
Chemoradiation-Altered Micromilieu of Glioblastoma Cells Particularly Impacts M1-like Macrophage Activation

 ,
,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
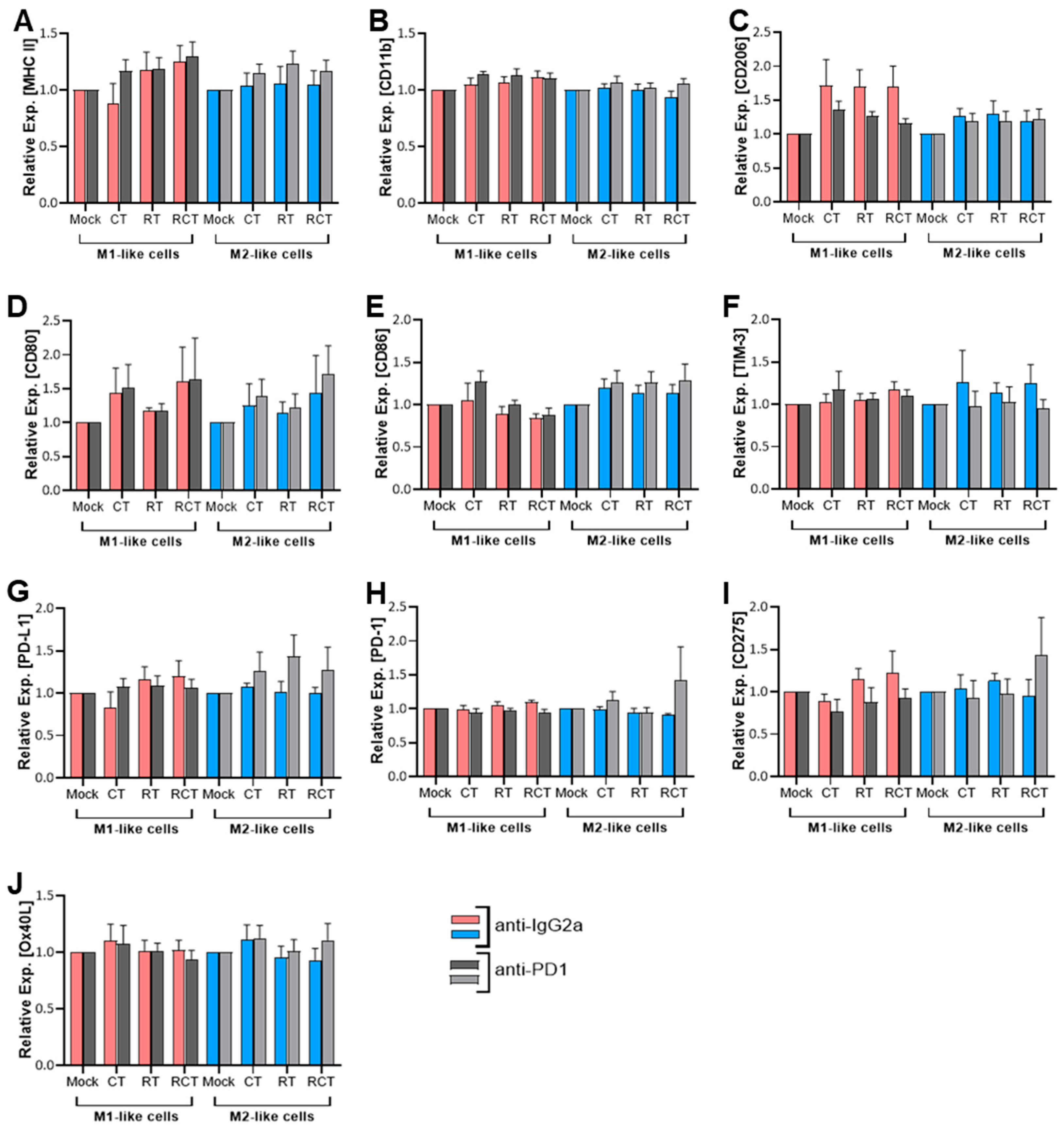
2.1. Neither Chemoradiation of Glioblastoma Cells nor PD-1 Blockade Affects the Differentiation Status or Immune Checkpoint Expression of M1- and M2-like Cells
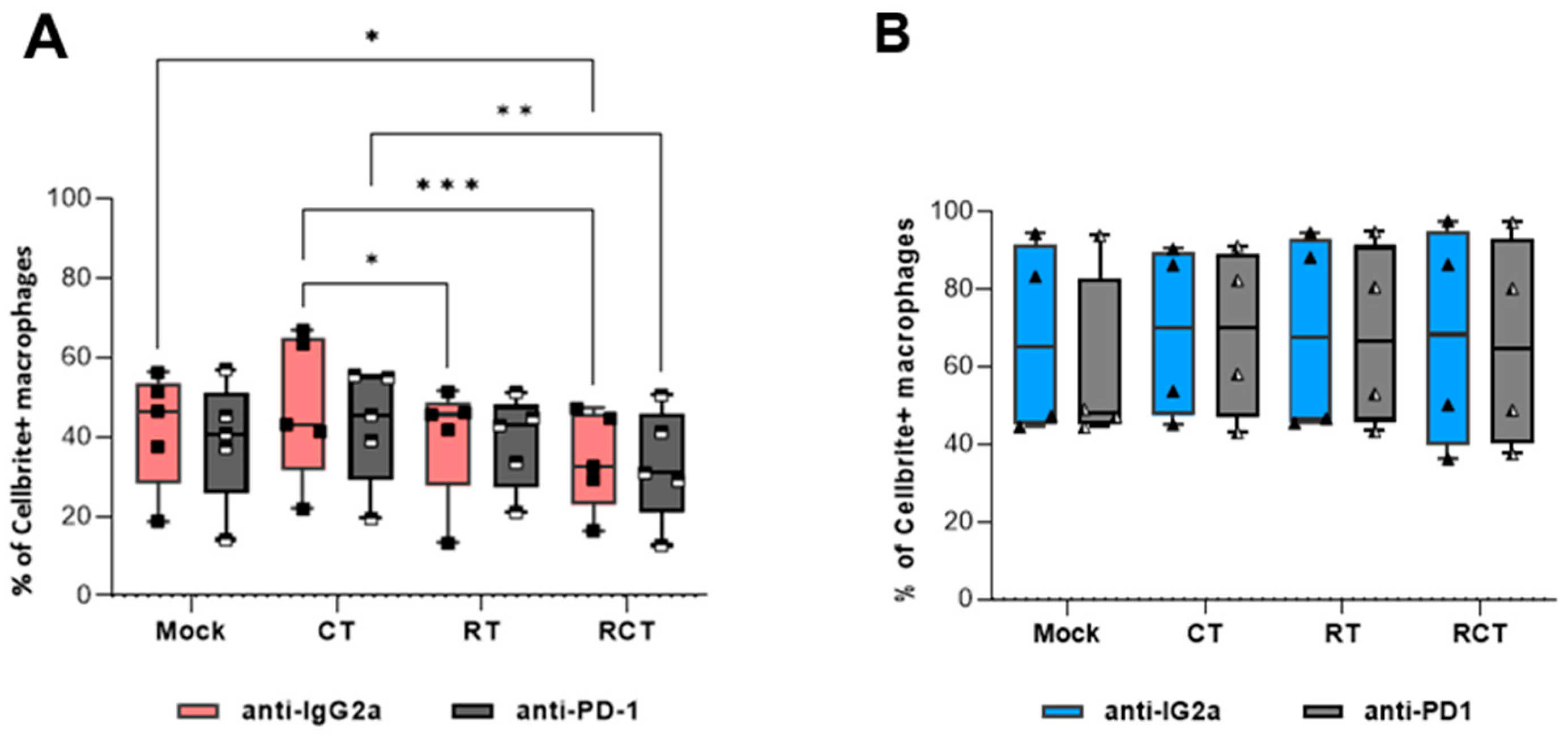
2.2. M2-like Macrophages Have a Higher Capacity to Phagocytize Tumor Cells than M1-like Macrophages, Regardless of Tumor Cell Treatment
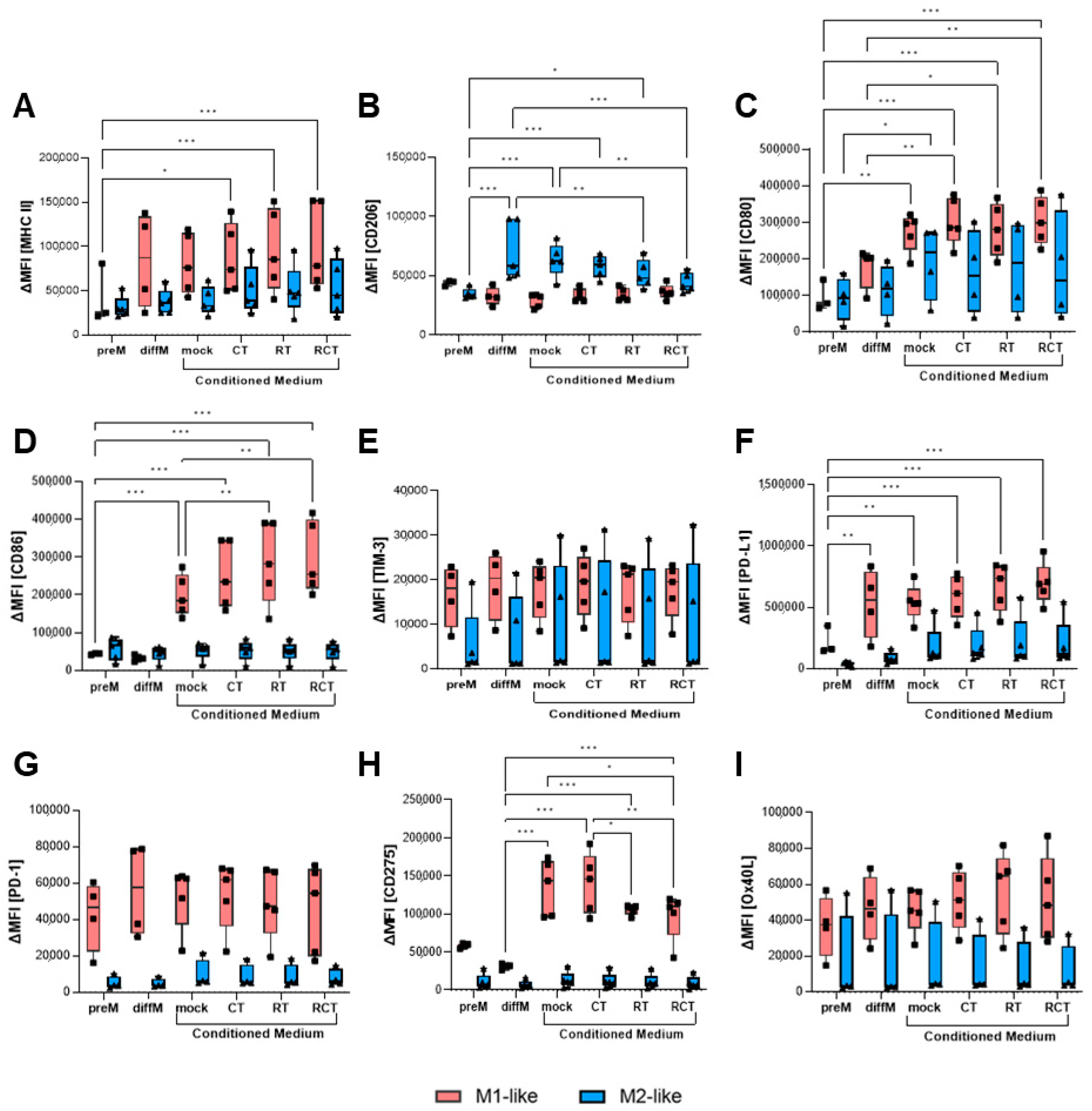
2.3. The Altered Micromilieu of Treated Tumor Cells Significantly Impacts M1-like Macrophage Activation and Immune Checkpoint Surface Expression
3. Discussion
4. Materials and Methods
4.1. Cell Line Culture and Treatment of Glioblastoma Cells
4.2. Animals
4.3. Generation of M1-like and M2-like Macrophages
4.4. Conditioned Medium Assay of M1-like and M2-like Macrophages
4.5. Phagocytosis Assay
4.6. Flow Cytometric Analyses
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BBB | blood–brain barrier |
| C57BL/6 | wildtype mouse strain |
| CD | cluster of differentiation |
| CNS | central nervous system |
| CT | chemotherapy |
| CTLA4 | Cytotoxic T Lymphocyte Antigen 4 |
| DC | dendritic cell |
| ICOS-L | Inducible costimulator-ligand |
| IFN-γ | interferon-gamma |
| LPS | lipopolysaccharide |
| MDSC | myeloid-derived suppressor cell |
| MFI | mean fluorescence intensity |
| MHC II | Major Histocompatibility Complex II |
| OS | overall survival |
| OX40L | CD252 |
| PD-1 | Programmed Cell Death Protein 1 |
| PD-L1 | Programmed Cell Death Protein Ligand 1 |
| PFS | progression-free survival |
| RCT | combination of RT and CT/chemoradiation |
| RT | radiotherapy |
| TAM | tumor-associated macrophage |
| TIM-3 | T cell immunoglobulin and mucin-domain containing-3 |
| TME | tumor microenvironment |
| TMZ | temozolomide |
| Treg | regulatory T cell |
References
- Politis, A.; Stavrinou, L.; Kalyvas, A.; Boviatsis, E.; Piperi, C. Glioblastoma: Molecular features, emerging molecular targets and novel therapeutic strategies. Crit. Rev. Oncol. Hematol. 2025, 212, 104764. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, P.; Penninger, J.M.; Timms, E.; Wakeham, A.; Shahinian, A.; Lee, K.P.; Thompson, C.B.; Griesser, H.; Mak, T.W. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science 1995, 270, 985–988. [Google Scholar] [CrossRef] [PubMed]
- Derer, A.; Spiljar, M.; Baumler, M.; Hecht, M.; Fietkau, R.; Frey, B.; Gaipl, U.S. Chemoradiation Increases PD-L1 Expression in Certain Melanoma and Glioblastoma Cells. Front. Immunol. 2016, 7, 610. [Google Scholar] [CrossRef] [PubMed]
- Schatz, J.; Ladinig, A.; Fietkau, R.; Putz, F.; Gaipl, U.S.; Frey, B.; Derer, A. Normofractionated irradiation and not temozolomide modulates the immunogenic and oncogenic phenotype of human glioblastoma cell lines. Strahlenther. Onkol. 2023, 199, 1140–1151. [Google Scholar] [CrossRef]
- Bhardwaj, J.S.; Paliwal, S.; Singhvi, G.; Taliyan, R. Immunological challenges and opportunities in glioblastoma multiforme: A comprehensive view from immune system lens. Life Sci. 2024, 357, 123089. [Google Scholar] [CrossRef]
- Chen, Z.; Feng, X.; Herting, C.J.; Garcia, V.A.; Nie, K.; Pong, W.W.; Rasmussen, R.; Dwivedi, B.; Seby, S.; Wolf, S.A.; et al. Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer Res. 2017, 77, 2266–2278. [Google Scholar] [CrossRef]
- Cai, X.; Beumer-Chuwonpad, A.; Westerman, B.; Garcia Vallejo, J. P02.05.A Comprehensive Analysis of Tumor-Associated Macrophages in Glioblastoma. Neuro-Oncology 2024, 26 (Suppl. S5), v35. [Google Scholar] [CrossRef]
- Kvisten, M.; Mikkelsen, V.E.; Stensjoen, A.L.; Solheim, O.; Van Der Want, J.; Torp, S.H. Microglia and macrophages in human glioblastomas: A morphological and immunohistochemical study. Mol. Clin. Oncol. 2019, 11, 31–36. [Google Scholar] [CrossRef]
- Guo, Q.; Jin, Z.; Yuan, Y.; Liu, R.; Xu, T.; Wei, H.; Xu, X.; He, S.; Chen, S.; Shi, Z.; et al. Corrigendum to “New Mechanisms of Tumor-Associated Macrophages on Promoting Tumor Progression: Recent Research Advances and Potential Targets for Tumor Immunotherapy”. J. Immunol. Res. 2018, 2018, 6728474. [Google Scholar] [CrossRef]
- Lecoultre, M.; Dutoit, V.; Walker, P.R. Phagocytic function of tumor-associated macrophages as a key determinant of tumor progression control: A review. J. Immunother. Cancer 2020, 8, e001408. [Google Scholar] [CrossRef]
- Boutilier, A.J.; Elsawa, S.F. Macrophage Polarization States in the Tumor Microenvironment. Int. J. Mol. Sci. 2021, 22, 6995. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.R.; Maute, R.L.; Dulken, B.W.; Hutter, G.; George, B.M.; McCracken, M.N.; Gupta, R.; Tsai, J.M.; Sinha, R.; Corey, D.; et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature 2017, 545, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Urbantat, R.M.; Jelgersma, C.; Brandenburg, S.; Nieminen-Kelha, M.; Kremenetskaia, I.; Zollfrank, J.; Mueller, S.; Rubarth, K.; Koch, A.; Vajkoczy, P.; et al. Tumor-Associated Microglia/Macrophages as a Predictor for Survival in Glioblastoma and Temozolomide-Induced Changes in CXCR2 Signaling with New Resistance Overcoming Strategy by Combination Therapy. Int. J. Mol. Sci. 2021, 22, 11180. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, F.; Augello, F.R.; Artone, S.; Ciafarone, A.; Topi, S.; Cifone, M.G.; Cinque, B.; Palumbo, P. Involvement of Cyclooxygenase-2 in Establishing an Immunosuppressive Microenvironment in Tumorspheres Derived from TMZ-Resistant Glioblastoma Cell Lines and Primary Cultures. Cells 2024, 13, 258. [Google Scholar] [CrossRef]
- Li, Z.; Fu, W.J.; Chen, X.Q.; Wang, S.; Deng, R.S.; Tang, X.P.; Yang, K.D.; Niu, Q.; Zhou, H.; Li, Q.R.; et al. Autophagy-based unconventional secretion of HMGB1 in glioblastoma promotes chemosensitivity to temozolomide through macrophage M1-like polarization. J. Exp. Clin. Cancer Res. 2022, 41, 74. [Google Scholar] [CrossRef]
- Zhang, G.; Tao, X.; Ji, B.; Gong, J. Hypoxia-Driven M2-Polarized Macrophages Facilitate Cancer Aggressiveness and Temozolomide Resistance in Glioblastoma. Oxid. Med. Cell Longev. 2022, 2022, 1614336. [Google Scholar] [CrossRef]
- Meng, W.; Li, L.; Hao, Y.; Tang, M.; Cao, C.; He, J.; Wang, L.; Cao, B.; Zhang, Y.; Li, L.; et al. NAD+ Metabolism Reprogramming Mediates Irradiation-Induced Immunosuppressive Polarization of Macrophages. Int. J. Radiat. Oncol. Biol. Phys. 2025, 121, 176–190. [Google Scholar] [CrossRef]
- Chen, X.; Wang, L.; Yu, H.; Shen, Q.; Hou, Y.; Xia, Y.X.; Li, L.; Chang, L.; Li, W.H. Irradiated lung cancer cell-derived exosomes modulate macrophage polarization by inhibiting MID1 via miR-4655-5p. Mol. Immunol. 2023, 155, 58–68. [Google Scholar] [CrossRef]
- Becherini, C.; Lancia, A.; Detti, B.; Lucidi, S.; Scartoni, D.; Ingrosso, G.; Carnevale, M.G.; Roghi, M.; Bertini, N.; Orsatti, C.; et al. Modulation of tumor-associated macrophage activity with radiation therapy: A systematic review. Strahlenther. Onkol. 2023, 199, 1173–1190. [Google Scholar] [CrossRef]
- Prakash, H.; Klug, F.; Nadella, V.; Mazumdar, V.; Schmitz-Winnenthal, H.; Umansky, L. Low doses of gamma irradiation potentially modifies immunosuppressive tumor microenvironment by retuning tumor-associated macrophages: Lesson from insulinoma. Carcinogenesis 2016, 37, 301–313. [Google Scholar] [CrossRef]
- Lu, Y.; Zhou, C.; Li, J.; Liu, L.; Liu, X.; Shen, L.; Lai, X.; Gu, H.; Zhang, Z.; Qian, J. Radiation induces M2 polarization of glioma-associated macrophages via upregulation of glutamine synthetases. Int. Immunopharmacol. 2025, 154, 114595. [Google Scholar] [CrossRef] [PubMed]
- Akkari, L.; Bowman, R.L.; Tessier, J.; Klemm, F.; Handgraaf, S.M.; de Groot, M.; Quail, D.F.; Tillard, L.; Gadiot, J.; Huse, J.T.; et al. Dynamic changes in glioma macrophage populations after radiotherapy reveal CSF-1R inhibition as a strategy to overcome resistance. Sci. Transl. Med. 2020, 12, eaaw7843. [Google Scholar] [CrossRef] [PubMed]
- Agosti, E.; Panciani, P.P.; Zeppieri, M.; De Maria, L.; Pasqualetti, F.; Tel, A.; Zanin, L.; Fontanella, M.M.; Ius, T. Tumor Microenvironment and Glioblastoma Cell Interplay as Promoters of Therapeutic Resistance. Biology 2023, 12, 736. [Google Scholar] [CrossRef] [PubMed]
- Lim, T.S.; Chew, V.; Sieow, J.L.; Goh, S.; Yeong, J.P.; Soon, A.L.; Ricciardi-Castagnoli, P. PD-1 expression on dendritic cells suppresses CD8(+) T cell function and antitumor immunity. Oncoimmunology 2016, 5, e1085146. [Google Scholar] [CrossRef]
- Xu, S.; Wang, C.; Yang, L.; Wu, J.; Li, M.; Xiao, P.; Xu, Z.; Xu, Y.; Wang, K. Targeting immune checkpoints on tumor-associated macrophages in tumor immunotherapy. Front. Immunol. 2023, 14, 1199631. [Google Scholar] [CrossRef]
- Pellegatta, S.; Valletta, L.; Corbetta, C.; Patane, M.; Zucca, I.; Riccardi Sirtori, F.; Bruzzone, M.G.; Fogliatto, G.; Isacchi, A.; Pollo, B.; et al. Effective immuno-targeting of the IDH1 mutation R132H in a murine model of intracranial glioma. Acta Neuropathol. Commun. 2015, 3, 4. [Google Scholar] [CrossRef]
- Hagemann, T.; Lawrence, T.; McNeish, I.; Charles, K.A.; Kulbe, H.; Thompson, R.G.; Robinson, S.C.; Balkwill, F.R. “Re-educating” tumor-associated macrophages by targeting NF-kappaB. J. Exp. Med. 2008, 205, 1261–1268. [Google Scholar] [CrossRef]
- Cutolo, M.; Campitiello, R.; Gotelli, E.; Soldano, S. The Role of M1/M2 Macrophage Polarization in Rheumatoid Arthritis Synovitis. Front. Immunol. 2022, 13, 867260. [Google Scholar] [CrossRef]
- Melichar, B.; Nash, M.A.; Lenzi, R.; Platsoucas, C.D.; Freedman, R.S. Expression of costimulatory molecules CD80 and CD86 and their receptors CD28, CTLA-4 on malignant ascites CD3+ tumour-infiltrating lymphocytes (TIL) from patients with ovarian and other types of peritoneal carcinomatosis. Clin. Exp. Immunol. 2000, 119, 19–27. [Google Scholar] [CrossRef]
- Ahmed, M.H.; Hernández-Verdin, I.; Bielle, F.; Verreault, M.; Lerond, J.; Alentorn, A.; Sanson, M.; Idbaih, A. Expression and Prognostic Value of CD80 and CD86 in the Tumor Microenvironment of Newly Diagnosed Glioblastoma. Can. J. Neurol. Sci. 2023, 50, 234–242. [Google Scholar] [CrossRef]
- Gabrilo, J.; Velde, S.V.; Henin, C.; Denanglaire, S.; Azouz, A.; Boon, L.; Van den Eynde, B.J.; Moser, M.; Goriely, S.; Leo, O. Interferon-γ driven differentiation of monocytes into PD-L1+ and MHC II+ macrophages and the frequency of Tim-3+ tumor-reactive CD8+ T cells within the tumor microenvironment predict a positive response to anti-PD-1-based therapy in tumor-bearing mice. bioRxiv 2024. bioRxiv:2024.2008.2001.606242. [Google Scholar]
- Palacios, L.M.; Peyret, V.; Viano, M.E.; Geysels, R.C.; Chocobar, Y.A.; Volpini, X.; Pellizas, C.G.; Nicola, J.P.; Motran, C.C.; Rodriguez-Galan, M.C.; et al. TIM3 Expression in Anaplastic-Thyroid-Cancer-Infiltrating Macrophages: An Emerging Immunotherapeutic Target. Biology 2022, 11, 1609. [Google Scholar] [CrossRef] [PubMed]
- Katagata, M.; Okayama, H.; Nakajima, S.; Saito, K.; Sato, T.; Sakuma, M.; Fukai, S.; Endo, E.; Sakamoto, W.; Saito, M.; et al. TIM-3 Expression and M2 Polarization of Macrophages in the TGFbeta-Activated Tumor Microenvironment in Colorectal Cancer. Cancers 2023, 15, 4943. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cai, P.; Liang, T.; Wang, L.; Hu, L. TIM-3 is a potential prognostic marker for patients with solid tumors: A systematic review and meta-analysis. Oncotarget 2017, 8, 31705–31713. [Google Scholar] [CrossRef]
- Qin, S.; Dong, B.; Yi, M.; Chu, Q.; Wu, K. Prognostic Values of TIM-3 Expression in Patients With Solid Tumors: A Meta-Analysis and Database Evaluation. Front. Oncol. 2020, 10, 1288. [Google Scholar] [CrossRef]
- Solinas, C.; Gu-Trantien, C.; Willard-Gallo, K. The rationale behind targeting the ICOS-ICOS ligand costimulatory pathway in cancer immunotherapy. ESMO Open 2020, 5, e000544. [Google Scholar] [CrossRef]
- Wang, J.; Shi, F.; Shan, A. Transcriptome profile and clinical characterization of ICOS expression in gliomas. Front. Oncol. 2022, 12, 946967. [Google Scholar] [CrossRef]
- Leibowitz-Amit, R.; Feldman, S.; Daana, B.; Elkis, L.; Mendelovic, S.; Avraham, A. 188P Expression of the co-stimulatory checkpoint protein OX40L (TNFSF4) in the melanoma micro-environment. Immuno-Oncol. Technol. 2023, 20, 100647. [Google Scholar] [CrossRef]
- Shibahara, I.; Saito, R.; Zhang, R.; Chonan, M.; Shoji, T.; Kanamori, M.; Sonoda, Y.; Kumabe, T.; Kanehira, M.; Kikuchi, T.; et al. OX40 ligand expressed in glioblastoma modulates adaptive immunity depending on the microenvironment: A clue for successful immunotherapy. Mol. Cancer 2015, 14, 41. [Google Scholar] [CrossRef]
- Badie, B.; Schartner, J.M. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery 2000, 46, 957–961. [Google Scholar] [CrossRef]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, A.C.; Badie, B. Microglia and macrophages in malignant gliomas: Recent discoveries and implications for promising therapies. Clin. Dev. Immunol. 2013, 2013, 264124. [Google Scholar] [CrossRef] [PubMed]
- Charles, N.A.; Holland, E.C.; Gilbertson, R.; Glass, R.; Kettenmann, H. The brain tumor microenvironment. Glia 2012, 60, 502–514. [Google Scholar] [CrossRef] [PubMed]
- Komohara, Y.; Ohnishi, K.; Kuratsu, J.; Takeya, M. Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J. Pathol. 2008, 216, 15–24. [Google Scholar] [CrossRef]
- Lee, C.; Lee, J.; Jeong, M.; Nam, D.; Rhee, I. Emerging strategies for targeting tumor-associated macrophages in glioblastoma: A focus on chemotaxis blockade. Life Sci. 2025, 376, 123762. [Google Scholar] [CrossRef]
- Ding, P.; Wang, W.; Wang, J.; Yang, Z.; Xue, L. Expression of tumor-associated macrophage in progression of human glioma. Cell Biochem. Biophys. 2014, 70, 1625–1631. [Google Scholar] [CrossRef]
- Wedekind, H.; Walz, K.; Buchbender, M.; Rieckmann, T.; Strasser, E.; Grottker, F.; Fietkau, R.; Frey, B.; Gaipl, U.S.; Ruckert, M. Head and neck tumor cells treated with hypofractionated irradiation die via apoptosis and are better taken up by M1-like macrophages. Strahlenther. Onkol. 2022, 198, 171–182. [Google Scholar] [CrossRef]
- Zhang, M.; Hutter, G.; Kahn, S.A.; Azad, T.D.; Gholamin, S.; Xu, C.Y.; Liu, J.; Achrol, A.S.; Richard, C.; Sommerkamp, P.; et al. Anti-CD47 Treatment Stimulates Phagocytosis of Glioblastoma by M1 and M2 Polarized Macrophages and Promotes M1 Polarized Macrophages In Vivo. PLoS ONE 2016, 11, e0153550. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-Associated Macrophages in Tumor Immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Joshi, T.; Butchar, J.P.; Tridandapani, S. Fcgamma Receptor Signaling in Phagocytes. Int. J. Hematol. 2006, 84, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Schaper, F.; de Leeuw, K.; Horst, G.; Bootsma, H.; Limburg, P.C.; Heeringa, P.; Bijl, M.; Westra, J. High mobility group box 1 skews macrophage polarization and negatively influences phagocytosis of apoptotic cells. Rheumatology 2016, 55, 2260–2270. [Google Scholar] [CrossRef] [PubMed]
- Canton, J.; Khezri, R.; Glogauer, M.; Grinstein, S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol. Biol. Cell 2014, 25, 3330–3341. [Google Scholar] [CrossRef] [PubMed]
- Iwata, R.; Hyoung Lee, J.; Hayashi, M.; Dianzani, U.; Ofune, K.; Maruyama, M.; Oe, S.; Ito, T.; Hashiba, T.; Yoshimura, K.; et al. ICOSLG-mediated regulatory T-cell expansion and IL-10 production promote progression of glioblastoma. Neuro Oncol. 2020, 22, 333–344. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shojaei, M.; Frey, B.; Putz, F.; Fietkau, R.; Gaipl, U.S.; Derer, A. Chemoradiation-Altered Micromilieu of Glioblastoma Cells Particularly Impacts M1-like Macrophage Activation. Int. J. Mol. Sci. 2025, 26, 6574. https://doi.org/10.3390/ijms26146574
Shojaei M, Frey B, Putz F, Fietkau R, Gaipl US, Derer A. Chemoradiation-Altered Micromilieu of Glioblastoma Cells Particularly Impacts M1-like Macrophage Activation. International Journal of Molecular Sciences. 2025; 26(14):6574. https://doi.org/10.3390/ijms26146574
Chicago/Turabian StyleShojaei, Mona, Benjamin Frey, Florian Putz, Rainer Fietkau, Udo S. Gaipl, and Anja Derer. 2025. "Chemoradiation-Altered Micromilieu of Glioblastoma Cells Particularly Impacts M1-like Macrophage Activation" International Journal of Molecular Sciences 26, no. 14: 6574. https://doi.org/10.3390/ijms26146574
APA StyleShojaei, M., Frey, B., Putz, F., Fietkau, R., Gaipl, U. S., & Derer, A. (2025). Chemoradiation-Altered Micromilieu of Glioblastoma Cells Particularly Impacts M1-like Macrophage Activation. International Journal of Molecular Sciences, 26(14), 6574. https://doi.org/10.3390/ijms26146574