. Toxicity of human monocytic THP-1 cells and microglia toward SH-SY5Y neuroblastoma cells is reduced by inhibitors of 5-lipoxygenase and its activating protein FLAP. J. Leukoc. Biol. 2003, 73, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Liu, Y.; Wang, T.; Wei, S.J.; Block, M.L.; Wilson, B.; Liu, B.; Hong, J.S. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J. Biol. Chem. 2004, 279, 1415–1421. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.S.; et al. Aggregated alpha-synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Cena, V.; Gallego, C.; Comella, J.X. Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Babatunde, K.A.; Wang, X.; Hopke, A.; Lannes, N.; Mantel, P.Y.; Irimia, D. Chemotaxis and swarming in differentiated HL-60 neutrophil-like cells. Sci. Rep. 2021, 11, 778. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group | Description of Treatment |
|---|---|
| RAW264.7 | Macrophage cell line-produced factors |
| RAW264.7 + LPS | Activated macrophage cell line-produced factors |
| BV2 | Microglial cell line-produced factors |
| BV2 + LPS | Activated microglial cell line-produced factors |
| HL60 | Undifferentiated promyeloblast cell line-produced factors |
| HL60 + LPS | Activated, undifferentiated promyeloblast cell line-produced factors |
| HL60 + DMSO | Neutrophil-like HL60 cell line-produced factors |
| HL60 + DMSO + LPS | Activated, neutrophil-like HL60 cell line-produced factors |
| HL60 + RA | Monocyte-like HL60 cell line-produced factors |
| HL60 + RA + LPS | Activated, monocyte-like HL60 cell line-produced factors |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Böröczky, C.; Paszternák, A.; Laufer, R.; Tarnóczi, K.; Sikur, N.; Bagaméry, F.; Szökő, É.; Varga, K.; Tábi, T. Neuroinflammation Based Neurodegenerative In Vitro Model of SH-SY5Y Cells—Differential Effects on Oxidative Stress and Insulin Resistance Relevant to Alzheimer’s Pathology. Int. J. Mol. Sci. 2025, 26, 6581. https://doi.org/10.3390/ijms26146581
Böröczky C, Paszternák A, Laufer R, Tarnóczi K, Sikur N, Bagaméry F, Szökő É, Varga K, Tábi T. Neuroinflammation Based Neurodegenerative In Vitro Model of SH-SY5Y Cells—Differential Effects on Oxidative Stress and Insulin Resistance Relevant to Alzheimer’s Pathology. International Journal of Molecular Sciences. 2025; 26(14):6581. https://doi.org/10.3390/ijms26146581
Chicago/Turabian StyleBöröczky, Csenge, Alexandra Paszternák, Rudolf Laufer, Katinka Tarnóczi, Noémi Sikur, Fruzsina Bagaméry, Éva Szökő, Kamilla Varga, and Tamás Tábi. 2025. "Neuroinflammation Based Neurodegenerative In Vitro Model of SH-SY5Y Cells—Differential Effects on Oxidative Stress and Insulin Resistance Relevant to Alzheimer’s Pathology" International Journal of Molecular Sciences 26, no. 14: 6581. https://doi.org/10.3390/ijms26146581
APA StyleBöröczky, C., Paszternák, A., Laufer, R., Tarnóczi, K., Sikur, N., Bagaméry, F., Szökő, É., Varga, K., & Tábi, T. (2025). Neuroinflammation Based Neurodegenerative In Vitro Model of SH-SY5Y Cells—Differential Effects on Oxidative Stress and Insulin Resistance Relevant to Alzheimer’s Pathology. International Journal of Molecular Sciences, 26(14), 6581. https://doi.org/10.3390/ijms26146581