
Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development
 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Results
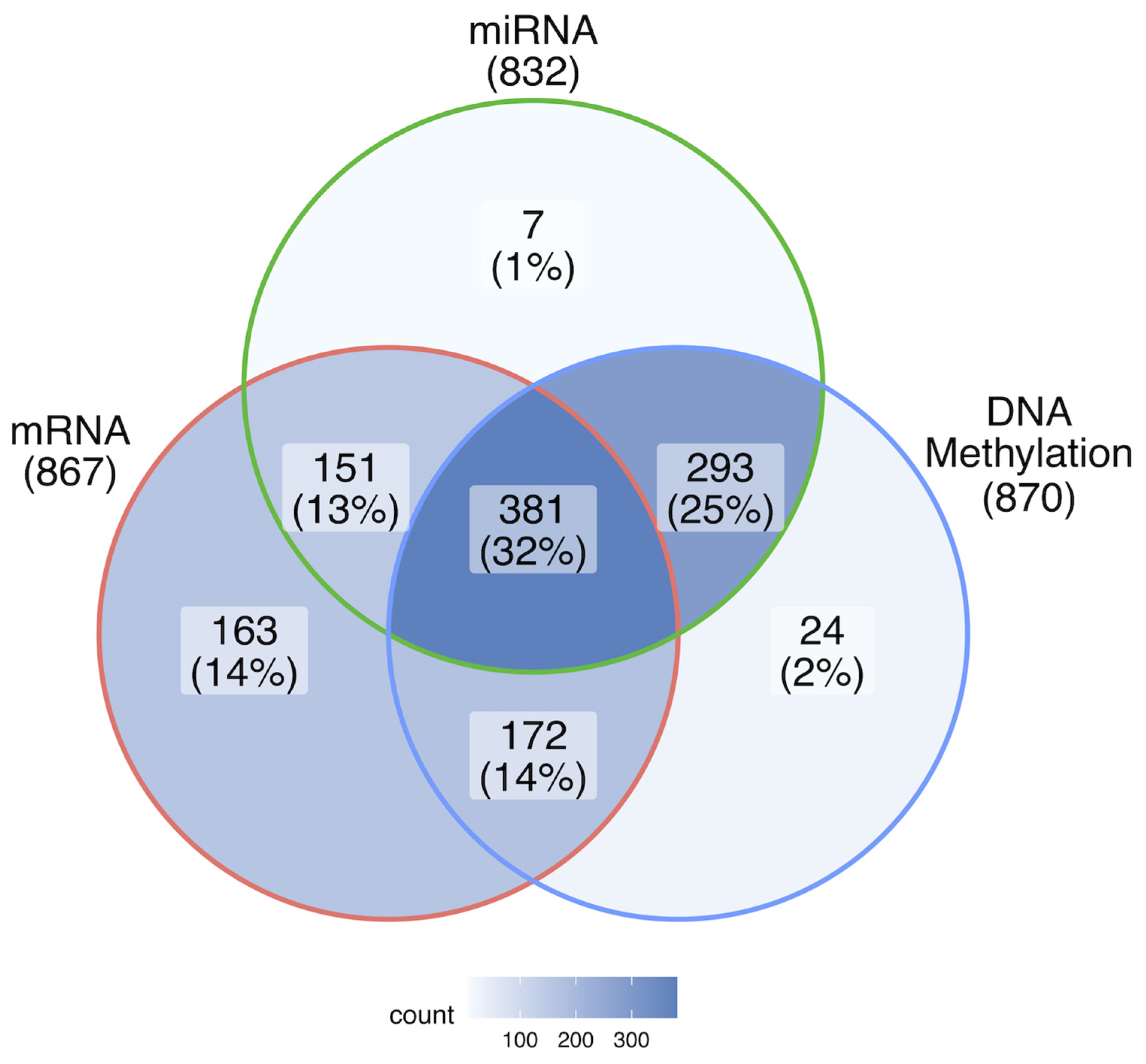
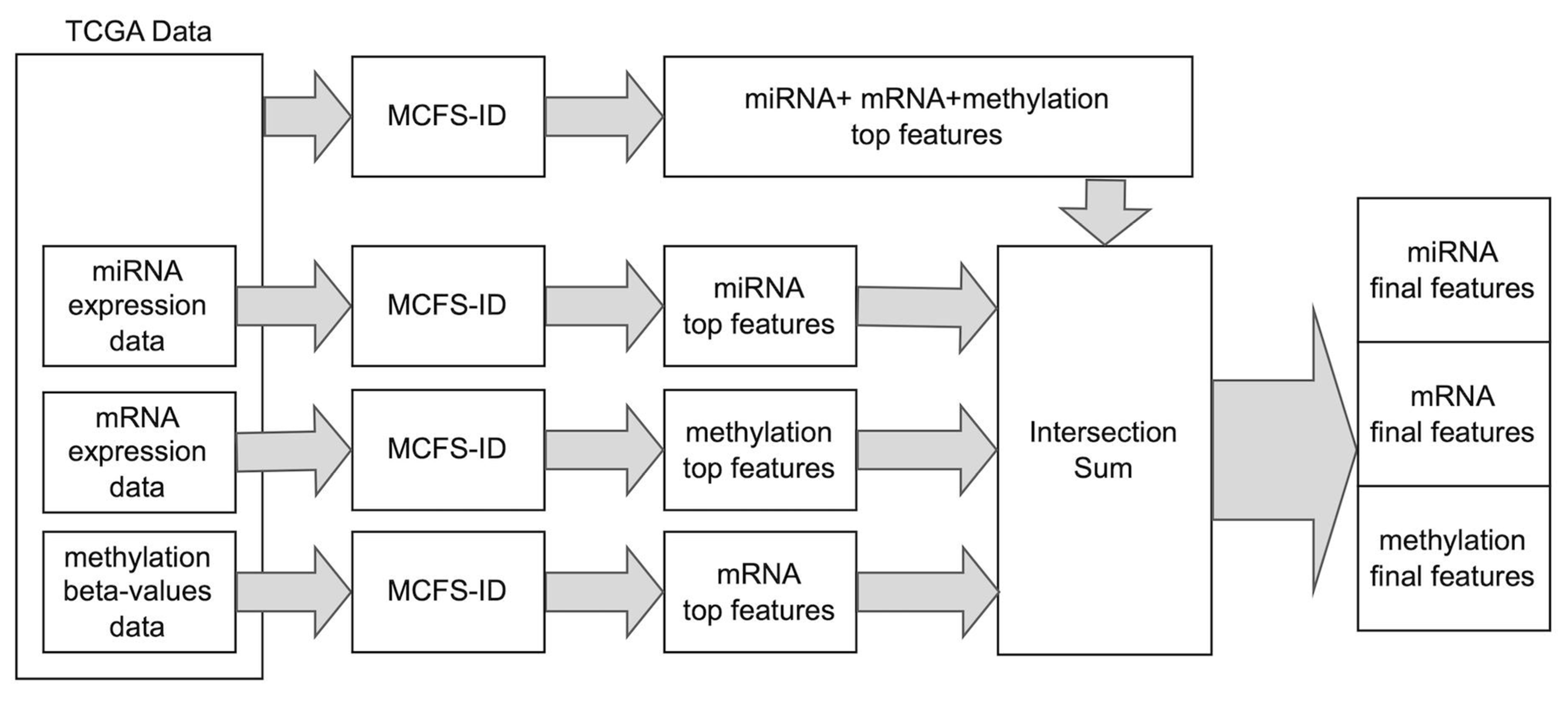
2.1. Detection of Potential Breast Cancer Biomarkers Using the MCFS-ID Algorithm
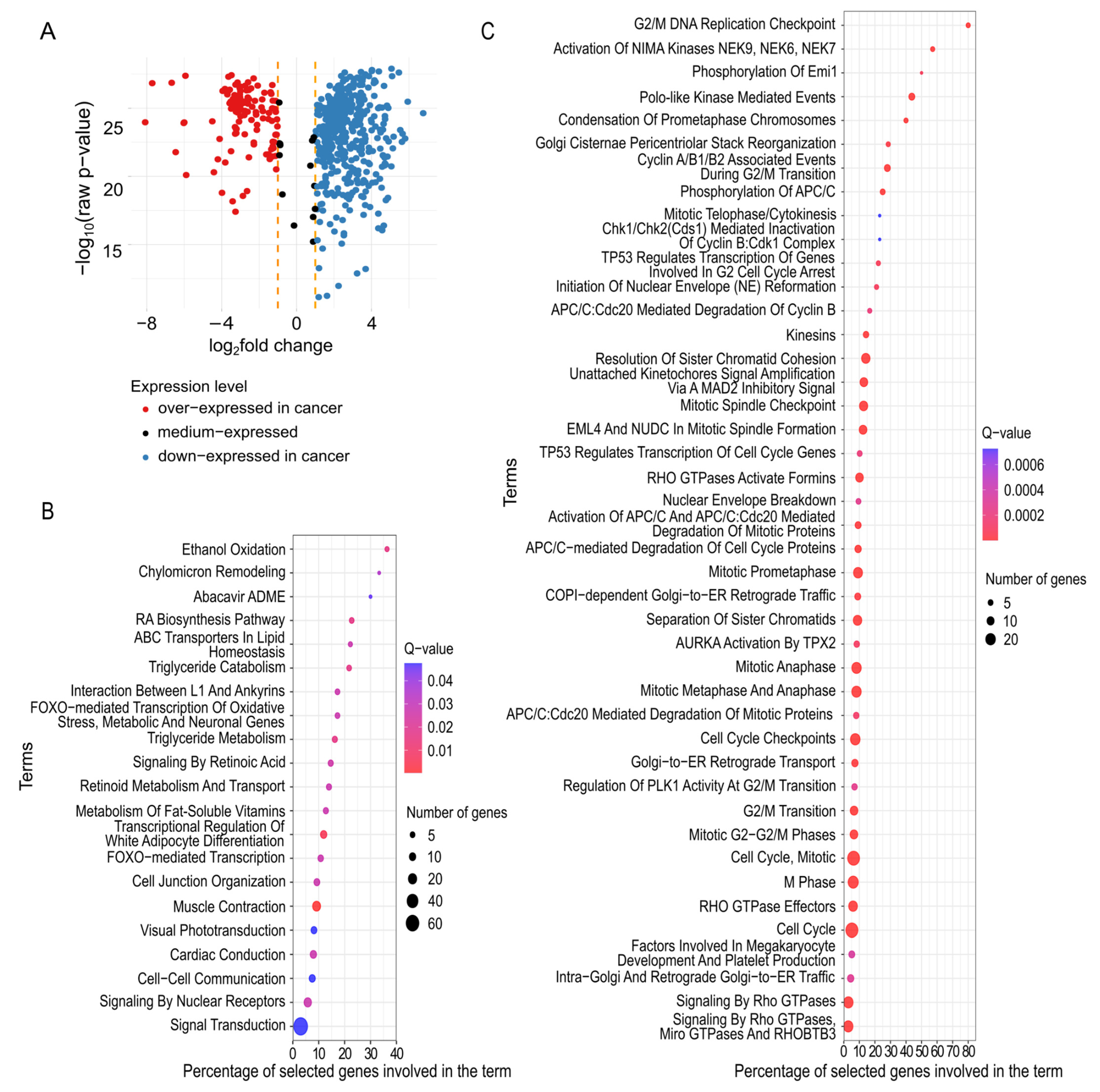
2.2. Descriptive Analysis of mRNAs Having a Significant Predictive Value
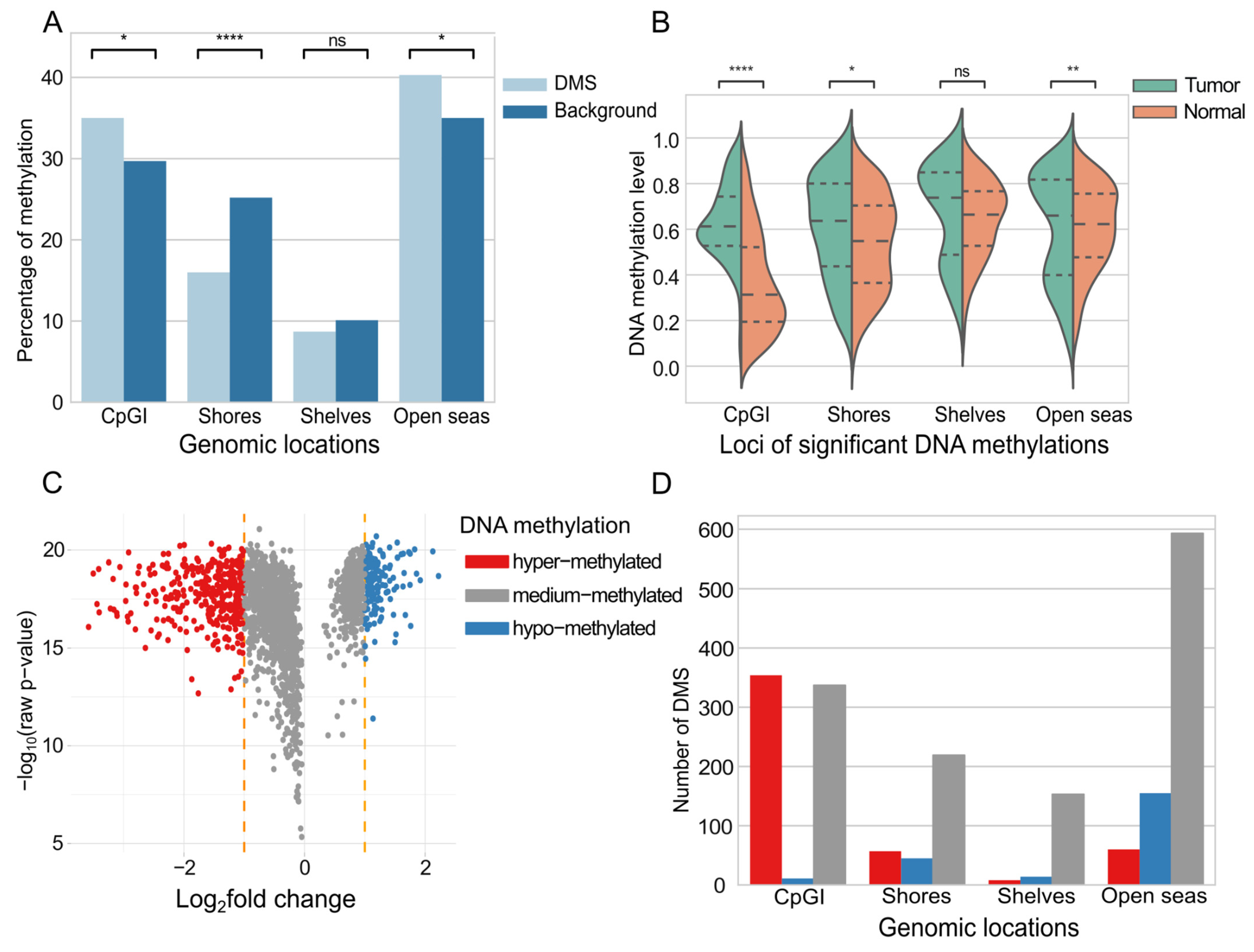
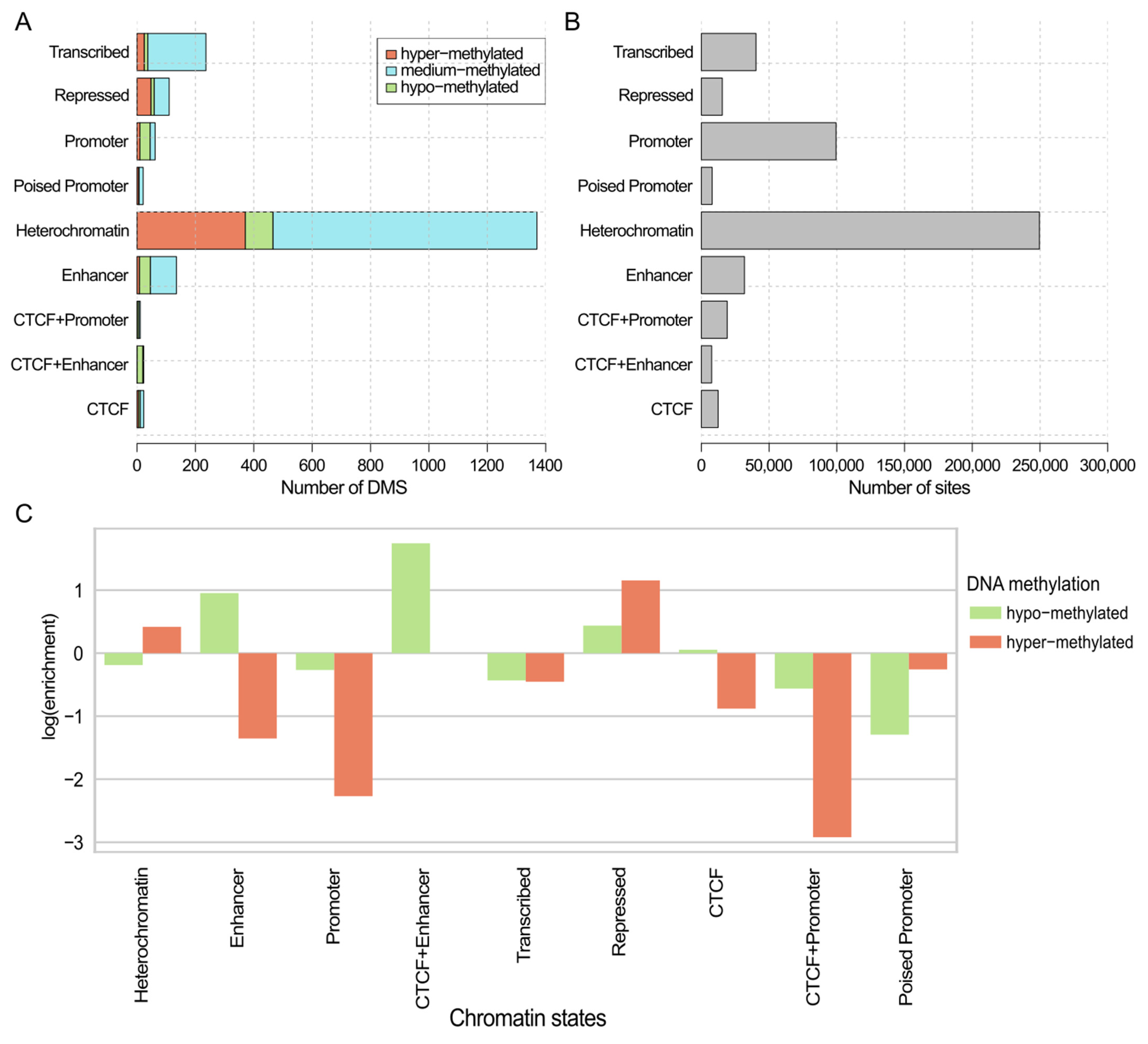
2.3. Genomic Context of DNA Methylations with Predictive Value
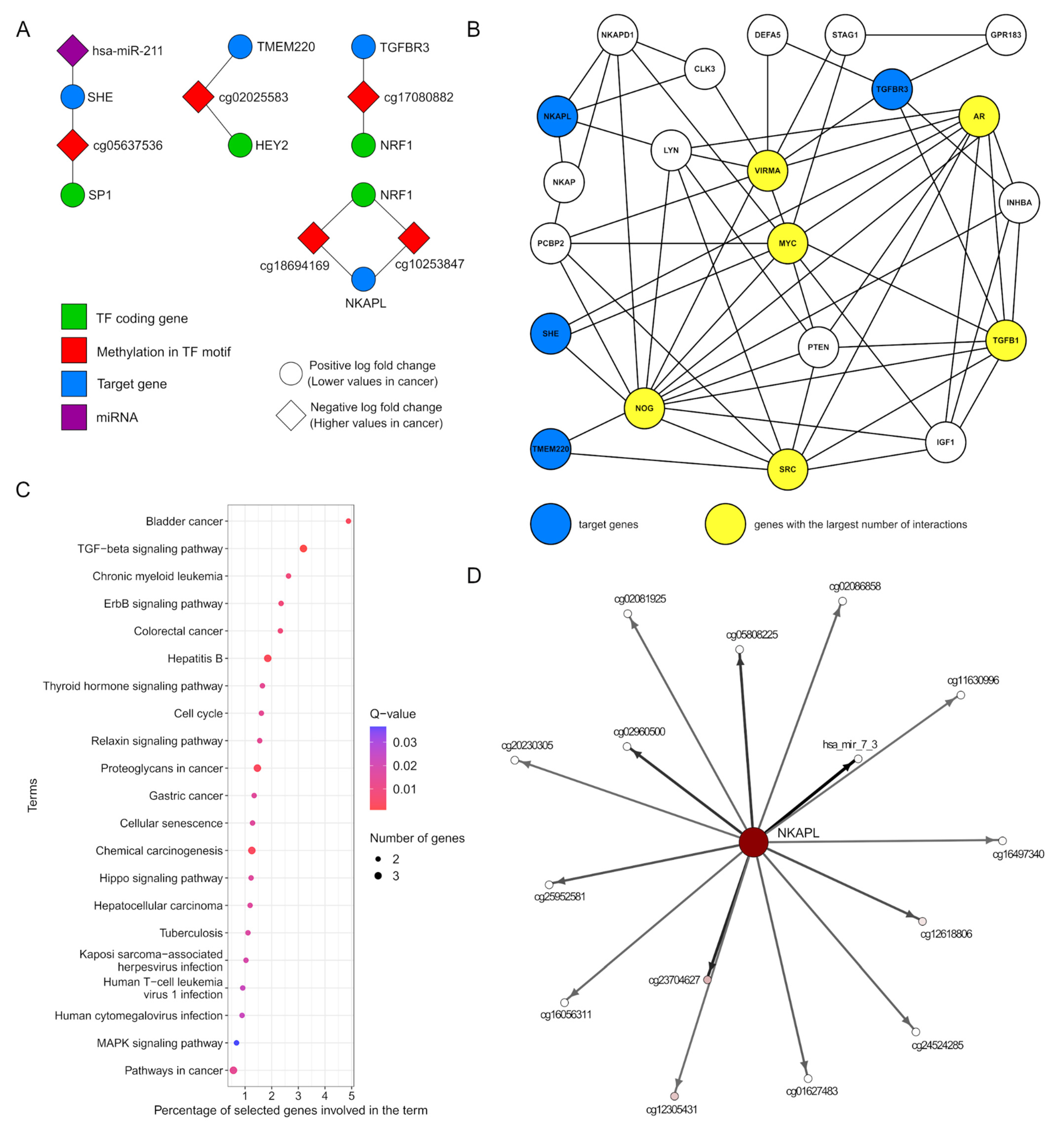
2.4. Biological Role of Significant miRNA Genes
2.5. Detection of miRNA and DNA Methylation Loci Significant in the Context of Predicting mRNA Expression Levels
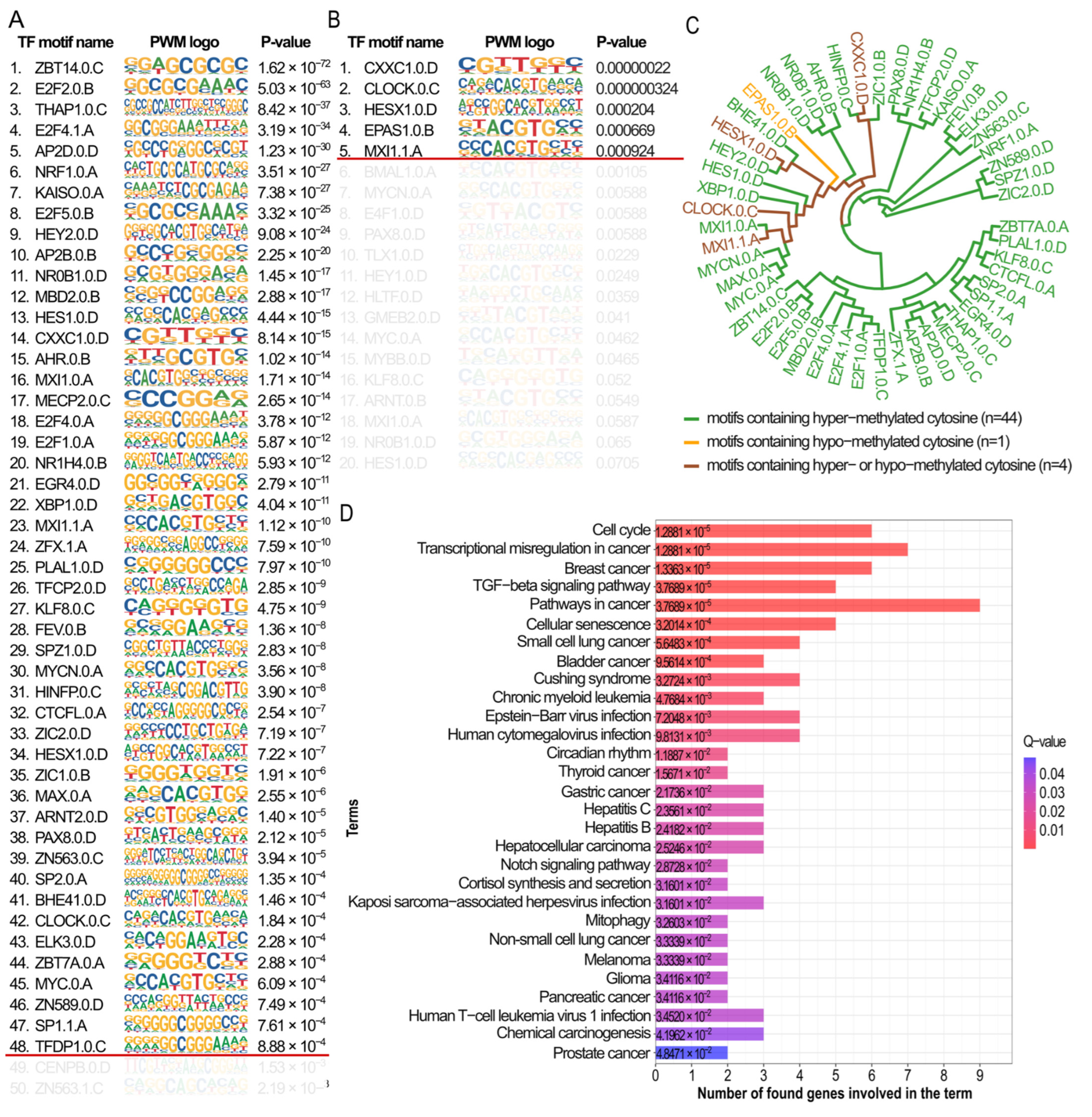
2.6. Tracking Associations Between DMSs and Detected TF Motifs
2.7. Models of Regulatory Networks
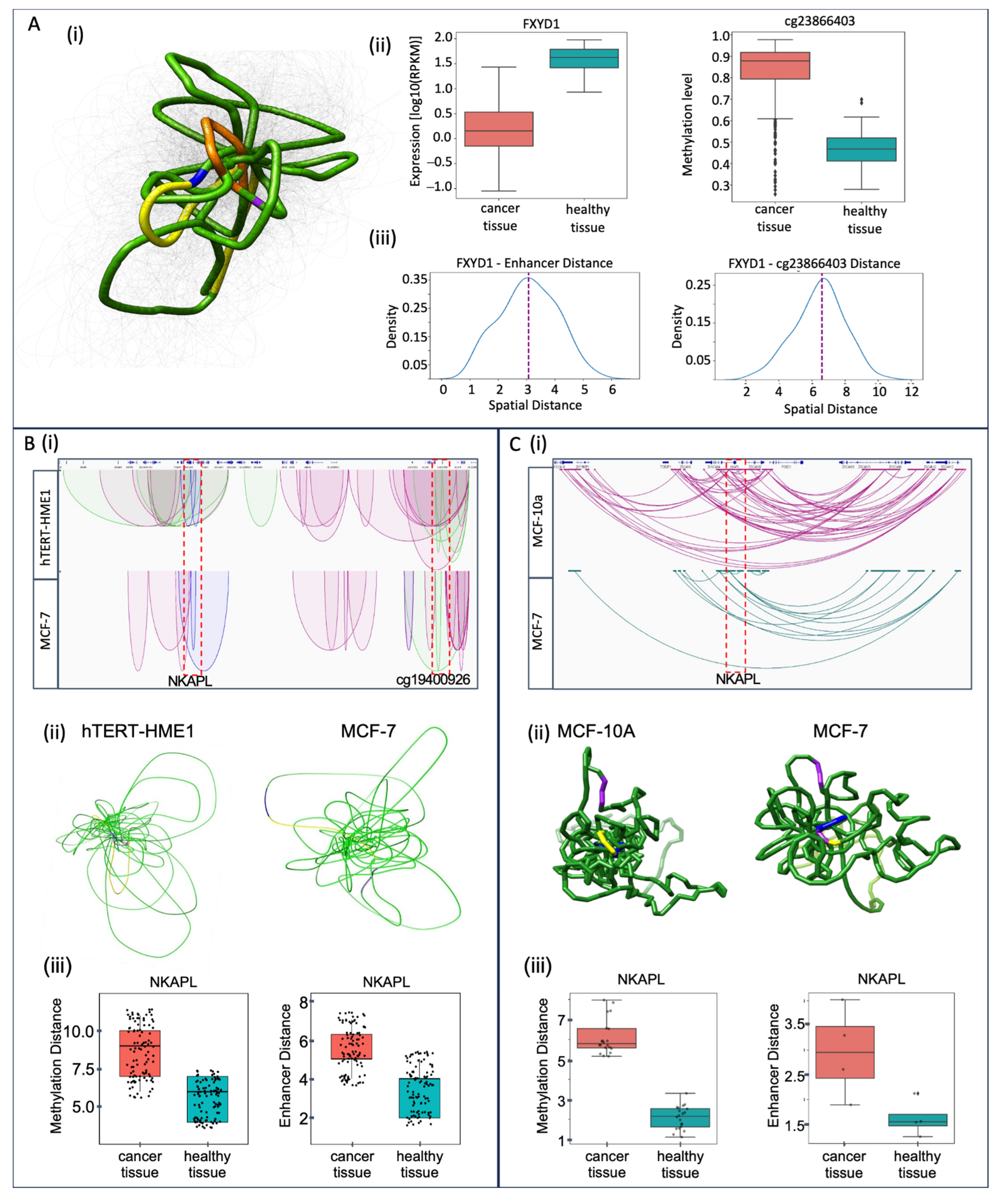
2.8. Epigenomic Regulatory Spatial Model
3. Discussion
4. Materials and Methods
4.1. Data Collection
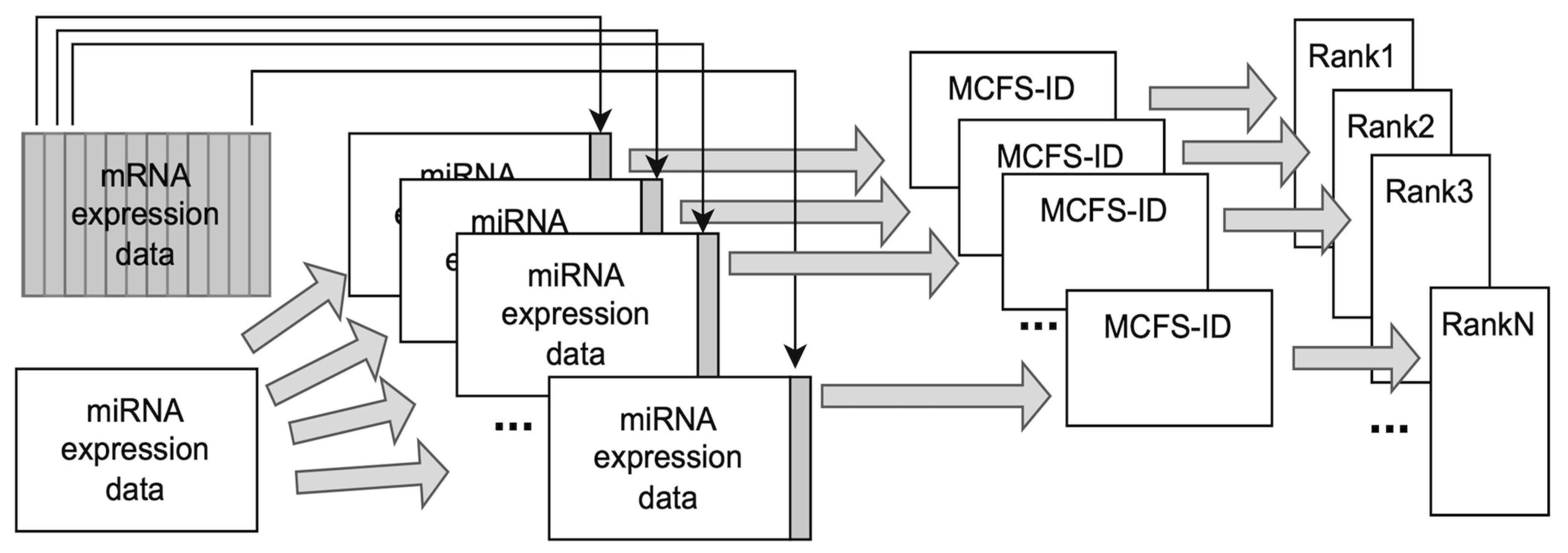
4.2. Detection of Significant Features Using MCFS-ID Algorithm
4.3. Descriptive Analysis of Significant mRNA Genes
4.4. Descriptive Analysis of Significant DNA Methylation Sites
4.5. Descriptive Analysis of Significant miRNA Genes
4.6. Detection of Significant miRNA and Methylations in the Context of Predicting mRNA Expression Levels
4.7. Descriptive Analysis of Associations Between DMS and TFs
4.8. Building Models of Regulatory Networks
4.9. The Visualization of Chromatin 3D Structure of Selected Loci
5. Conclusions
6. Limitations of the Study
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ChIA-PET | Chromatin Interaction Analysis by Paired-End Tag Sequencing |
| CpG | Cytosine Phosphate Guanine |
| CpGI | Cpg Island |
| DEG | Differentially Expressed Gene |
| DMS | Differentially Methylated Sites |
| DScore | Drug Score |
| GScore | Gene Score |
| GO BP | Gene Ontology Biological Processes |
| H3K27ac | Acetylation of the Lysine 27 of the Histone H3 Protein |
| HOCOMOCO | Homo Sapiens Comprehensive Model Collection |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| log2FC | Log2 Fold Change |
| MCFS-ID | Monte Carlo Feature Selection and Interdependencies Discovery |
| NLP | Natural Language Processing Methods |
| NCBI | National Center for Biotechnology Information |
| PCHi-C | Promoter Capture Hi-C |
| PWM | Position Weighted Matrix |
| RF | Random Forest |
| RI | Relative Importance |
| SM | Spring Model |
| SVM | Support Vector Machine |
| TCGA | The Cancer Genome Atlas |
| TF | Transcription Factor |
| TF-IDF | Term Frequency-Inverse Document Frequency |
| TFBS | Transcription Factor Binding Site |
| TSS | Transcription Start Site |
| wAcc | Weighted Accuracy |
| WHO | World Health Organization |
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 Countries. CA A Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dumbrava, E.I.; Meric-Bernstam, F. Personalized cancer therapy—Leveraging a knowledge base for clinical decision-making. Mol. Case Stud. 2018, 4, a001578. [Google Scholar] [CrossRef] [PubMed]
- Chopra, S.; Khosla, M.; Vidya, R. Innovations and challenges in breast cancer care: A review. Medicina 2023, 59, 957. [Google Scholar] [CrossRef]
- Bean, G.R.; Lin, C.Y. Breast neuroendocrine neoplasms: Practical applications and continuing challenges in the era of the 5th edition of the WHO classification of breast tumours. Diagn. Histopathol. 2021, 27, 139–147. [Google Scholar] [CrossRef]
- Cree, I.A.; White, V.A.; Indave, B.I.; Lokuhetty, D. Revising the WHO classification: Female genital tract tumours. Histopathology 2019, 76, 151–156. [Google Scholar] [CrossRef]
- Blumer, A.; Ehrenfeucht, A.; Haussler, D.; Warmuth, M.K. Learnability and the Vapnik-Chervonenkis dimension. J. ACM 1989, 36, 929–965. [Google Scholar] [CrossRef]
- Dramiński, M.; Koronacki, J. rmcfs: An R Package for Monte Carlo Feature Selection and Interdependency Discovery. J. Stat. Softw. 2018, 85, 1–28. [Google Scholar] [CrossRef]
- Dramiński, M.; Da̧browski, M.J.; Diamanti, K.; Koronacki, J.; Komorowski, J. Discovering networks of interdependent features in high-dimensional problems. In Big Data Analysis: New Algorithms for a New Society. Studies in Big Data; Japkowicz, N., Stefanowski, J., Eds.; Springer International Publishing: Cham, Switzerland, 2015; Volume 16, pp. 285–304. [Google Scholar]
- Chen, L.; Zhou, X.; Zeng, T.; Pan, X.; Zhang, Y.H.; Huang, T.; Fang, Z.; Cai, Y.D. Recognizing pattern and rule of mutation signatures corresponding to cancer types. Front. Cell Dev. Biol. 2021, 9, 712931. [Google Scholar] [CrossRef]
- Li, J.; Xu, Q.; Wu, M.; Huang, T.; Wang, Y. Pan-cancer classification based on self-normalizing neural networks and feature selection. Front. Bioeng. Biotechnol. 2020, 8, 766. [Google Scholar] [CrossRef]
- Li, Z.; Mei, Z.; Ding, S.; Chen, L.; Li, H.; Feng, K.; Huang, T.; Cai, Y.D. Identifying methylation signatures and rules for COVID-19 with machine learning methods. Front. Mol. Biosci. 2022, 9, 908080. [Google Scholar] [CrossRef]
- Chen, L.; Zhang, S.; Pan, X.; Hu, X.; Zhang, Y.H.; Yuan, F.; Huang, T.; Cai, Y.D. HIV infection alters the human epigenetic landscape. Gene Ther. 2018, 26, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lin, H.; Li, L. Multiple feature selection strategies identified novel cardiac gene expression signature for heart failure. Front. Physiol. 2020, 11, 604241. [Google Scholar] [CrossRef] [PubMed]
- Paratala, B.S.; Dolfi, S.C.; Khiabanian, H.; Rodríguez-Rodríguez, L.; Ganesan, S.; Hirshfield, K.M. Emerging role of genomic rearrangements in breast cancer: Applying knowledge from other cancers. Biomark. Cancer 2016, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Kagohara, L.T.; Stein-O’Brien, G.L.; Kelley, D.; Flam, E.; Wick, H.C.; Danilova, L.V.; Easwaran, H.; Favorov, A.V.; Qian, J.; Gaykalova, D.A.; et al. Epigenetic regulation of gene expression in cancer: Techniques; resources and analysis. Brief. Funct. Genom. 2017, 17, 49–63. [Google Scholar] [CrossRef]
- Dąbrowski, M.J.; Dramiński, M.; Diamanti, K.; Stępniak, K.; Mozolewska, M.A.; Teisseyre, P.; Koronacki, J.; Komorowski, J.; Kaminska, B.; Wojtas, B. Unveiling new interdependencies between significant DNA methylation sites; gene expression profiles and glioma patients survival. Sci. Rep. 2018, 8, 4390. [Google Scholar] [CrossRef]
- Cao, J.; Yan, Q. Cancer epigenetics; tumor immunity; and immunotherapy. Trends Cancer 2020, 6, 580–592. [Google Scholar] [CrossRef]
- Zhong, Q.; Fan, J.; Chu, H.; Pang, M.; Li, J.; Fan, Y.; Liu, P.; Wu, C.; Qiao, J.; Li, R.; et al. Integrative analysis of genomic and epigenetic regulation of endometrial cancer. Aging 2020, 12, 9260–9274. [Google Scholar] [CrossRef]
- Vezzani, B.; Carinci, M.; Previati, M.; Giacovazzi, S.; Della Sala, M.; Gafà, R.; Lanza, G.; Wieckowski, M.R.; Pinton, P.; Giorgi, C. Epigenetic regulation: A link between inflammation and carcinogenesis. Cancers 2022, 14, 1221. [Google Scholar] [CrossRef]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef]
- Skvortsova, K.; Stirzaker, C.; Taberlay, P. The DNA methylation landscape in cancer. Essays Biochem. 2019, 63, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Yan, W.; Herman, J.G.; Guo, M. Epigenome-based personalized medicine in human cancer. Epigenomics 2016, 8, 119–133. [Google Scholar] [CrossRef] [PubMed]
- Héberlé, É.; Bardet, A. Sensitivity of transcription factors to DNA methylation. Essays Biochem. 2019, 63, 727–741. [Google Scholar] [CrossRef]
- Blattler, A.; Farnham, P.J. Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 2013, 288, 34287–34294. [Google Scholar] [CrossRef]
- Costa, F.F.; Paixão, V.A.; Cavalher, F.P.; Ribeiro, K.B.; Cunha, I.W.; Rinck, J.A., Jr.; O’Hare, M.; Mackay, A.; Soares, F.A.; Brentani, R.R.; et al. SATR-1 hypomethylation is a common and early event in breast cancer. Cancer Genet. Cytogenet. 2006, 165, 135–143. [Google Scholar] [CrossRef]
- Yi, J.; Gao, R.; Chen, Y.; Yang, Z.; Han, P.; Zhang, H.; Dou, Y.; Liu, W.; Wang, W.; Du, G.; et al. Overexpression of NSUN2 by DNA hypomethylation is associated with metastatic progression in human breast cancer. Oncotarget 2016, 8, 20751–20765. [Google Scholar] [CrossRef]
- Choi, J.Y.; James, S.R.; Link, P.A.; McCann, S.E.; Hong, C.C.; Davis, W.; Nesline, M.K.; Ambrosone, C.B.; Karpf, A.R. Association between global DNA hypomethylation in leukocytes and risk of breast cancer. Carcinogenesis 2009, 30, 1889–1897. [Google Scholar] [CrossRef]
- Martin, T.A.; Goyal, A.; Watkins, G.; Jiang, W.G. Expression of the transcription factors Snail; Slug; and Twist and their clinical significance in human breast cancer. Ann. Surg. Oncol. 2005, 12, 488–496. [Google Scholar] [CrossRef]
- Dulaimi, E.; Hillinck, J.; Ibanez de Caceres, I.; Al-Saleem, T.; Cairns, P. Tumor suppressor gene promoter hypermethylation in serum of breast cancer patients. Clin. Cancer Res. 2004, 10, 6189–6193. [Google Scholar] [CrossRef]
- Alvarez, C.; Tapia, T.; Cornejo, V.; Fernandez, W.; Muñoz, A.; Camus, M.; Alvarez, M.; Devoto, L.; Carvallo, P. Silencing of tumor suppressor genes RASSF1A.; SLIT2; and WIF1 by promoter hypermethylation in hereditary breast cancer. Mol. Carcinog. 2012, 52, 475–487. [Google Scholar] [CrossRef]
- Su, J.; Huang, Y.H.; Cui, X.; Wang, X.; Zhang, X.; Lei, Y.; Xu, J.; Lin, X.; Chen, K.; Lv, J.; et al. Homeobox oncogene activation by pan-cancer DNA hypermethylation. Genome Biol. 2018, 19, 108. [Google Scholar] [CrossRef] [PubMed]
- Spainhour, J.C.; Lim, H.S.; Yi, S.V.; Qiu, P. Correlation patterns between DNA methylation and gene expression in the cancer genome atlas. Cancer Inform. 2019, 18, 1176935119828776. [Google Scholar] [CrossRef] [PubMed]
- Yao, Q.; Chen, Y.; Zhou, X. The roles of microRNAs in epigenetic regulation. Curr. Opin. Chem. Biol. 2019, 51, 11–17. [Google Scholar] [CrossRef]
- Ying, S.Y.; Chang, D.C.; Lin, S.L. The microRNA (miRNA): Overview of the RNA genes that modulate gene function. Mol. Biotechnol. 2008, 38, 257–268. [Google Scholar] [CrossRef]
- Pavlíková, L.; Šereš, M.; Breier, A.; Sulová, Z. The roles of microRNAs in cancer multidrug resistance. Cancers 2022, 14, 1090. [Google Scholar] [CrossRef]
- Muñoz, J.P.; Pérez-Moreno, P.; Pérez, Y.; Calaf, G.M. The role of microRNAs in breast cancer and the challenges of their clinical application. Diagnostics 2023, 13, 3072. [Google Scholar] [CrossRef]
- Umer, H.M.; Cavalli, M.; Dabrowski, M.; Diamanti, K.; Kruczyk, M.; Pan, G.; Komorowski, J.; Wadelius, C. A significant regulatory mutation burden at a high-affinity position of the CTCF motif in gastrointestinal cancers. Hum. Mutat. 2016, 37, 904–913. [Google Scholar] [CrossRef]
- Monteagudo-Sánchez, A.; Noordermeer, D.; Greenberg, M.V.C. The impact of DNA methylation on CTCF-mediated 3D genome organization. Nat. Struct. Mol. Biol. 2024, 31, 404–412. [Google Scholar] [CrossRef]
- Grabowicz, I.E.; Wilczyński, B.; Kamińska, B.; Roura, A.J.; Wojtaś, B.; Dąbrowski, M.J. The role of epigenetic modifications; long-range contacts; enhancers and topologically associating domains in the regulation of glioma grade-specific genes. Sci. Rep. 2021, 11, 15668. [Google Scholar] [CrossRef]
- Poulos, R.C.; Sloane, M.A.; Hesson, L.B.; Wong, J.W.H. The search for cis-regulatory driver mutations in cancer genomes. Oncotarget 2015, 6, 32509–32525. [Google Scholar] [CrossRef]
- Cestarelli, V.; Fiscon, G.; Felici, G.; Bertolazzi, P.; Weitschek, E. CAMUR: Knowledge extraction from RNA-seq cancer data through equivalent classification rules. Bioinformatics 2015, 32, 697–704. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sun, Y.; Chen, H.; Liao, Y.; Li, S.; Chen, C.; Yang, Z. ADAMTS5 acts as a tumor suppressor by inhibiting migration; invasion and angiogenesis in human gastric cancer. Gastric Cancer 2018, 22, 287–301. [Google Scholar] [CrossRef]
- Choi, B.; Han, T.S.; Min, J.; Hur, K.; Lee, S.M.; Lee, H.J.; Kim, Y.J.; Yang, H.K. MAL and TMEM220 are novel DNA methylation markers in human gastric cancer. Biomarkers 2016, 22, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Li, J.; Zhang, C.Y.; Huang, D.Y.; Xu, J.W. ARHGAP20 Expression inhibited HCC progression by regulating the PI3K-AKT signaling pathway. J. Hepatocell. Carcinoma 2021, 8, 271–284. [Google Scholar] [CrossRef]
- Bai, Y.; Wei, C.; Zhong, Y.; Zhang, Y.; Long, J.; Huang, S.; Xie, F.; Tian, Y.; Wang, X.; Zhao, H. Development and validation of a prognostic nomogram for gastric cancer based on DNA methylation-driven differentially expressed genes. Int. J. Biol. Sci. 2020, 16, 1153–1165. [Google Scholar] [CrossRef]
- Giussani, M.; Landoni, E.; Merlino, G.; Turdo, F.; Veneroni, S.; Paolini, B.; Cappelletti, V.; Miceli, R.; Orlandi, R.; Triulzi, T.; et al. Extracellular matrix proteins as diagnostic markers of breast carcinoma. J. Cell. Physiol. 2018, 233, 6280–6290. [Google Scholar] [CrossRef]
- Zhuang, Y.; Li, X.; Zhan, P.; Pi, G.; Wen, G. MMP11 promotes the proliferation and progression of breast cancer through stabilizing Smad2 protein. Oncol. Rep. 2021, 45, 16. [Google Scholar] [CrossRef]
- Ozturk, S.; Papageorgis, P.; Wong, C.K.; Lambert, A.W.; Abdolmaleky, H.M.; Thiagalingam, A.; Cohen, H.T.; Thiagalingam, S. SDPR functions as a metastasis suppressor in breast cancer by promoting apoptosis. Proc. Natl. Acad. Sci. USA 2016, 113, 638–643. [Google Scholar] [CrossRef]
- Nema, R.; Kumar, A. Sphingosine-1-phosphate catabolizing enzymes predict better prognosis in triple-negative breast cancer patients and correlates with tumor-infiltrating immune cells. Front. Mol. Biosci. 2021, 8, 697922. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, H.; Qi, Y.; Li, S.; Geng, C. Epigenetic study of early breast cancer (EBC) based on DNA methylation and gene integration analysis. Sci. Rep. 2022, 12, 1989. [Google Scholar] [CrossRef]
- Bao, Y.; Wang, L.; Shi, L.; Yun, F.; Liu, X.; Chen, Y.; Chen, C.; Ren, Y.; Jia, Y. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 2019, 24, 38. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The reactome pathway knowledgebase 2022. Nucleic Acids Res. 2021, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.K.S.; Lau, C.P.Y.; Lam, E.K.Y.; Li, S.S.K.; Lui, V.W.Y.; Yeo, W.; Ng, Y.K.; Lai, P.B.S.; Tsui, S.K.W. Hypermethylation of NF-κB-Activating Protein-Like (NKAPL) promoter in hepatocellular carcinoma suppresses Its expression and predicts a poor prognosis. Dig. Dis. Sci. 2018, 63, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Dai, H.; Gallagher, D.; Schmitt, S.; Pessetto, Z.Y.; Fan, F.; Godwin, A.K.; Tawfik, O. Role of miR-139 as a surrogate marker for tumor aggression in breast cancer. Hum. Pathol. 2016, 61, 68–77. [Google Scholar] [CrossRef]
- Halim, A.; Al-Qadi, N.; Kenyon, E.; Conner, K.N.; Mondal, S.K.; Medarova, Z.; Moore, A. Inhibition of miR-10b treats metastatic breast cancer by targeting stem cell-like properties. Oncotarget 2024, 15, 591–606. [Google Scholar] [CrossRef]
- Wang, H.; Tan, Z.; Hu, H.; Liu, H.; Wu, T.; Zheng, C.; Wang, X.; Luo, Z.; Wang, J.; Liu, S.; et al. microRNA-21 promotes breast cancer proliferation and metastasis by targeting LZTFL1. BMC Cancer 2019, 19, 738. [Google Scholar] [CrossRef]
- Mohammaddoust, S.; Sadeghizadeh, M. Mir-183 functions as an oncogene via decreasing PTEN in breast cancer cells. Sci. Rep. 2023, 13, 8086. [Google Scholar] [CrossRef]
- Hajibabaei, S.; Sotoodehnejadnematalahi, F.; Nafissi, N.; Zeinali, S.; Azizi, M. Aberrant promoter hypermethylation of miR-335 and miR-145 is involved in breast cancer PD-L1 overexpression. Sci. Rep. 2023, 13, 1003. [Google Scholar] [CrossRef]
- Long, X.; Shi, Y.; Ye, P.; Guo, J.; Zhou, Q.; Tang, Y. MicroRNA-99a Suppresses Breast Cancer Progression by Targeting FGFR3. Front. Oncol. 2020, 9, 1473. [Google Scholar] [CrossRef]
- Sameti, P.; Tohidast, M.; Amini, M.; Bahojb Mahdavi, S.Z.; Najafi, S.; Mokhtarzadeh, A. The emerging role of MicroRNA-182 in tumorigenesis; a promising therapeutic target. Cancer Cell Int. 2023, 23, 134. [Google Scholar] [CrossRef]
- Hong, Y.; Liang, H.; Urrehman, U.; Wang, Y.; Zhang, W.; Zhou, Y.; Chen, S.; Yu, M.; Cui, S.; Liu, M.; et al. miR-96 promotes cell proliferation, migration and invasion by targeting PTPN9 in breast cancer. Sci. Rep. 2016, 6, 37421. [Google Scholar] [CrossRef] [PubMed]
- Gharehdaghchi, Z.; Baradaran, B.; Salehzadeh, A.; Kazemi, T. miR-486-5p regulates cell proliferation and migration in breast cancer. Meta Gene 2019, 23, 100643. [Google Scholar] [CrossRef]
- Li, P.; Xu, T.; Zhou, X.; Liao, L.; Pang, G.; Luo, W.; Han, L.; Zhang, J.; Luo, X.; Xie, X.; et al. Downregulation of miRNA-141 in breast cancer cells is associated with cell migration and invasion: Involvement of ANP32E targeting. Cancer Med. 2017, 6, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Hart, L.; Campone, M.; Petrakova, K.; Winer, E.P.; Janni, W.; et al. Overall survival with ribociclib plus letrozole in advanced breast cancer. N. Engl. J. Med. 2022, 386, 942–950. [Google Scholar] [CrossRef]
- Tolaney, S.M.; Beeram, M.; Beck, J.T.; Conlin, A.; Dees, E.C.; Puhalla, S.L.; Rexer, B.N.; Burris, H.A.; Jhaveri, K.; Helsten, T.; et al. Abemaciclib in combination with endocrine therapy for patients with hormone receptor-positive; HER2-negative metastatic breast cancer: A phase 1b study. Front. Oncol. 2022, 11, 810023. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2020, 49, D545–D551. [Google Scholar] [CrossRef]
- Colak, S.; ten Dijke, P. Targeting TGF-β Signaling in Cancer. Trends Cancer 2017, 3, 56–71. [Google Scholar] [CrossRef]
- Michaelis, M.; Doerr, H.W.; Cinatl, J. The story of human cytomegalovirus and cancer: Increasing evidence and open questions. Neoplasia 2009, 11, 1–9. [Google Scholar] [CrossRef]
- Zhao, J.; Xu, Y. PITX1 plays essential functions in cancer. Front. Oncol. 2023, 13, 1253238. [Google Scholar] [CrossRef]
- Vidula, N.; Yau, C.; Wolf, D.; Rugo, H.S. Androgen receptor gene expression in primary breast cancer. npj Breast Cancer 2019, 5, 47. [Google Scholar] [CrossRef]
- Fallah, Y.; Brundage, J.; Allegakoen, P.; Shajahan-Haq, A.N. MYC-driven pathways in breast cancer subtypes. Biomolecules 2017, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- Pietri, E.; Conteduca, V.; Andreis, D.; Massa, I.; Melegari, E.; Sarti, S.; Cecconetto, L.; Schirone, A.; Bravaccini, S.; Serra, P.; et al. Androgen receptor signaling pathways as a target for breast cancer treatment. Endocr.-Relat. Cancer 2016, 23, R485–R498. [Google Scholar] [CrossRef] [PubMed]
- Hardy, K.M.; Booth, B.W.; Hendrix, M.J.C.; Salomon, D.S.; Strizzi, L. ErbB/EGF signaling and EMT in mammary development and breast cancer. J. Mammary Gland Biol. Neoplasia 2010, 15, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Thanasupawat, T.; Glogowska, A.; Nivedita-Krishnan, S.; Wilson, B.; Klonisch, T.; Hombach-Klonisch, S. Emerging roles for the relaxin/RXFP1 system in cancer therapy. Mol. Cell. Endocrinol. 2019, 487, 85–93. [Google Scholar] [CrossRef]
- Yang, S.; Chen, K.; Cao, K.; Xu, S.; Ma, C.; Cai, Y.; Hu, Y.; Zhou, Y. miR-182-5p Inhibits NKAPL expression and Promotes the Proliferation of Osteosarcoma. Biotechnol. Bioprocess Eng. 2021, 26, 758–766. [Google Scholar] [CrossRef]
- Zhang, X.; Kang, X.; Jin, L.; Bai, J.; Zhang, H.; Liu, W.; Wang, Z. ABCC9; NKAPL; and TMEM132C are potential diagnostic and prognostic markers in triple-negative breast cancer. Cell Biol. Int. 2020, 44, 2002–2010. [Google Scholar] [CrossRef]
- Silva, R.; Glennon, K.; Metoudi, M.; Moran, B.; Salta, S.; Slattery, K.; Treacy, A.; Martin, T.; Shaw, J.; Doran, P.; et al. Unveiling the epigenomic mechanisms of acquired platinum-resistance in high-grade serous ovarian cancer. Int. J. Cancer 2023, 153, 120–132. [Google Scholar] [CrossRef]
- Morales-Martínez, M.; Vega, M.I. Role of MicroRNA-7 (MiR-7) in Cancer Physiopathology. Int. J. Mol. Sci. 2022, 23, 9091. [Google Scholar] [CrossRef]
- Schneider, E.; Pliushch, G.; El Hajj, N.; Galetzka, D.; Puhl, A.; Schorsch, M.; Frauenknecht, K.; Riepert, T.; Tresch, A.; Müller, A.M.; et al. Spatial, temporal and interindividual epigenetic variation of functionally important DNA methylation patterns. Nucleic Acids Res. 2010, 38, 3880–3890. [Google Scholar] [CrossRef]
- Cedar, H. DNA methylation and gene activity. Cell 1988, 53, 3–4. [Google Scholar] [CrossRef]
- Szalaj, P.; Michalski, P.J.; Wróblewski, P.; Tang, Z.; Kadlof, M.; Mazzocco, G.; Ruan, Y.; Plewczynski, D. 3D-GNOME: An integrated web service for structural modeling of the 3D genome. Nucleic Acids Res. 2016, 44, W288–W293. [Google Scholar] [CrossRef] [PubMed]
- Wlasnowolski, M.; Sadowski, M.; Czarnota, T.; Jodkowska, K.; Szalaj, P.; Tang, Z.; Ruan, Y.; Plewczynski, D. 3D-GNOME 2.0: A three-dimensional genome modeling engine for predicting structural variation-driven alterations of chromatin spatial structure in the human genome. Nucleic Acids Res. 2020, 48, W170–W176. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Grubert, F.; Srivas, R.; Spacek, D.; Kasowski, M.; Ruiz-Velasco, M.; Sinnott-Armstrong, N.; Greenside, P.; Narasimha, A.; Liu, Q.; Geller, B.; et al. Landscape of cohesin-mediated chromatin loops in the human genome. Nature 2020, 583, 737–743. [Google Scholar] [CrossRef]
- Javierre, B.M.; Burren, O.S.; Wilder, S.P.; Kreuzhuber, R.; Hill, S.M.; Sewitz, S.; Cairns, J.; Wingett, S.W.; Várnai, C.; Thiecke, M.J.; et al. Lineage-Specific Genome Architecture Links Enhancers and Non-coding Disease Variants to Target Gene Promoters. Cell 2016, 167, 1369–1384.e19. [Google Scholar] [CrossRef]
- Beesley, J.; Sivakumaran, H.; Moradi Marjaneh, M.; Lima, L.G.; Hillman, K.M.; Kaufmann, S.; Tuano, N.; Hussein, N.; Ham, S.; Mukhopadhyay, P.; et al. Chromatin interactome mapping at 139 independent breast cancer risk signals. Genome Biol. 2020, 21, 8. [Google Scholar] [CrossRef]
- Achinger-Kawecka, J.; Stirzaker, C.; Portman, N.; Campbell, E.; Chia, K.M.; Du, Q.; Laven-Law, G.; Nair, S.S.; Yong, A.; Wilkinson, A.; et al. The potential of epigenetic therapy to target the 3D epigenome in endocrine-resistant breast cancer. Nat. Struct. Mol. Biol. 2024, 31, 498–512. [Google Scholar] [CrossRef]
- Kadlof, M.; Rozycka, J.; Plewczynski, D. Spring Model—Chromatin modeling tool based on OpenMM. Methods 2020, 181–182, 62–69. [Google Scholar] [CrossRef]
- Mei, J.; Zhao, J.; Fu, Y. Molecular classification of breast cancer using the mRNA expression profiles of immune-related genes. Sci. Rep. 2020, 10, 4800. [Google Scholar] [CrossRef]
- Deng, J.L.; Xu, Y.; Wang, G. Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front. Genet. 2019, 10, 695. [Google Scholar] [CrossRef]
- de Cárcer, G.; Venkateswaran, S.V.; Salgueiro, L.; El Bakkali, A.; Somogyi, K.; Rowald, K.; Montañés, P.; Sanclemente, M.; Escobar, B.; de Martino, A.; et al. Plk1 overexpression induces chromosomal instability and suppresses tumor development. Nat. Commun. 2018, 9, 3012. [Google Scholar] [CrossRef] [PubMed]
- Lashen, A.; Toss, M.S.; Wootton, L.L.; Green, A.; Mongan, N.P.; Madhusudan, S.; Rakha, E. Characteristics and prognostic significance of polo-like kinase-1 (PLK1) expression in breast cancer. Histopathology 2023, 83, 414–425. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Shen, W.; Pan, A.; Sun, F.; Zhang, J.; Gao, P.; Li, L. Decreased Nek9 expression correlates with aggressive behaviour and predicts unfavourable prognosis in breast cancer. Pathology 2020, 52, 329–335. [Google Scholar] [CrossRef]
- Lesko, A.C.; Goss, K.H.; Yang, F.F.; Schwertner, A.; Hulur, I.; Onel, K.; Prosperi, J.R. The APC tumor suppressor is required for epithelial cell polarization and three-dimensional morphogenesis. Biochim. Biophys. Acta (BBA)—Mol. Cell Res. 2015, 1853, 711–723. [Google Scholar] [CrossRef]
- Seitz, H.K.; Stickel, F. Acetaldehyde as an underestimated risk factor for cancer development: Role of genetics in ethanol metabolism. Genes Nutr. 2010, 5, 121–128. [Google Scholar] [CrossRef]
- Fu, Y.; Zou, T.; Shen, X.; Nelson, P.J.; Li, J.; Wu, C.; Yang, J.; Zheng, Y.; Bruns, C.; Zhao, Y.; et al. Lipid metabolism in cancer progression and therapeutic strategies. MedComm. 2020, 2, 27–59. [Google Scholar] [CrossRef]
- Long, J.; Zhang, C.J.; Zhu, N.; Du, K.; Yin, Y.F.; Tan, X.; Liao, D.F.; Qin, L. Lipid metabolism and carcinogenesis; cancer development. Am. J. Cancer Res. 2018, 8, 778–791. [Google Scholar]
- Chen, M.C.; Hsu, S.L.; Lin, H.; Yang, T.Y. Retinoic acid and cancer treatment. BioMedicine 2014, 4, 22. [Google Scholar] [CrossRef]
- Jan, N.; Sofi, S.; Qayoom, H.; Haq, B.U.; Shabir, A.M.; Mir, M.A. Targeting breast cancer stem cells through retinoids: A new hope for treatment. Crit. Rev. Oncol./Hematol. 2023, 192, 104156. [Google Scholar] [CrossRef]
- Costantini, L.; Molinari, R.; Farinon, B.; Merendino, N. Retinoic acids in the treatment of most lethal solid cancers. J. Clin. Med. 2020, 9, 360. [Google Scholar] [CrossRef]
- Li, R.Q.; Zhao, X.H.; Zhu, Q.; Liu, T.; Hondermarck, H.; Thorne, R.F.; Zhang, X.D.; Gao, J.N. Exploring neurotransmitters and their receptors for breast cancer prevention and treatment. Theranostics 2023, 13, 1109–1129. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, D.M.; Rasmussen, S.G.F.; Kobilka, B.K. The structure and function of G-protein-coupled receptors. Nature 2009, 459, 356–363. [Google Scholar] [CrossRef] [PubMed]
- Dorsam, R.T.; Gutkind, J.S. G-protein-coupled receptors and cancer. Nat. Rev. Cancer 2007, 7, 79–94. [Google Scholar] [CrossRef]
- De Francesco, E.; Sotgia, F.; Clarke, R.; Lisanti, M.; Maggiolini, M. G Protein-Coupled receptors at the crossroad between physiologic and pathologic angiogenesis: Old paradigms and emerging concepts. Int. J. Mol. Sci. 2017, 18, 2713. [Google Scholar] [CrossRef]
- Singh, A.; Nunes, J.J.; Ateeq, B. Role and therapeutic potential of G-protein coupled receptors in breast cancer progression and metastases. Eur. J. Pharmacol. 2015, 763, 178–183. [Google Scholar] [CrossRef]
- Lappano, R.; Jacquot, Y.; Maggiolini, M. GPCR modulation in breast cancer. Int. J. Mol. Sci. 2018, 19, 3840. [Google Scholar] [CrossRef]
- He, J.; Fortunati, E.; Liu, D.X.; Yan, L. Pleiotropic roles of ABC transporters in breast cancer. Int. J. Mol. Sci. 2021, 22, 3199. [Google Scholar] [CrossRef]
- Chen, Y.; Gera, L.; Zhang, S.; Li, X.; Yang, Y.; Mamouni, K.; Wu, A.Y.; Liu, H.; Kucuk, O.; Wu, D. Small molecule BKM1972 inhibits human prostate cancer growth and overcomes docetaxel resistance in intraosseous models. Cancer Lett. 2019, 446, 62–72. [Google Scholar] [CrossRef]
- Muriithi, W.; Wanjiku Macharia, L.W.; Pilotto Heming, C.; Lima Echevarria, J.; Nyachieo, A.; Niemeyer Filho, P.; Moura Neto, V. ABC transporters and the hallmarks of cancer: Roles in cancer aggressiveness beyond multidrug resistance. Cancer Biol. Med. 2020, 17, 253–269. [Google Scholar] [CrossRef]
- Demidenko, R.; Razanauskas, D.; Daniunaite, K.; Lazutka, J.R.; Jankevicius, F.; Jarmalaite, S. Frequent down-regulation of ABC transporter genes in prostate cancer. BMC Cancer 2015, 15, 683. [Google Scholar] [CrossRef]
- Elsnerova, K.; Mohelnikova-Duchonova, B.; Cerovska, E.; Ehrlichova, M.; Gut, I.; Rob, L.; Skapa, P.; Hruda, M.; Bartakova, A.; Bouda, J.; et al. Gene expression of membrane transporters: Importance for prognosis and progression of ovarian carcinoma. Oncol. Rep. 2016, 35, 2159–2170. [Google Scholar] [CrossRef] [PubMed]
- Lv, C.; Yang, H.; Yu, J.; Dai, X. ABCA8 inhibits breast cancer cell proliferation by regulating the AMP activated protein kinase/mammalian target of rapamycin signaling pathway. Environ. Toxicol. 2022, 37, 1423–1431. [Google Scholar] [CrossRef] [PubMed]
- Agius, R.; Parviz, M.; Niemann, C.U. Artificial intelligence models in chronic lymphocytic leukemia—Recommendations toward state-of-the-art. Leuk. Lymphoma 2021, 63, 265–278. [Google Scholar] [CrossRef]
- Wilting, R.H.; Dannenberg, J.H. Epigenetic mechanisms in tumorigenesis; tumor cell heterogeneity and drug resistance. Drug Resist. Updates 2012, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Li, C.; Yin, H.; Huang, J.; Yu, S.; Zhao, J.; Tang, Y.; Yu, M.; Lin, J.; Ding, L.; et al. The mechanism of DNA methylation and miRNA in breast cancer. Int. J. Mol. Sci. 2023, 24, 9360. [Google Scholar] [CrossRef]
- Li, Y.; Lu, W.; Chen, D.; Boohaker, R.J.; Zhai, L.; Padmalayam, I.; Wennerberg, K.; Xu, B.; Zhang, W. KIFC1 is a novel potential therapeutic target for breast cancer. Cancer Biol. Ther. 2015, 16, 1316–1322. [Google Scholar] [CrossRef]
- Xiao, Y.X.; Yang, W.X. KIFC1: A promising chemotherapy target for cancer treatment? Oncotarget 2016, 7, 48656–48670. [Google Scholar] [CrossRef]
- Jiao, D.; Zhang, J.; Chen, P.; Guo, X.; Qiao, J.; Zhu, J.; Wang, L.; Lu, Z.; Liu, Z. HN1L promotes migration and invasion of breast cancer by up-regulating the expression of HMGB1. J. Cell. Mol. Med. 2020, 25, 397–410. [Google Scholar] [CrossRef]
- Domcke, S.; Bardet, A.F.; Adrian Ginno, P.; Hartl, D.; Burger, L.; Schübeler, D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature 2015, 528, 575–579. [Google Scholar] [CrossRef]
- Schmolka, N.; Karemaker, I.D.; Cardoso, R.; Recchia, D.C.; Spegg, V.; Bhaskaran, J.; Teske, M.; de Wagenaar, N.P.; Altmeyer, M.; Baubec, T. Dissecting the roles of MBD2 isoforms and domains in regulating NuRD complex function during cellular differentiation. Nat. Commun. 2023, 14, 3848. [Google Scholar] [CrossRef]
- Osborne, C.K. Tamoxifen in the treatment of breast cancer. N. Engl. J. Med. 1998, 339, 1609–1618. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Wan, J.; Su, Y.; Song, Q.; Zeng, Y.; Nguyen, H.N.; Shin, J.; Cox, E.; Rho, H.S.; Woodard, C.; et al. DNA methylation presents distinct binding sites for human transcription factors. eLife 2013, 2, e00726. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Bai, Z.; Feng, F.; Song, E.; Du, F.; Zhao, J.; Shen, G.; Ji, F.; Li, G.; Ma, X.; et al. Cross-talk between EPAS-1/HIF-2α and PXR signaling pathway regulates multi-drug resistance of stomach cancer cell. Int. J. Biochem. Cell Biol. 2016, 72, 73–88. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, W.; Zhang, J.; Ren, D.; Zhao, P.; Ying, Y. EPAS1; a hypoxia- and ferroptosis-related gene; promotes malignant behaviour of cervical cancer by ceRNA and super-enhancer. J. Cell. Mol. Med. 2024, 28, e18361. [Google Scholar] [CrossRef]
- Abdollahi, A.; Pisarcik, D.A.; Roberts, D.D.; Weinstein, J.K.; Cairns, P.; Hamilton, T.A. LOT1 (PLAGL1/ZAC1); the candidate tumor suppressor gene at chromosome 6q24–25; is epigenetically regulated in cancer. J. Biol. Chem. 2003, 278, 6041–6049. [Google Scholar] [CrossRef]
- Lanzino, M.; Maris, P.; Sirianni, R.; Barone, I.; Casaburi, I.; Chimento, A.; Giordano, C.; Morelli, C.; Sisci, D.; Rizza, P.; et al. DAX-1; as an androgen-target gene; inhibits aromatase expression: A novel mechanism blocking estrogen-dependent breast cancer cell proliferation. Cell Death Dis. 2013, 4, e724. [Google Scholar] [CrossRef]
- Chae, B.J.; Lee, A.; Bae, J.S.; Song, B.J.; Jung, S.S. Expression of nuclear receptor DAX-1 and androgen receptor in human breast cancer. J. Surg. Oncol. 2011, 103, 768–772. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, Q.; Wang, C.; Zhao, N.; Liu, Y.; Wang, E. BHLHE41 suppresses MCF-7 cell invasion via MAPK/JNK pathway. J. Cell. Mol. Med. 2020, 24, 4001–4010. [Google Scholar] [CrossRef]
- Keith, B.; Johnson, R.S.; Simon, M.C. HIF1α and HIF2α: Sibling rivalry in hypoxic tumor growth and progression. Nat. Rev. Cancer 2011, 12, 9–22. [Google Scholar] [CrossRef]
- Okuda, H.; Kiuchi, H.; Takao, T.; Miyagawa, Y.; Tsujimura, A.; Nonomura, N.; Miyata, H.; Okabe, M.; Ikawa, M.; Kawakami, Y.; et al. A novel transcriptional factor Nkapl is a germ cell-specific suppressor of notch signaling and is indispensable for spermatogenesis. PLoS ONE 2015, 10, e0124293. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Sun, Q.; Wang, X. Transcriptional landscape of human cancers. Oncotarget 2017, 8, 34534–34551. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.A.; Gray, P.C.; Blount, A.L.; MacConell, L.A.; Wiater, E.; Bilezikjian, L.M.; Vale, W. Betaglycan binds inhibin and can mediate functional antagonism of activin signalling. Nature 2000, 404, 411–414. [Google Scholar] [CrossRef]
- Bao, S.; He, G. Identification of key genes and key pathways in breast cancer based on machine learning. Med. Sci. Monit. 2022, 28, e935515. [Google Scholar] [CrossRef]
- Dong, M.; How, T.; Kirkbride, K.C.; Gordon, K.J.; Lee, J.T.; Hempel, N.; Kelly, P.; Moeller, B.J.; Marks, J.R.; Blobe, G.C. The type III TGF-β receptor suppresses breast cancer progression. J. Clin. Investig. 2007, 117, 206–217. [Google Scholar] [CrossRef]
- Mayer, B.J.; Baltimore, D. Signalling through SH2 and SH3 domains. Trends Cell Biol. 1993, 3, 8–13. [Google Scholar] [CrossRef]
- Li, T.; Guan, L.; Tang, G.; He, B.; Huang, L.; Wang, J.; Li, M.; Bai, Y.; Li, X.; Zhang, H. Downregulation of TMEM220 promotes tumor progression in Hepatocellular Carcinoma. Cancer Gene Ther. 2021, 29, 835–844. [Google Scholar] [CrossRef]
- Jung, S.Y.; Kim, D.Y.; Yune, T.Y.; Shin, D.H.; Baek, S.B.; Kim, C.J. Treadmill exercise reduces spinal cord injury-induced apoptosis by activating the PI3K/Akt pathway in rats. Exp. Ther. Med. 2013, 7, 587–593. [Google Scholar] [CrossRef]
- Su, P.H.; Hsu, Y.C.; Huang, R.; Weng, Y.C.; Wang, H.C.; Chen, Y.; Tsai, Y.J.; Yuan, C.C.; Lai, H.C. Methylomics of nitroxidative stress on precancerous cells reveals DNA methylation alteration at the transition from in situ to invasive cervical cancer. Oncotarget 2017, 8, 65281–65291. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, H.; Fan, C.; Hildesjö, C.; Shen, B.; Sun, X.F. Loss of CHGA protein as a potential biomarker for colon cancer diagnosis: A study on biomarker discovery by machine learning and confirmation by immunohistochemistry in colorectal cancer tissue microarrays. Cancers 2022, 14, 2664. [Google Scholar] [CrossRef]
- Kirouac, D.C.; Du, J.; Lahdenranta, J.; Onsum, M.D.; Nielsen, U.B.; Schoeberl, B.; McDonagh, C.F. HER2+ cancer cell dependence on PI3K vs. MAPK signaling axes is determined by expression of EGFR.; ERBB3 and CDKN1B. PLoS Comput. Biol. 2016, 12, e1004827. [Google Scholar] [CrossRef] [PubMed]
- Arteaga, C.L.; Engelman, J.A. ERBB receptors: From oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell 2014, 25, 282–303. [Google Scholar] [CrossRef]
- Paplomata, E.; O’Regan, R. The PI3K/AKT/mTOR pathway in breast cancer: Targets; trials and biomarkers. Ther. Adv. Med. Oncol. 2014, 6, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Prever, L.; Hirsch, E.; Gulluni, F. Targeting PI3K/AKT/mTOR signaling pathway in breast cancer. Cancers 2021, 13, 3517. [Google Scholar] [CrossRef]
- Shi, P.; Feng, J.; Chen, C. Hippo pathway in mammary gland development and breast cancer. Acta Biochim. Biophys. Sin. 2014, 47, 53–59. [Google Scholar] [CrossRef]
- Han, Y. Analysis of the role of the Hippo pathway in cancer. J. Transl. Med. 2019, 17, 116. [Google Scholar] [CrossRef]
- Zhao, M.; Mishra, L.; Deng, C.X. The role of TGF-β/SMAD4 signaling in cancer. Int. J. Biol. Sci. 2018, 14, 111–123. [Google Scholar] [CrossRef]
- Labibi, B.; Bashkurov, M.; Wrana, J.L.; Attisano, L. Modeling the control of TGF-β/Smad nuclear accumulation by the Hippo pathway effectors; Taz/Yap. iScience 2020, 23, 101416. [Google Scholar] [CrossRef]
- Band, A.M.; Laiho, M. Crosstalk of TGF-β and estrogen receptor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2011, 16, 109–115. [Google Scholar] [CrossRef]
- Moses, H.; Barcellos-Hoff, M.H. TGF-β biology in mammary development and breast cancer. Cold Spring Harb. Perspect. Biol. 2010, 3, a003277. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β family signaling in the control of cell proliferation and survival. Cold Spring Harb. Perspect. Biol. 2016, 9, a022145. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, Z. The emerging role of microRNAs in breast cancer. J. Oncol. 2020, 2020, 9160905. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, F.; Wang, J.; Wu, H.; Yang, H.; Yang, Z.; Huang, H. Role and mechanism of miR-211 in human cancer. J. Cancer 2022, 13, 2933–2944. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Kunhiraman, H.; Perera, R.J. The Paradoxical Behavior of microRNA-211 in Melanomas and Other Human Cancers. Front. Oncol. 2021, 10, 628367. [Google Scholar] [CrossRef]
- Dąbrowski, M.J.; Wojtaś, B. Global DNA methylation patterns in human gliomas and their interplay with other epigenetic modifications. Int. J. Mol. Sci. 2019, 20, 3478. [Google Scholar] [CrossRef]
- Líu, H.; Tsai, H.W.; Yang, M.; Li, G.; Bian, Q.; Ding, G.; Wu, D.; Dai, J. Three-dimensional genome structure and function. MedComm. 2023, 4, e326. [Google Scholar] [CrossRef]
- Calderon, L.; Weiss, F.D.; Beagan, J.A.; Oliveira, M.S.; Georgieva, R.; Wang, Y.F.; Carroll, T.S.; Dharmalingam, G.; Gong, W.; Tossell, K.; et al. Cohesin-dependence of neuronal gene expression relates to chromatin loop length. eLife 2022, 11, e76539. [Google Scholar] [CrossRef]
- Bateman, J.R.; Johnson, J.E. Altering enhancer–promoter linear distance impacts promoter competition in cis and in trans. Genetics 2022, 222, iyac098. [Google Scholar] [CrossRef]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Sehgal, P.; Chaturvedi, P. Chromatin and cancer: Implications of disrupted chromatin organization in tumorigenesis and its diversification. Cancers 2023, 15, 466. [Google Scholar] [CrossRef]
- Bompadre, O.; Andrey, G. Chromatin topology in development and disease. Curr. Opin. Genet. Dev. 2019, 55, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Stephens, A.D. Chromatin rigidity provides mechanical and genome protection. Mutat. Res. 2020, 821, 111712. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Hayn, A.; Mierke, C.T. Effect of Nuclear Stiffness on Cell Mechanics and Migration of Human Breast Cancer Cells. Front. Cell Dev. Biol. 2020, 8, 393. [Google Scholar] [CrossRef]
- Meghani, K.; Folgosa Cooley, L.; Piunti, A.; Meeks, J.J. Role of chromatin modifying complexes and therapeutic opportunities in bladder cancer. Bladder Cancer 2022, 8, 101–112. [Google Scholar] [CrossRef]
- Yun, J.W.; Song, S.H.; Kim, H.P.; Han, S.W.; Yi, E.C.; Kim, T.Y. Dynamic cohesin-mediated chromatin architecture controls epithelial–mesenchymal plasticity in cancer. EMBO Rep. 2016, 17, 1343–1359. [Google Scholar] [CrossRef]
- Hao, Y.; Baker, D.; ten Dijke, P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. Int. J. Mol. Sci. 2019, 20, 2767. [Google Scholar] [CrossRef]
- Dramiński, M.; Rada-Iglesias, A.; Enroth, S.; Wadelius, C.; Koronacki, J.; Komorowski, J. Monte Carlo feature selection for supervised classification. Bioinformatics 2007, 24, 110–117. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Weisenberger, D.J.; Van Den Berg, D.; Pan, F.; Berman, B.P.; Laird, P.W. Comprehensive DNA Methylation Analysis on the Illumina ® Infinium ® Assay Platform. 2008. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/appnotes/appnote_dna_methylation_analysis_infinium.pdf (accessed on 21 September 2024).
- Pian, C.; Zhang, G.; Gao, L.; Fan, X.; Li, F. miR+Pathway: The integration and visualization of miRNA and KEGG pathways. Brief. Bioinform. 2019, 21, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wang, X. miRDB: An online database for prediction of functional microRNA targets. Nucleic Acids Res. 2020, 48, D127–D131. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [Google Scholar] [CrossRef]
- Pongubala, J.M.R.; Murre, C. Spatial organization of chromatin: Transcriptional control of adaptive immune cell development. Front. Immunol. 2021, 12, 633825. [Google Scholar] [CrossRef]
- Kulakovskiy, I.V.; Vorontsov, I.E.; Yevshin, I.S.; Sharipov, R.N.; Fedorova, A.D.; Rumynskiy, E.I.; Medvedeva, Y.A.; Magana-Mora, A.; Bajic, V.B.; Papatsenko, D.A.; et al. HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2017, 46, D252–D259. [Google Scholar] [CrossRef]
- Stojnic, R.; Diez, D. PWMEnrich: PWM Enrichment Analysis. Bioconductor 2024. Available online: https://bioconductor.org/packages/release/bioc/html/PWMEnrich.html (accessed on 9 August 2024).
- Grant, C.E.; Bailey, T.L.; Noble, W.S. FIMO: Scanning for occurrences of a given motif. Bioinformatics 2011, 27, 1017–1018. [Google Scholar] [CrossRef]
- Mahony, S.; Benos, P.V. STAMP: A web tool for exploring DNA-binding motif similarities. Nucleic Acids Res. 2007, 35, W253–W258. [Google Scholar] [CrossRef]
- Rodchenkov, I.; Babur, O.; Luna, A.; Aksoy, B.A.; Wong, J.V.; Fong, D.; Franz, M.; Siper, M.C.; Cheung, M.; Wrana, M.; et al. Pathway Commons 2019 Update: Integration; analysis and exploration of pathway data. Nucleic Acids Res. 2019, 48, D489–D497. [Google Scholar] [CrossRef]
- Szałaj, P.; Tang, Z.; Michalski, P.; Pietal, M.J.; Luo, O.J.; Sadowski, M.; Li, X.; Radew, K.; Ruan, Y.; Plewczynski, D. An integrated 3-Dimensional Genome Modeling Engine for data-driven simulation of spatial genome organization. Genome Res. 2016, 26, 1697–1709. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Joined-Set | Individual-Set | Intersection | Sum | RF wAcc | SVM wAcc | |
|---|---|---|---|---|---|---|
| DNA methylation | 1504 | 1987 | 1485 | 2006 | 0.9068 | 0.9398 |
| mRNA | 432 | 588 | 430 | 590 | 0.9347 | 0.9347 |
| miRNA | 6 | 105 | 6 | 105 | 0.9848 | 0.9370 |
| Cluster ID | Cluster Size | Top Words Associated with Genes in Cluster | Keywords Interpretation | Mean Log Fold Change for Verification Set | Direction of Change in Expression |
|---|---|---|---|---|---|
| 1 | 393 | regulation, process, metabolism, negative, negative_regulation, response, positive, positive regulation, gene, metabolic | regulation and metabolic processes | 1.5894 | over-expressed: 78 down-expressed: 313 |
| 2 | 38 | transport, ion, transmembrane, transmembrane_transport, calcium, abc, muscle, cardiac, ion_transmembrane, contraction | ion trans- membrane transport | 2.5636 | over-expressed: 4 down-expressed: 34 |
| 3 | 18 | receptor, g, coupled, g_protein, protein_coupled, gpcr, coupled_receptor, receptors, protein, ligand | receptor proteins | 2.8793 | over-expressed: 0 down-expressed: 18 |
| 4 | 24 | transcription, polymerase, rna_polymerase, rna, polymerase_ii, ii, regulation_transcription, transcription_rna, differentiation, development | transcription process | 1.5496 | over-expressed: 4 down-expressed: 20 |
| 5 | 31 | mitotic, cell_cycle, cycle, g, cell, apc, transition, apc_c, c, g_transition | cell cycle regulation | −2.5362 | over-expressed: 28 down-expressed: 3 |
| 6 | 16 | golgi, transport, er, golgi_er, retrograde, vesicle, vesicle_mediated, mediated_transport, mediated, traffic | golgi apparatus related | −0.9197 | over-expressed: 11 down-expressed: 5 |
| 7 | 4 | biological_process, biological, process | biological processes | 2.7369 | over-expressed: 0 down-expressed: 4 |
| miRNA Gene | Freq | Sum RI | Mean RI | MCFS-ID Rank | DNA Methylation | Freq | Sum RI | Mean RI | MCFS-ID Rank | |
|---|---|---|---|---|---|---|---|---|---|---|
| hsa_mir_139 | 73 | 65.628 | 0.899 | 1 | cg07267550 | 7 | 3.044 | 0.435 | 1234 | |
| hsa_mir_141 | 73 | 38.519 | 0.528 | 10 | cg00914963 | 7 | 3.036 | 0.434 | 2048 | |
| hsa_mir_10b | 73 | 36.690 | 0.503 | 2 | cg19533977 | 7 | 3.024 | 0.432 | 25 | |
| hsa_mir_183 | 73 | 33.020 | 0.452 | 4 | cg08113562 | 7 | 2.714 | 0.388 | 12,061 | |
| hsa_mir_140 | 73 | 32.825 | 0.450 | 11 | cg17901038 | 7 | 2.620 | 0.374 | 186 | |
| hsa_mir_200a | 73 | 31.182 | 0.427 | 15 | cg18253799 | 7 | 2.584 | 0.369 | 2217 | |
| hsa_mir_96 | 73 | 28.417 | 0.389 | 8 | cg20417953 | 7 | 2.504 | 0.358 | 6490 | |
| hsa_mir_429 | 73 | 25.452 | 0.349 | 20 | cg20524128 | 7 | 2.488 | 0.355 | 2066 | |
| hsa_mir_204 | 73 | 23.546 | 0.323 | 12 | cg10520594 | 7 | 2.393 | 0.342 | 4407 | |
| hsa_mir_99a | 73 | 23.041 | 0.316 | 6 | cg20701457 | 7 | 2.159 | 0.308 | 1487 | |
| hsa_mir_592 | 73 | 22.352 | 0.306 | 16 | cg16009970 | 7 | 2.090 | 0.299 | 13,163 | |
| hsa_mir_378 | 73 | 22.320 | 0.306 | 38 | cg06976025 | 7 | 2.012 | 0.287 | 3878 | |
| hsa_mir_145 | 73 | 22.218 | 0.304 | 5 | cg15601264 | 7 | 1.993 | 0.285 | 188 | |
| hsa_mir_21 | 73 | 21.700 | 0.297 | 3 | cg22608492 | 7 | 1.932 | 0.276 | 151 | |
| hsa_let_7c | 73 | 21.686 | 0.297 | 13 | cg11441693 | 7 | 1.913 | 0.273 | 12,369 |
| Data Type | Unit of Measurement | Number of Total Samples | Number of Normal Samples | Number of Features |
|---|---|---|---|---|
| mRNA expression | reads per kilobase million | 867 | 99 | 20,524 |
| DNA methylation | beta-value | 870 | 97 | 396,065 |
| miRNA expressions | reads per million miRNA mapped | 832 | 86 | 897 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jardanowska-Kotuniak, M.; Dramiński, M.; Wlasnowolski, M.; Łapiński, M.; Sengupta, K.; Agarwal, A.; Filip, A.; Ghosh, N.; Pancaldi, V.; Grynberg, M.; et al. Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development. Int. J. Mol. Sci. 2025, 26, 6558. https://doi.org/10.3390/ijms26146558
Jardanowska-Kotuniak M, Dramiński M, Wlasnowolski M, Łapiński M, Sengupta K, Agarwal A, Filip A, Ghosh N, Pancaldi V, Grynberg M, et al. Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development. International Journal of Molecular Sciences. 2025; 26(14):6558. https://doi.org/10.3390/ijms26146558
Chicago/Turabian StyleJardanowska-Kotuniak, Marta, Michał Dramiński, Michal Wlasnowolski, Marcin Łapiński, Kaustav Sengupta, Abhishek Agarwal, Adam Filip, Nimisha Ghosh, Vera Pancaldi, Marcin Grynberg, and et al. 2025. "Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development" International Journal of Molecular Sciences 26, no. 14: 6558. https://doi.org/10.3390/ijms26146558
APA StyleJardanowska-Kotuniak, M., Dramiński, M., Wlasnowolski, M., Łapiński, M., Sengupta, K., Agarwal, A., Filip, A., Ghosh, N., Pancaldi, V., Grynberg, M., Saha, I., Plewczynski, D., & Dąbrowski, M. J. (2025). Unveiling Epigenetic Regulatory Elements Associated with Breast Cancer Development. International Journal of Molecular Sciences, 26(14), 6558. https://doi.org/10.3390/ijms26146558