

Novel (1S,3R)-RSL3-Encapsulated Polyunsaturated Fatty Acid Rich Liposomes Sensitise Multiple Myeloma Cells to Ferroptosis-Mediated Cell Death
 , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Background
2. Results
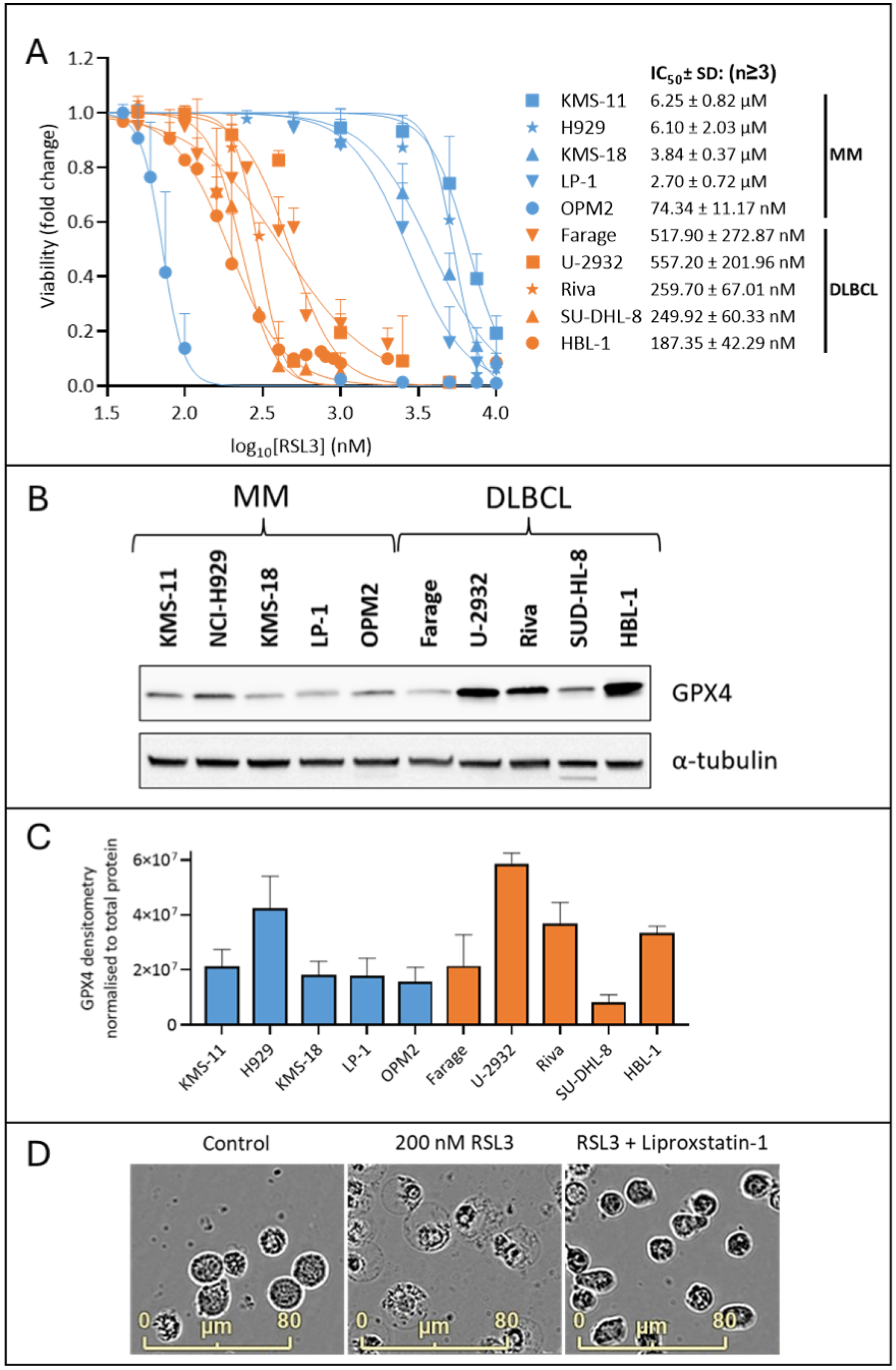
2.1. MM Cells Are Less Sensitive to the GPX4 Inhibitor, RSL3, than DLBCL Cells
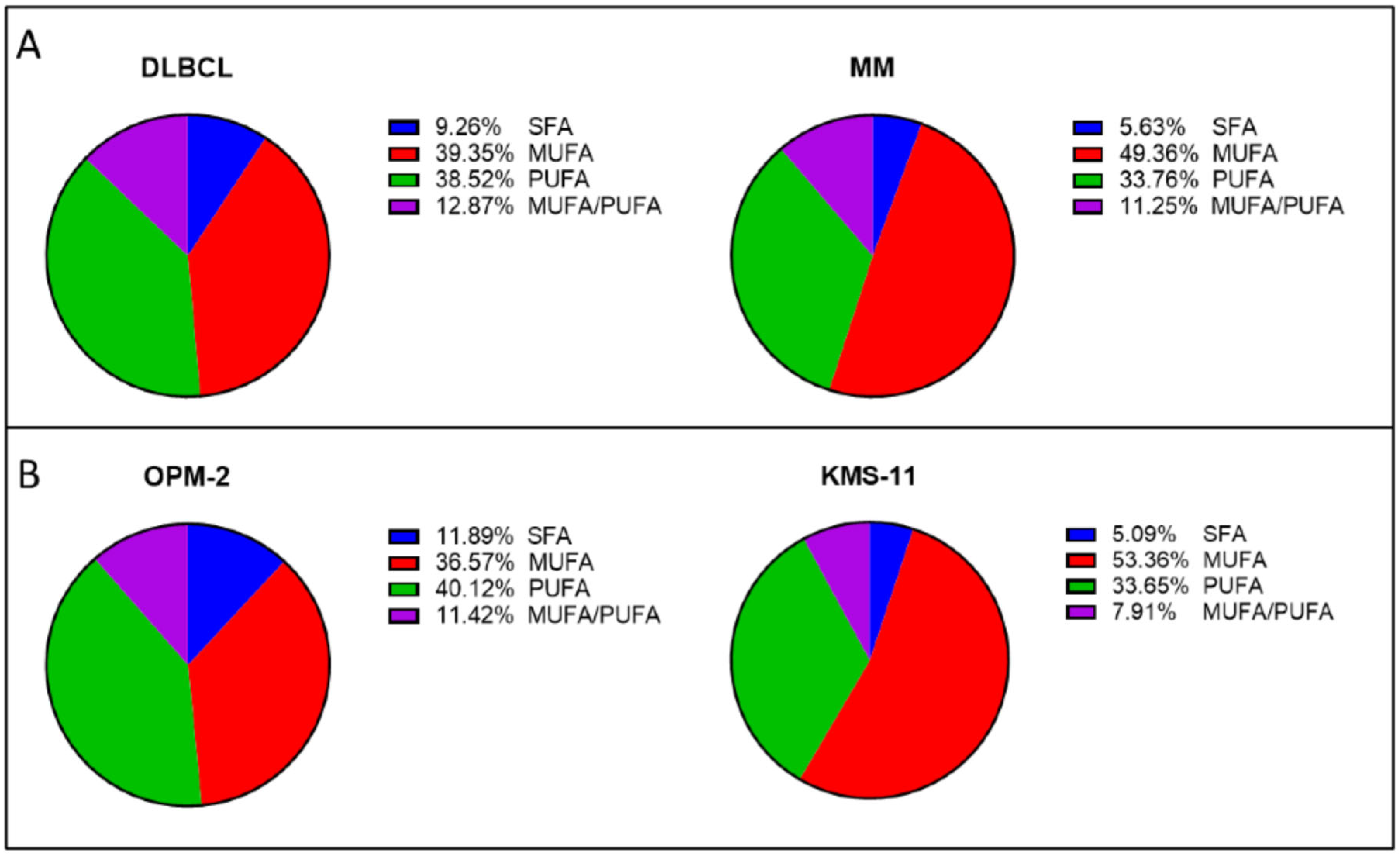
2.2. MM Cells Generally Contain Higher Proportions of PL-MUFA than DLBCL Cell Lines
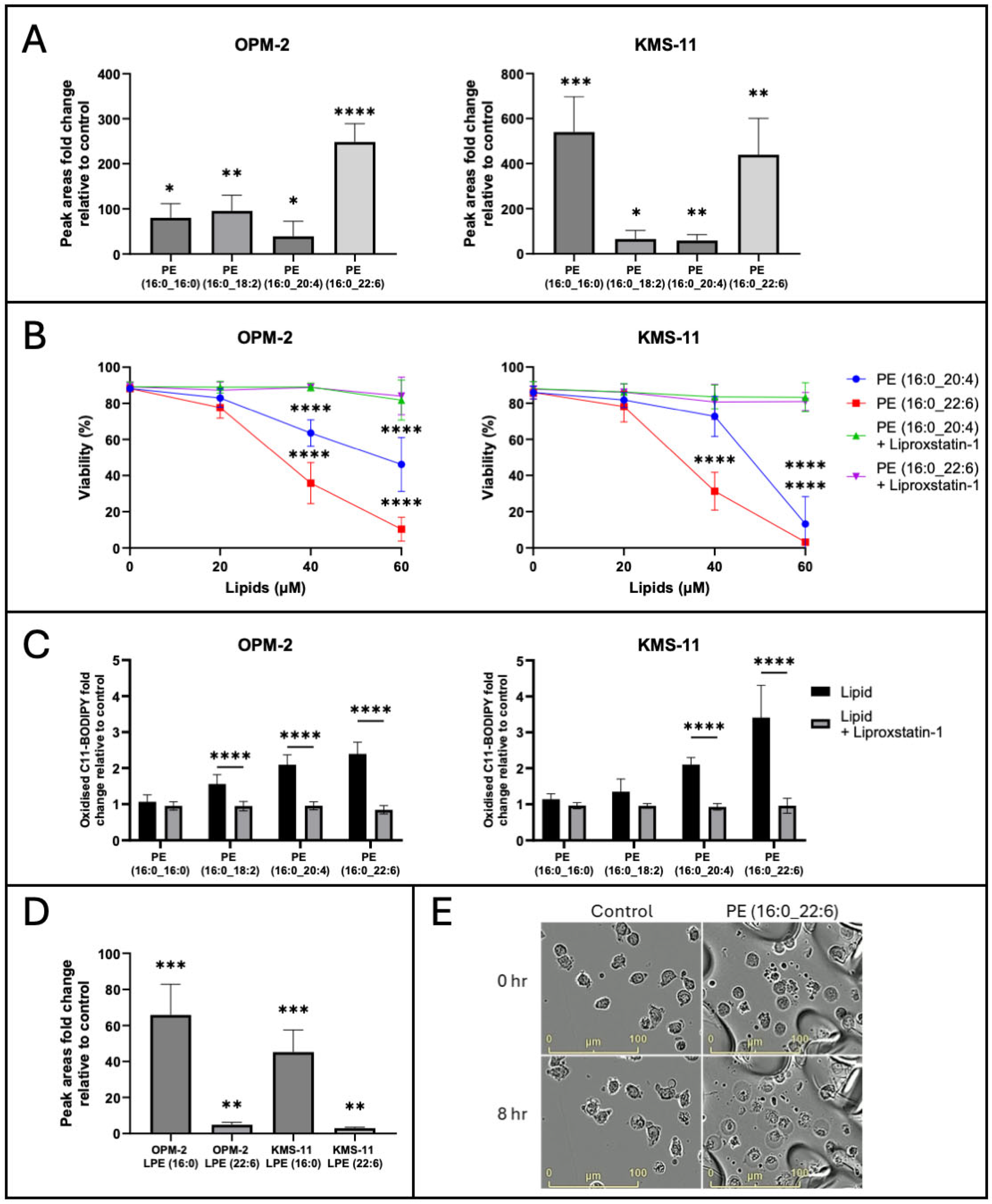
2.3. Exogenous PL-PUFA Induces Ferroptosis in MM Cells Proportional to the Degree of Acyl Chain Saturation
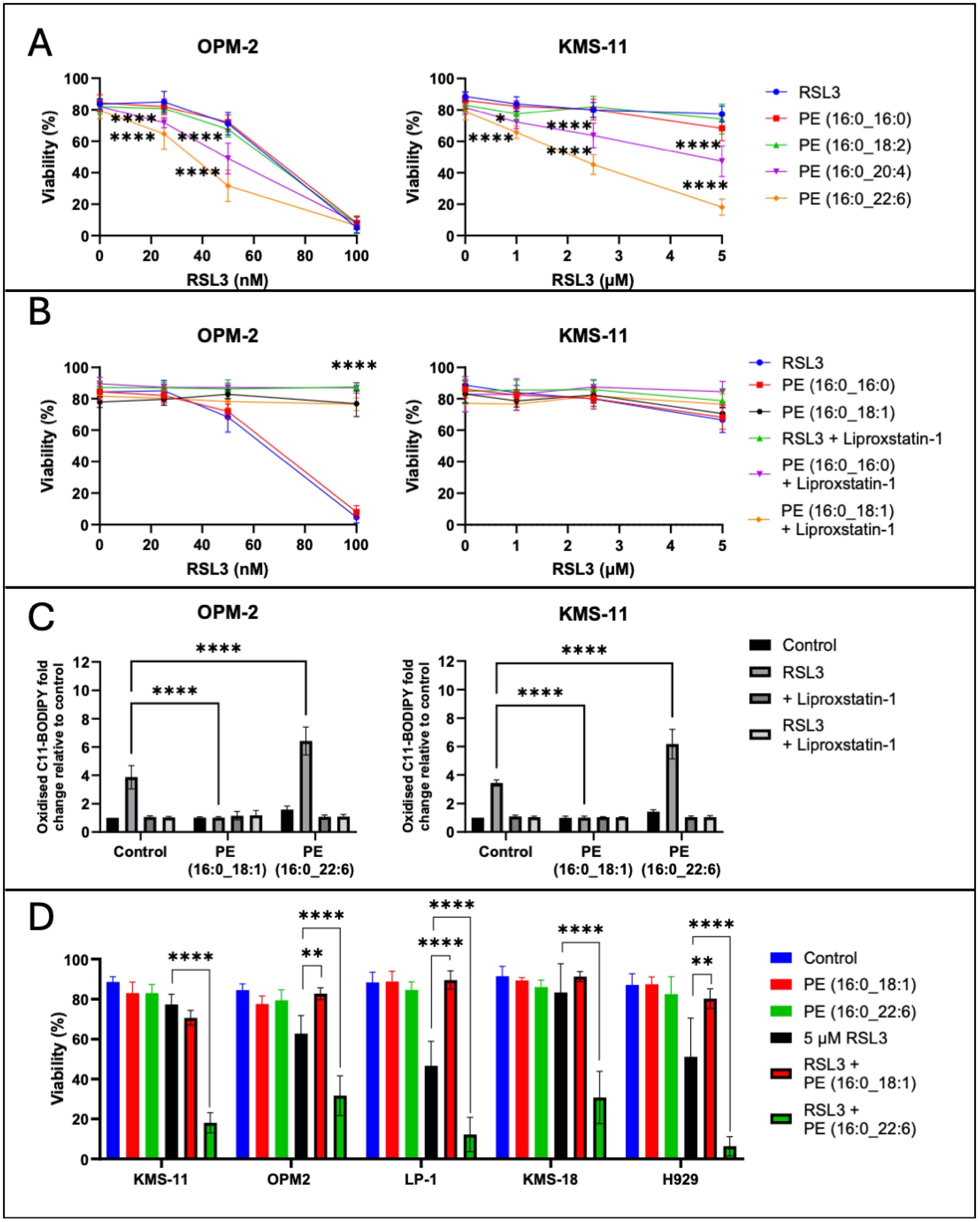
2.4. Exogenous PL-PUFA and RSL3 Synergise, Inducing Ferroptosis-Mediated Cell Death
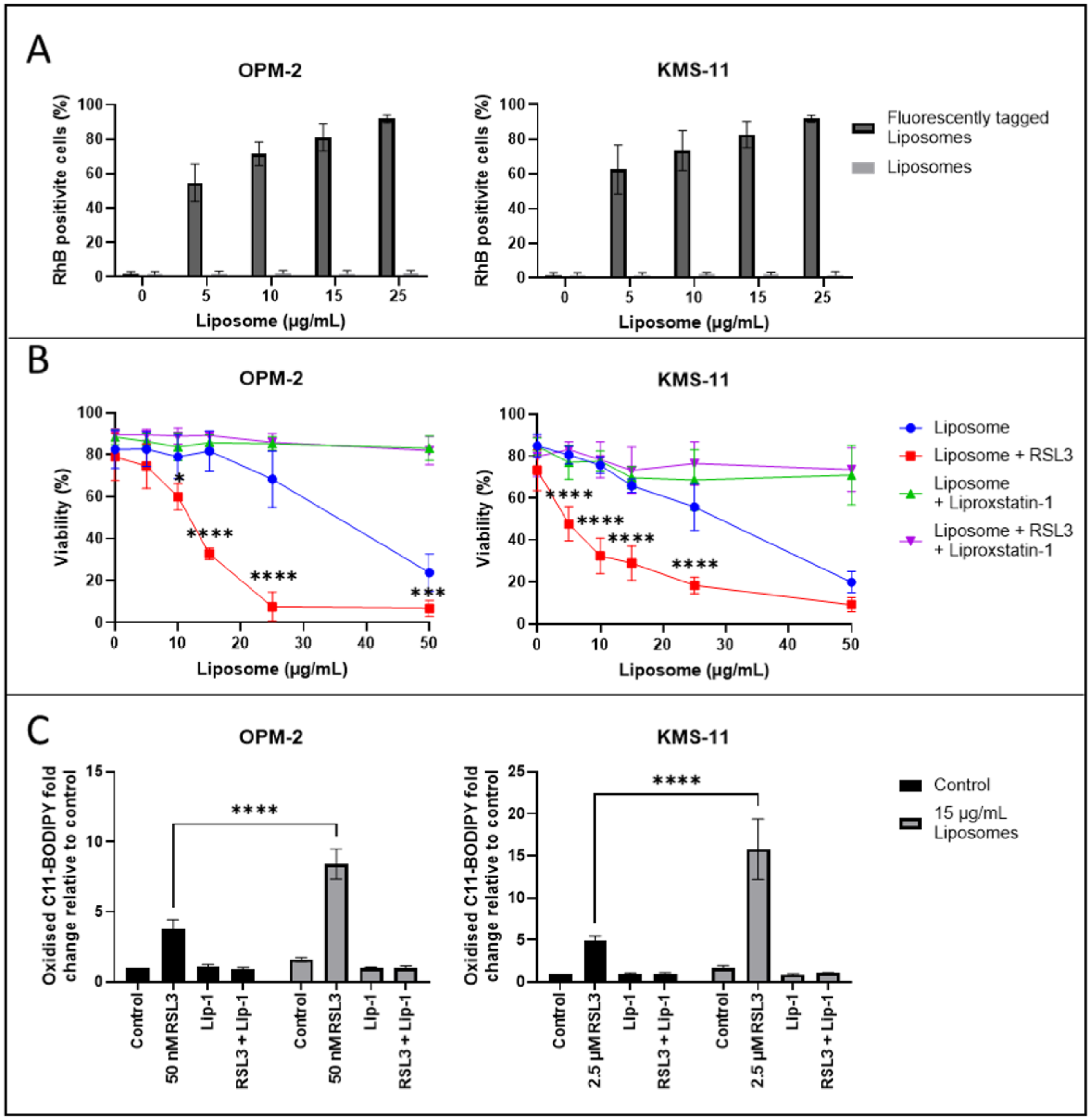
2.5. Induction of Ferroptosis in MM Cells by PL-PUFA-Rich Liposomes
2.6. PL-PUFA-Rich Liposomes Containing RSL3 Induce Ferroptosis-Mediated Cell Death of MM Cells
3. Discussion
4. Methods
4.1. Drugs, Chemicals, and Other Reagents
4.2. Cell Culture
4.3. Assessment of Cell Viability
4.4. Assessment of Lipid ROS
4.5. Sample Preparation for Lipidomic Analyses
4.6. Live Cell Imaging
4.7. Western Blotting
4.8. Liposome Preparation
4.8.1. Micro-Fluidics Synthesis
4.8.2. Liposome Characterisation
4.8.3. Assessment of Liposome Uptake
4.8.4. Assessment of Liposome RSL3 Encapsulation
4.8.5. High-Performance Liquid Chromatography
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
List of Abbreviations
| DLBCL | Diffuse large B cell lymphoma |
| FA | Fatty acid |
| GPX4 | Glutathione peroxidase 4 |
| HPLC | High-performance liquid chromatography |
| LC-MS | Liquid chromatography–mass spectrometry |
| LIP-1 | Liproxstatin-1 |
| LOX | Lipooxygenase |
| LPC | Lysophosphatidylcholine |
| LPE | Lysophosphatidylethanolamine |
| MM | Multiple myeloma |
| MUFA | Monounsaturated fatty acid |
| NEC-1 | Necrostatin-1 |
| PC | Phosphatidylcholine |
| PCD | Programmed cell death |
| PDI | Polydispersity index |
| PE | Phosphatidylethanolamine |
| PI | Propidium iodide |
| PL | Phospholipid |
| PS | Phosphatidylserine |
| PUFA | Polyunsaturated fatty acid |
| RIPK | Receptor-interacting protein kinase |
| ROS | Reactive oxygen species |
| RSL3 | (1S,3R)-RSL3 |
| SFA | Saturated fatty acid |
References
- Rasche, L.; Chavan, S.S.; Stephens, O.W.; Patel, P.H.; Tytarenko, R.; Ashby, C.; Bauer, M.; Stein, C.; Deshpande, S.; Wardell, C.; et al. Spatial genomic heterogeneity in multiple myeloma revealed by multi-region sequencing. Nat. Commun. 2017, 8, 268. [Google Scholar] [CrossRef]
- Cowan, A.J.; Green, D.J.; Kwok, M.; Lee, S.; Coffey, D.G.; Holmberg, L.A.; Tuazon, S.; Gopal, A.K.; Libby, E.N. Diagnosis and Management of Multiple Myeloma: A Review. JAMA 2022, 327, 464–477. [Google Scholar] [CrossRef]
- Australian Institute of Health and Welfare. Cancer Data in Australia. Available online: https://www.aihw.gov.au/reports/cancer/cancer-data-in-australia/contents/survival (accessed on 20 March 2025).
- Shah, U.A.; Mailankody, S. Emerging immunotherapies in multiple myeloma. BMJ 2020, 370, 3176. [Google Scholar] [CrossRef]
- Kazandjian, D.; Landgren, O. A look backward and forward in the regulatory and treatment history of multiple myeloma: Approval of novel-novel agents, new drug development, and longer patient survival. Semin. Oncol. 2016, 43, 682–689. [Google Scholar] [CrossRef] [PubMed]
- Al-Odat, O.S.; Guirguis, D.A.; Schmalbach, N.K.; Yao, G.; Budak-Alpdogan, T.; Jonnalagadda, S.C.; Pandey, M.K. Autophagy and Apoptosis: Current Challenges of Treatment and Drug Resistance in Multiple Myeloma. Int. J. Mol. Sci. 2022, 24, 644. [Google Scholar] [CrossRef]
- Safa, A.R. Drug and apoptosis resistance in cancer stem cells: A puzzle with many pieces. Cancer Drug Resist. 2022, 5, 850–872. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef]
- Ahmed, S.; Shah, P.; Ahmed, O. Biochemistry, Lipids; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Rodencal, J.; Dixon, S.J. A tale of two lipids: Lipid unsaturation commands ferroptosis sensitivity. Proteomics 2023, 23, e2100308. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, e1800311. [Google Scholar] [CrossRef]
- Hassannia, B.; Van Coillie, S.; Vanden Berghe, T. Ferroptosis: Biological Rust of Lipid Membranes. Antioxid. Redox Signal 2021, 35, 487–509. [Google Scholar] [CrossRef] [PubMed]
- Henneberry, A.L.; Wright, M.M.; McMaster, C.R. The major sites of cellular phospholipid synthesis and molecular determinants of Fatty Acid and lipid head group specificity. Mol. Biol. Cell 2002, 13, 3148–3161. [Google Scholar] [CrossRef]
- Fahy, E.; Cotter, D.; Sud, M.; Subramaniam, S. Lipid classification, structures and tools. Biochim. Biophys. Acta 2011, 1811, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Mortensen, M.S.; Ruiz, J.; Watts, J.L. Polyunsaturated Fatty Acids Drive Lipid Peroxidation during Ferroptosis. Cells 2023, 12, 804. [Google Scholar] [CrossRef] [PubMed]
- Mynott, R.L.; Habib, A.; Best, O.G.; Wallington-Gates, C.T. Ferroptosis in Haematological Malignancies and Associated Therapeutic Nanotechnologies. Int. J. Mol. Sci. 2023, 24, 7661. [Google Scholar] [CrossRef]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascon, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Seibt, T.M.; Proneth, B.; Conrad, M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radic. Biol. Med. 2019, 133, 144–152. [Google Scholar] [CrossRef]
- Sim, S.; Wong, N.K. Nanotechnology and its use in imaging and drug delivery (Review). Biomed. Rep. 2021, 14, 42. [Google Scholar] [CrossRef]
- Alshehri, A.; Grabowska, A.; Stolnik, S. Pathways of cellular internalisation of liposomes delivered siRNA and effects on siRNA engagement with target mRNA and silencing in cancer cells. Sci. Rep. 2018, 8, 3748. [Google Scholar] [CrossRef]
- Akiyama, H.; Zhao, R.; Rahhal, A.; Nishida, Y.; Ayoub, E.; Ostermann, L.B.; Andreeff, M.; Ishizawa, J. Therapeutic Targeting of Ferroptosis Pathway in Combination with Mitochondrial Oxidative Stress Induction in Acute Myeloid Leukemia. Blood 2021, 138, 1162. [Google Scholar] [CrossRef]
- Koppula, P.; Lei, G.; Zhang, Y.; Yan, Y.; Mao, C.; Kondiparthi, L.; Shi, J.; Liu, X.; Horbath, A.; Das, M.; et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat. Commun. 2022, 13, 2206. [Google Scholar]
- Wu, F.; Du, Y.; Yang, J.; Shao, B.; Mi, Z.; Yao, Y.; Cui, Y.; He, F.; Zhang, Y.; Yang, P. Peroxidase-like Active Nanomedicine with Dual Glutathione Depletion Property to Restore Oxaliplatin Chemosensitivity and Promote Programmed Cell Death. ACS Nano 2022, 16, 3647–3663. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Gao, W.; Zhang, C.; Wang, P.; Wang, X.; Yan, M.; Jiang, W.; Wu, Z.; Wei, P.; Tian, G.; et al. A Biodegradable High-Efficiency Magnetic Nanoliposome Promotes Tumor Microenvironment-Responsive Multimodal Tumor Therapy Along with Switchable T2 Magnetic Resonance Imaging. ACS Appl. Mater. Interfaces 2022, 14, 24160–24173. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Deng, J.; Liu, F.; Fan, A.; Wang, Y.; Wu, H.; Ding, D.; Kong, D.; Wang, Z.; Peer, D.; et al. Triggered ferroptotic polymer micelles for reversing multidrug resistance to chemotherapy. Biomaterials 2019, 223, 119486. [Google Scholar] [CrossRef]
- Cao, Y.; Li, Y.; He, C.; Yan, F.; Li, J.R.; Xu, H.Z.; Zhuang, J.F.; Zhou, H.; Peng, Y.C.; Fu, X.J.; et al. Selective Ferroptosis Inhibitor Liproxstatin-1 Attenuates Neurological Deficits and Neuroinflammation After Subarachnoid Hemorrhage. Neurosci. Bull. 2021, 37, 535–549. [Google Scholar] [CrossRef]
- Kou, L.; Sun, R.; Jiang, X.; Lin, X.; Huang, H.; Bao, S.; Zhang, Y.; Li, C.; Chen, R.; Yao, Q. Tumor Microenvironment-Responsive, Multistaged Liposome Induces Apoptosis and Ferroptosis by Amplifying Oxidative Stress for Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2020, 12, 30031–30043. [Google Scholar] [CrossRef] [PubMed]
- Ji, P.; Wang, X.; Yin, J.; Yao, Y.; Du, W. Amplification of ferroptosis with a liposomal nanoreactor cooperates with low-toxicity doxorubicin apoptosis for enhanced tumor chemotherapy. Biomater. Sci. 2022, 10, 1544–1553. [Google Scholar] [CrossRef]
- Panaroni, C.; Fulzele, K.; Soucy, R.; Siu, K.T.; Mukaihara, K.; Huang, C.; Chattopadhyay, S.; Raje, N. Arachidonic Acid Induces Ferroptosis-Mediated Cell-Death in Multiple Myeloma. Blood 2018, 132, 4498. [Google Scholar] [CrossRef]
- Bordini, J.; Morisi, F.; Cerruti, F.; Cascio, P.; Camaschella, C.; Ghia, P.; Campanella, A. Iron Causes Lipid Oxidation and Inhibits Proteasome Function in Multiple Myeloma Cells: A Proof of Concept for Novel Combination Therapies. Cancers Basel 2020, 12, 970. [Google Scholar] [CrossRef]
- Toyokuni, S.; Ito, F.; Yamashita, K.; Okazaki, Y.; Akatsuka, S. Iron and thiol redox signaling in cancer: An exquisite balance to escape ferroptosis. Free Radic. Biol. Med. 2017, 108, 610–626. [Google Scholar] [CrossRef] [PubMed]
- Wallington-Beddoe, C.T.; Bennett, M.K.; Vandyke, K.; Davies, L.; Zebol, J.R.; Moretti, P.A.B.; Pitman, M.R.; Hewett, D.R.; Zannettino, A.C.W.; Pitson, S.M. Sphingosine kinase 2 inhibition synergises with bortezomib to target myeloma by enhancing endoplasmic reticulum stress. Oncotarget 2017, 8, 43602–43616. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Kim, Y.-S. Roles of RIPK3 in necroptosis, cell signaling, and disease. Exp. Mol. Med. 2022, 54, 1695–1704. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Manni, M.M.; Tiberti, M.L.; Pagnotta, S.; Barelli, H.; Gautier, R.; Antonny, B. Acyl chain asymmetry and polyunsaturation of brain phospholipids facilitate membrane vesiculation without leakage. eLife 2018, 7, e34394. [Google Scholar] [CrossRef] [PubMed]
- Magtanong, L.; Ko, P.J.; To, M.; Cao, J.Y.; Forcina, G.C.; Tarangelo, A.; Ward, C.C.; Cho, K.; Patti, G.J.; Nomura, D.K.; et al. Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem. Biol. 2019, 26, 420–432. [Google Scholar] [CrossRef]
- Das, U.N. Saturated Fatty Acids, MUFAs and PUFAs Regulate Ferroptosis. Cell Chem. Biol. 2019, 26, 309–311. [Google Scholar] [CrossRef]
- Wang, N.; Ma, H.; Li, J.; Meng, C.; Zou, J.; Wang, H.; Liu, K.; Liu, M.; Xiao, X.; Zhang, H.; et al. HSF1 functions as a key defender against palmitic acid-induced ferroptosis in cardiomyocytes. J. Mol. Cell Cardiol. 2021, 150, 65–76. [Google Scholar] [CrossRef]
- Kuang, H.; Sun, X.; Liu, Y.; Tang, M.; Wei, Y.; Shi, Y.; Li, R.; Xiao, G.; Kang, J.; Wang, F.; et al. Palmitic acid-induced ferroptosis via CD36 activates ER stress to break calcium-iron balance in colon cancer cells. FEBS J. 2023, 290, 3664–3687. [Google Scholar] [CrossRef]
- Miao, S.; Zhang, Q.; Ding, W.; Hou, B.; Su, Z.; Li, M.; Yang, L.; Zhang, J.; Chang, W.; Wang, J. Platelet Internalization Mediates Ferroptosis in Myocardial Infarction. Arterioscler. Thromb. Vasc. Biol. 2023, 43, 218–230. [Google Scholar] [CrossRef]
- Dierge, E.; Debock, E.; Guilbaud, C.; Corbet, C.; Mignolet, E.; Mignard, L.; Bastien, E.; Dessy, C.; Larondelle, Y.; Feron, O. Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab. 2021, 33, 1701–1715. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Heringdorf, D. Lysophospholipids. In Encyclopedia of Molecular Pharmacology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 710–716. [Google Scholar] [CrossRef]
- Bersuker, K.; Hendricks, J.M.; Li, Z.; Magtanong, L.; Ford, B.; Tang, P.H.; Roberts, M.A.; Tong, B.; Maimone, T.J.; Zoncu, R.; et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019, 575, 688–692. [Google Scholar] [CrossRef]
- Battaglia, A.M.; Chirillo, R.; Aversa, I.; Sacco, A.; Costanzo, F.; Biamonte, F. Ferroptosis and Cancer: Mitochondria Meet the “Iron Maiden” Cell Death. Cells 2020, 9, 1505. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.L. Effect of more than one inhibitor. In Enzymes and Metabolic Inhibitors; Hochster, R., Quastel, J., Eds.; Academic Press: New York, NY, USA, 1963; Volume 1, pp. 487–512. [Google Scholar]
- Tian, H.; Zhao, X.; Zhang, Y.; Xia, Z. Abnormalities of glucose and lipid metabolism in myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2023, 163, 114827. [Google Scholar] [CrossRef]
- Nosova, A.S.; Koloskova, O.O.; Nikonova, A.A.; Simonova, V.A.; Smirnov, V.V.; Kudlay, D.; Khaitov, M.R. Diversity of PEGylation methods of liposomes and their influence on RNA delivery. Medchemcomm 2019, 10, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Xin, S.; Mueller, C.; Pfeiffer, S.; Kraft, V.A.N.; Merl-Pham, J.; Bao, X.; Feederle, R.; Jin, X.; Hauck, S.M.; Schmitt-Kopplin, P.; et al. MS4A15 drives ferroptosis resistance through calcium-restricted lipid remodeling. Cell Death Differ. 2022, 29, 670–686. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. 1), S6–S20. [Google Scholar] [CrossRef]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Ayala, A.; Munoz, M.F.; Arguelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef]
- Shintoku, R.; Takigawa, Y.; Yamada, K.; Kubota, C.; Yoshimoto, Y.; Takeuchi, T.; Koshiishi, I.; Torii, S. Lipoxygenase-mediated generation of lipid peroxides enhances ferroptosis induced by erastin and RSL3. Cancer Sci. 2017, 108, 2187–2194. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, C.; Wang, J.; Hu, W.; Feng, Z. The Regulation of Ferroptosis by Tumor Suppressor p53 and its Pathway. Int. J. Mol. Sci. 2020, 21, 8387. [Google Scholar] [CrossRef]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966. [Google Scholar] [CrossRef]
- Zeissig, M.N.; Hewett, D.R.; Mrozik, K.M.; Panagopoulos, V.; Wallington-Gates, C.T.; Spencer, A.; Dold, S.M.; Engelhardt, M.; Vandyke, K.; Zannettino, A.C.W. Expression of the chemokine receptor CCR1 decreases sensitivity to bortezomib in multiple myeloma cell lines. Leuk. Res. 2024, 139, 107469. [Google Scholar] [CrossRef] [PubMed]
- Moosavian, S.A.; Bianconi, V.; Pirro, M.; Sahebkar, A. Challenges and pitfalls in the development of liposomal delivery systems for cancer therapy. Semin. Cancer Biol. 2021, 69, 337–348. [Google Scholar] [CrossRef] [PubMed]
- White, J.B.; Trim, P.J.; Salagaras, T.; Long, A.; Psaltis, P.J.; Verjans, J.W.; Snel, M.F. Equivalent Carbon Number and Interclass Retention Time Conversion Enhance Lipid Identification in Untargeted Clinical Lipidomics. Anal. Chem. 2022, 94, 3476–3484. [Google Scholar] [CrossRef]
- Pino, L.K.; Searle, B.C.; Bollinger, J.G.; Nunn, B.; MacLean, B.; MacCoss, M.J. The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass. Spectrom. Rev. 2020, 39, 229–244. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Habib, A.; Mynott, R.L.; Best, O.G.; Revesz, I.A.; Prestidge, C.A.; Wallington-Gates, C.T. Novel (1S,3R)-RSL3-Encapsulated Polyunsaturated Fatty Acid Rich Liposomes Sensitise Multiple Myeloma Cells to Ferroptosis-Mediated Cell Death. Int. J. Mol. Sci. 2025, 26, 6579. https://doi.org/10.3390/ijms26146579
Habib A, Mynott RL, Best OG, Revesz IA, Prestidge CA, Wallington-Gates CT. Novel (1S,3R)-RSL3-Encapsulated Polyunsaturated Fatty Acid Rich Liposomes Sensitise Multiple Myeloma Cells to Ferroptosis-Mediated Cell Death. International Journal of Molecular Sciences. 2025; 26(14):6579. https://doi.org/10.3390/ijms26146579
Chicago/Turabian StyleHabib, Ali, Rachel L. Mynott, Oliver G. Best, Isabella A. Revesz, Clive A. Prestidge, and Craig T. Wallington-Gates. 2025. "Novel (1S,3R)-RSL3-Encapsulated Polyunsaturated Fatty Acid Rich Liposomes Sensitise Multiple Myeloma Cells to Ferroptosis-Mediated Cell Death" International Journal of Molecular Sciences 26, no. 14: 6579. https://doi.org/10.3390/ijms26146579
APA StyleHabib, A., Mynott, R. L., Best, O. G., Revesz, I. A., Prestidge, C. A., & Wallington-Gates, C. T. (2025). Novel (1S,3R)-RSL3-Encapsulated Polyunsaturated Fatty Acid Rich Liposomes Sensitise Multiple Myeloma Cells to Ferroptosis-Mediated Cell Death. International Journal of Molecular Sciences, 26(14), 6579. https://doi.org/10.3390/ijms26146579