

Dual Inhibition of SRC Family Kinases and Sorafenib Enhances Anti-Tumor Activity in Hepatocellular Carcinoma Cells

 , and
, and
Abstract
1. Introduction
2. Results
2.1. Identification of SFKs as Targets for Therapy
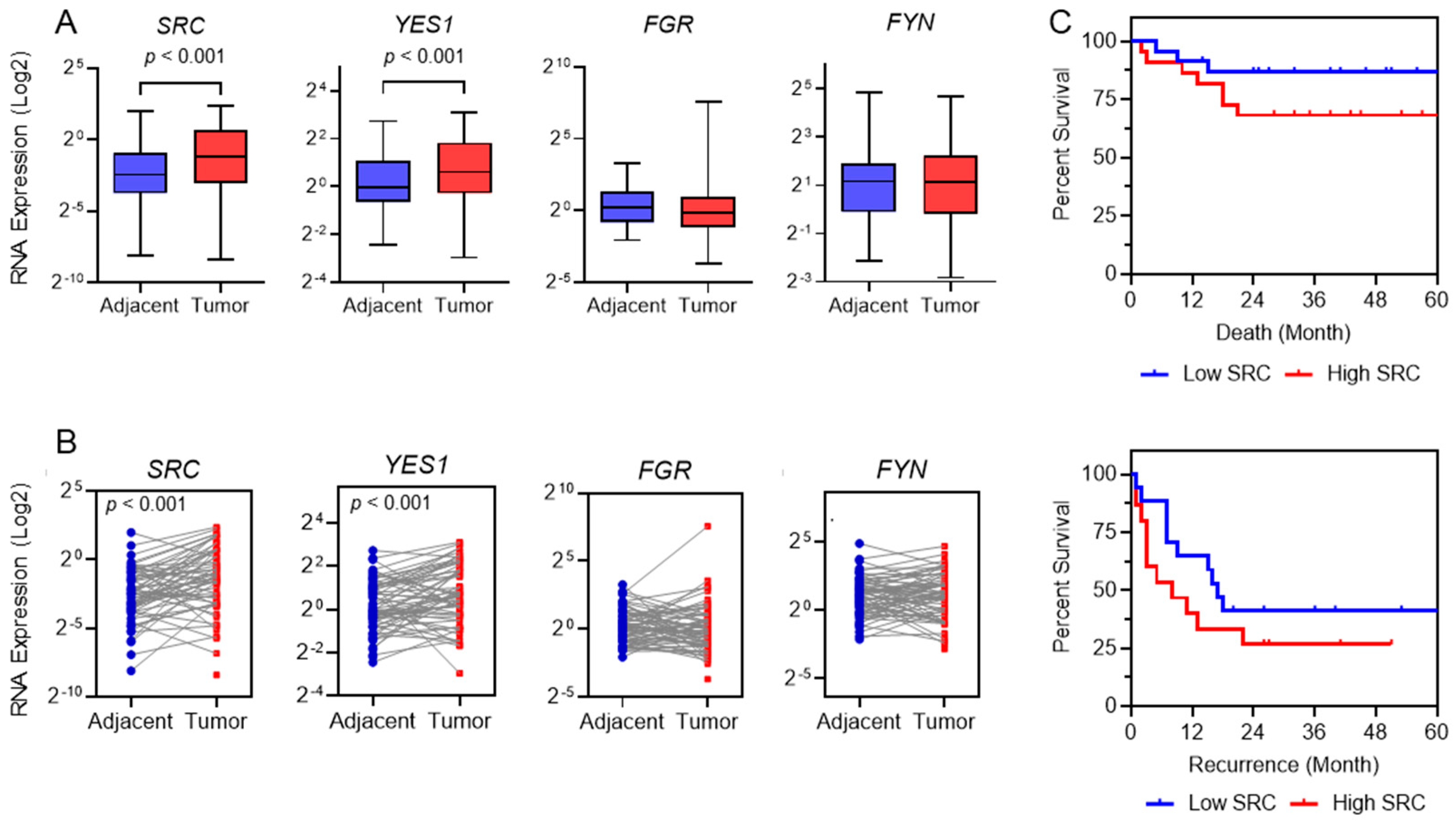
2.2. SRC as Prognostic Tool in HCC Patients
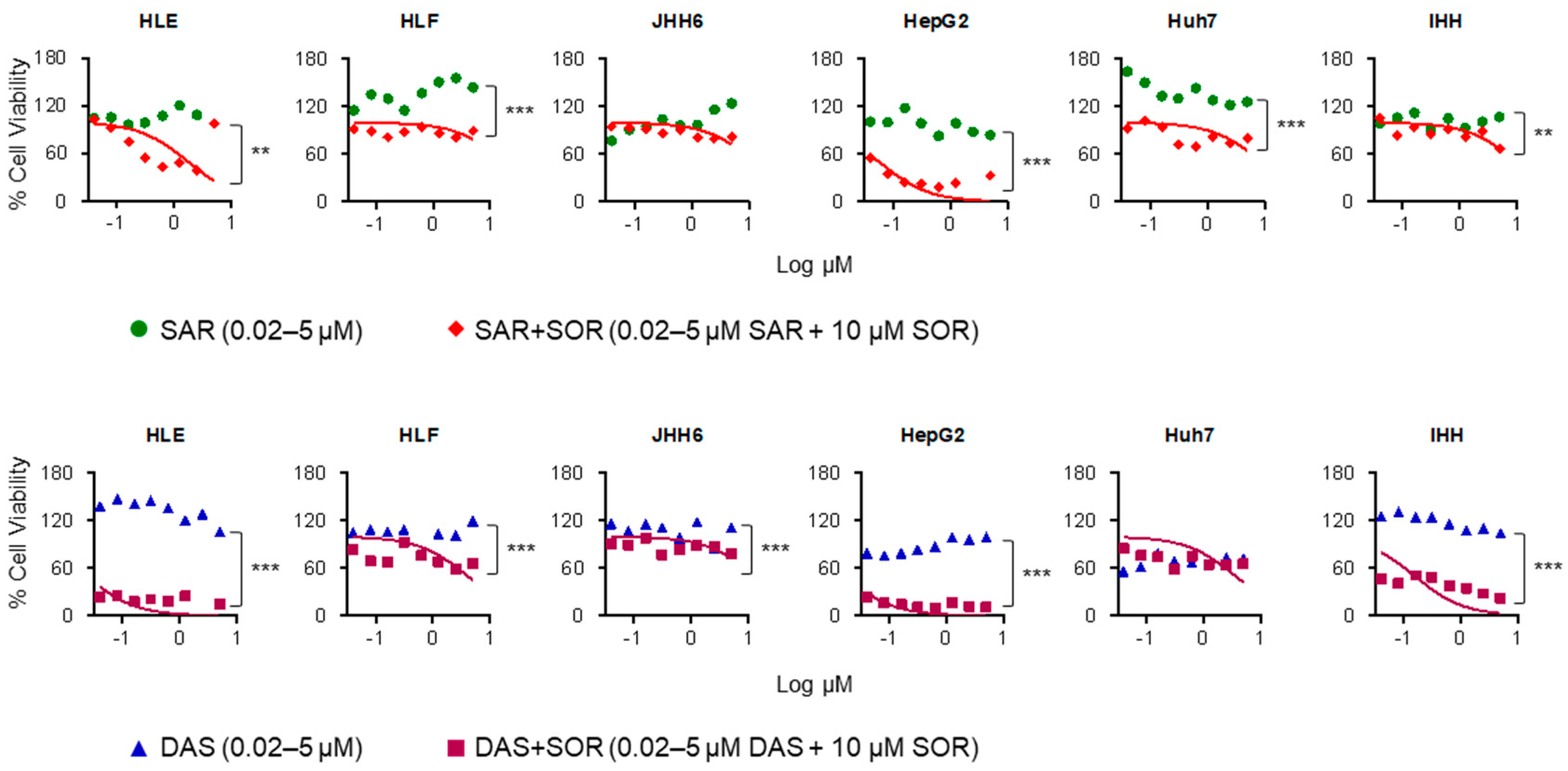
2.3. Combination Therapy Increases Sorafenib Cytotoxicity in HCC Cell Lines
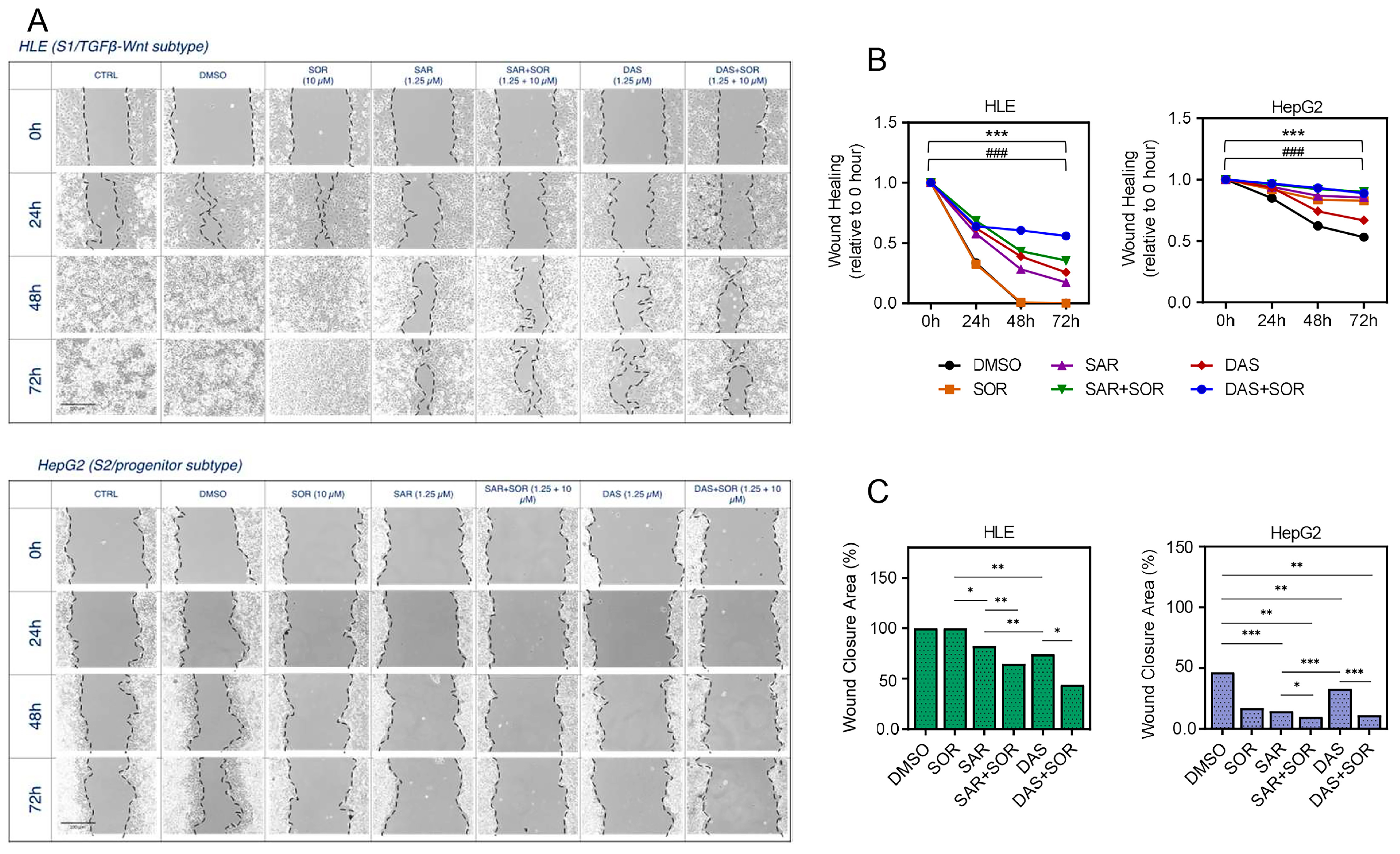
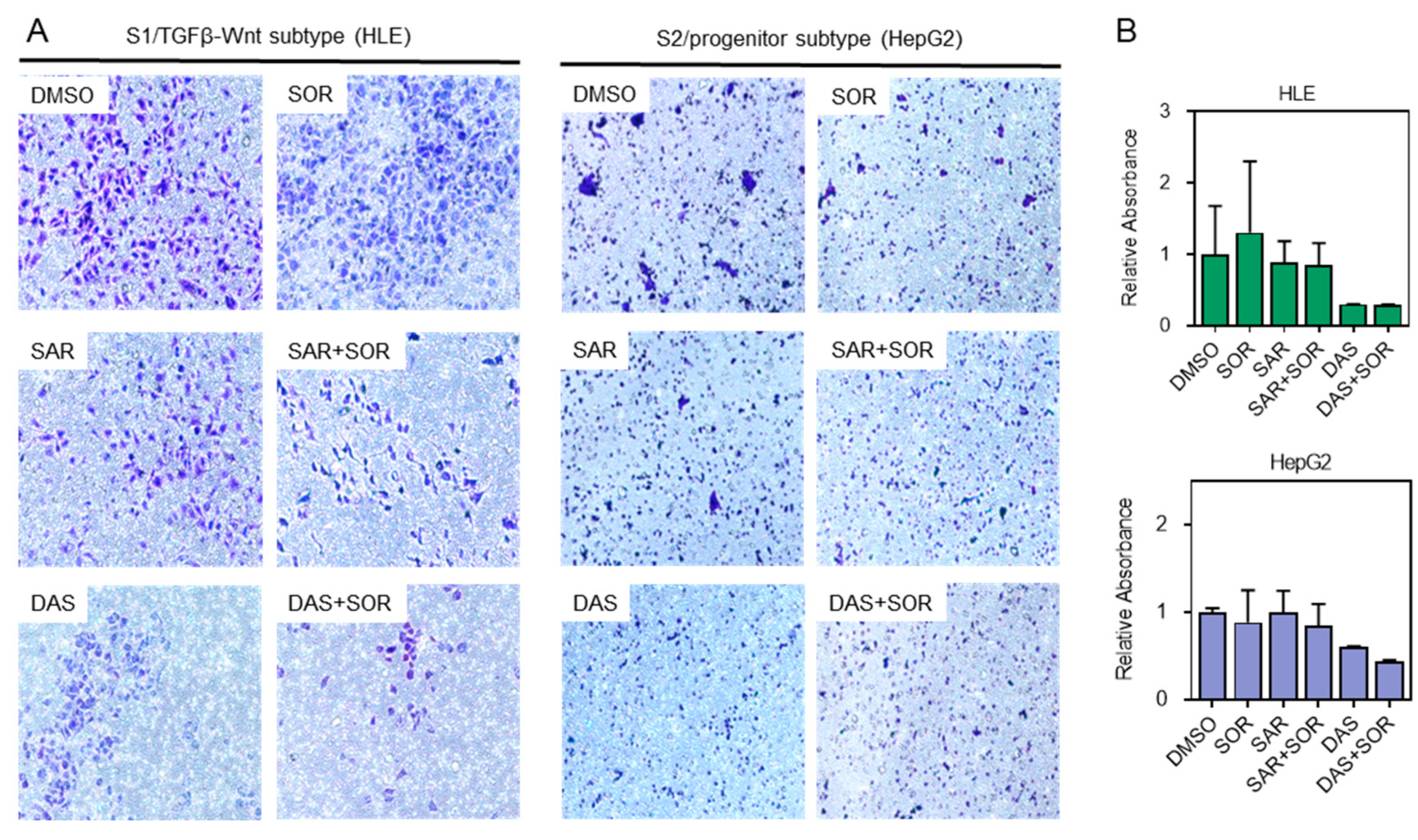
2.4. Combination Therapy Reduces Cell Aggressiveness and Migration Capacity
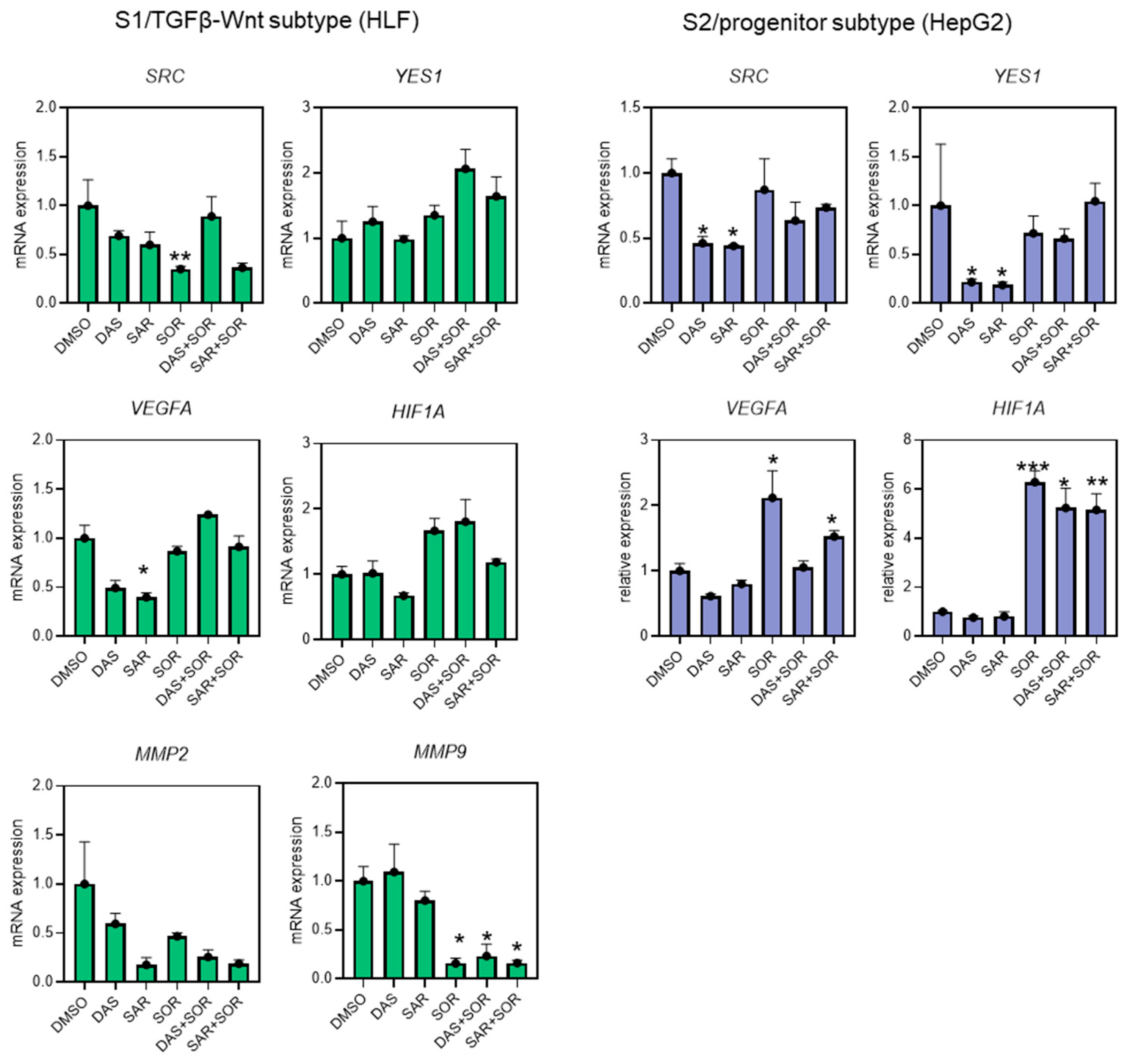
2.5. Combination Therapy Dysregulated Gene Expression Related to Cell Migration and Angiogenesis
3. Discussion
4. Materials and Methods
4.1. Bioinformatics Analysis
4.2. Human HCC Specimens
4.3. SFK Genes Expression and Prognostic Significance
4.4. Cell Lines
4.5. Cytotoxicity of Combination Therapy
4.6. Total RNA Extraction from Solid Tissue Samples and Cell Lines
4.7. RT-qPCR
4.8. Wound Healing Assay
4.9. Transwell Migration Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| HCC | Hepatocellular Carcinoma |
| SFKs | Src Family Kinases |
| NRTKs | Non-Receptor Tyrosine Kinases |
| RT-qPCR | Reverse Transcription-Quantitative Real-Time Polymerase Chain Reaction |
| RT | Reverse Transcription |
| cDNA | Complementary DNA |
| PCR | Polymerase Chain Reaction |
| TCGA | The Cancer Genome Atlas |
| GTEx | Genotype-Tissue Expression |
| GEPIA | Gene Expression Profiling Interactive Analysis |
| LIHC | Liver Hepatocellular Carcinoma (TCGA dataset) |
| EGFR | Epidermal Growth Factor Receptor |
| ErbB2 | Erb-B2 Receptor Tyrosine Kinase 2 (also known as HER2) |
| BCR-Abl | Breakpoint Cluster Region-Abelson Murine Leukemia Viral Oncogene Homolog 1 |
| PI3K/Akt | Phosphoinositide 3-Kinase/Protein Kinase B Signaling Pathway |
| RTKs | Receptor Tyrosine Kinases |
| FDA | Food and Drug Administration |
| NSCLC | Non-Small Cell Lung Cancer |
| MMP2 | Matrix Metallopeptidase 2 |
References
- Li, Q.; Ding, C.; Cao, M.; Yang, F.; Yan, X.; He, S.; Cao, M.; Zhang, S.; Teng, Y.; Tan, N.; et al. Global Epidemiology of Liver Cancer 2022: An Emphasis on Geographic Disparities. Chin. Med. J. 2024, 137, 2334–2342. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef] [PubMed]
- Sukowati, C.H.C. Heterogeneity of Hepatic Cancer Stem Cells. Adv. Exp. Med. Biol. 2019, 1139, 59–81. [Google Scholar] [CrossRef]
- Lee, J.-S.; Thorgeirsson, S.S. Genetic Profiling of Human Hepatocellular Carcinoma. Semin. Liver Dis. 2005, 25, 125–132. [Google Scholar] [CrossRef]
- Boyault, S.; Rickman, D.S.; de Reyniès, A.; Balabaud, C.; Rebouissou, S.; Jeannot, E.; Hérault, A.; Saric, J.; Belghiti, J.; Franco, D.; et al. Transcriptome Classification of HCC Is Related to Gene Alterations and to New Therapeutic Targets. Hepatology 2007, 45, 42–52. [Google Scholar] [CrossRef]
- Caruso, S.; Calatayud, A.-L.; Pilet, J.; La Bella, T.; Rekik, S.; Imbeaud, S.; Letouzé, E.; Meunier, L.; Bayard, Q.; Rohr-Udilova, N.; et al. Analysis of Liver Cancer Cell Lines Identifies Agents with Likely Efficacy Against Hepatocellular Carcinoma and Markers of Response. Gastroenterology 2019, 157, 760–776. [Google Scholar] [CrossRef]
- Llovet, J.M.; Kelley, R.K.; Villanueva, A.; Singal, A.G.; Pikarsky, E.; Roayaie, S.; Lencioni, R.; Koike, K.; Zucman-Rossi, J.; Finn, R.S. Hepatocellular Carcinoma. Nat. Rev. Dis. Prim. 2021, 7, 6. [Google Scholar] [CrossRef]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Sun, M.; Zhang, Z.; Chen, C.; Zhong, J.; Long, Z.; Shen, L.; Huang, H.; Lu, J. Exploring the Potential Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma Cell Lines Based on RNA Sequencing. Cancer Cell Int. 2025, 25, 91. [Google Scholar] [CrossRef]
- Cabral, L.K.D.; Tiribelli, C.; Sukowati, C.H.C. Sorafenib Resistance in Hepatocellular Carcinoma: The Relevance of Genetic Heterogeneity. Cancers 2020, 12, 1576. [Google Scholar] [CrossRef]
- Pelaz, S.G.; Tabernero, A. Src: Coordinating Metabolism in Cancer. Oncogene 2022, 41, 4917–4928. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Fang, J.; Ding, X.; Chen, Q. Role and Inhibition of Src Signaling in the Progression of Liver Cancer. Open Life Sci. 2016, 11, 513–518. [Google Scholar] [CrossRef]
- Cabral, L.K.D.; Giraudi, P.J.; Giannelli, G.; Dituri, F.; Negro, R.; Tiribelli, C.; Sukowati, C.H.C. Network Analysis for the Discovery of Common Oncogenic Biomarkers in Liver Cancer Experimental Models. Biomedicines 2023, 11, 342. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING Database in 2023: Protein–Protein Association Networks and Functional Enrichment Analyses for Any Sequenced Genome of Interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Raudvere, U.; Kolberg, L.; Kuzmin, I.; Arak, T.; Adler, P.; Peterson, H.; Vilo, J. G: Profiler: A Web Server for Functional Enrichment Analysis and Conversions of Gene Lists (2019 Update). Nucleic Acids Res. 2019, 47, W191–W198. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- GTEx Consortium Human Genomics. The Genotype-Tissue Expression (GTEx) Pilot Analysis: Multitissue Gene Regulation in Humans. Science 2015, 348, 648–660. [Google Scholar] [CrossRef]
- Tang, Z.; Li, C.; Kang, B.; Gao, G.; Li, C.; Zhang, Z. GEPIA: A Web Server for Cancer and Normal Gene Expression Profiling and Interactive Analyses. Nucleic Acids Res. 2017, 45, W98–W102. [Google Scholar] [CrossRef]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef]
- Blagosklonny, M.V. Analysis of FDA Approved Anticancer Drugs Reveals the Future of Cancer Therapy. Cell Cycle 2004, 3, 1033–1040. [Google Scholar] [CrossRef]
- Yap, T.A.; Omlin, A.; de Bono, J.S. Development of Therapeutic Combinations Targeting Major Cancer Signaling Pathways. J. Clin. Oncol. 2013, 31, 1592–1605. [Google Scholar] [CrossRef] [PubMed]
- Simatou, A.; Simatos, G.; Goulielmaki, M.; Spandidos, D.A.; Baliou, S.; Zoumpourlis, V. Historical Retrospective of the SRC Oncogene and New Perspectives (Review). Mol. Clin. Oncol. 2020, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Feng, H. Activation of C-Yes in Hepatocellular Carcinoma: A Preliminary Study. World J. Gastroenterol. 2006, 12, 5743. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R. Src Protein-Tyrosine Kinase Structure, Mechanism, and Small Molecule Inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef]
- Thomas, S.M.; Brugge, J.S. Cellular Functions Regulated by Src Family Kinases. Annu. Rev. Cell Dev. Biol. 1997, 13, 513–609. [Google Scholar] [CrossRef]
- Takahashi, M.; Araki, T.; Yashima, H.; Nagamine, A.; Nagano, D.; Yamamoto, K. Increased c-SRC Expression Is Involved in Acquired Resistance to Lenvatinib in Hepatocellular Carcinoma. Oncol. Lett. 2023, 26, 529. [Google Scholar] [CrossRef]
- Yao, D.; Deng, Y.; Zhang, S.; Liang, L.; Zhang, L.; Weng, S.; Chen, S. Comprehensive Analysis of Prognostic Value and Immune Infiltration of Src Family Kinases in Hepatocellular Carcinoma. Front. Biosci. (Landmark Ed.) 2023, 28, 90. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, X.; Liu, L.; Yang, X.; Qian, Q.; Du, B. C-Src Promotes the Growth and Tumorigenesis of Hepatocellular Carcinoma via the Hippo Signaling Pathway. Life Sci. 2021, 264, 118711. [Google Scholar] [CrossRef]
- Voisin, L.; Lapouge, M.; Saba-El-Leil, M.K.; Gombos, M.; Javary, J.; Trinh, V.Q.; Meloche, S. Syngeneic Mouse Model of YES-Driven Metastatic and Proliferative Hepatocellular Carcinoma. Dis. Model. Mech. 2024, 17, dmm050553. [Google Scholar] [CrossRef]
- Steinberg, M. Dasatinib: A Tyrosine Kinase Inhibitor for the Treatment of Chronic Myelogenous Leukemia and Philadelphia Chromosome-Positive Acute Lymphoblastic Leukemia. Clin. Ther. 2007, 29, 2289–2308. [Google Scholar] [CrossRef]
- Tokarski, J.S.; Newitt, J.A.; Chang, C.Y.J.; Cheng, J.D.; Wittekind, M.; Kiefer, S.E.; Kish, K.; Lee, F.Y.F.; Borzillerri, R.; Lombardo, L.J.; et al. The Structure of Dasatinib (BMS-354825) Bound to Activated ABL Kinase Domain Elucidates Its Inhibitory Activity against Imatinib-Resistant ABL Mutants. Cancer Res. 2006, 66, 5790–5797. [Google Scholar] [CrossRef] [PubMed]
- Puls, L.N.; Eadens, M.; Messersmith, W. Current Status of SRC Inhibitors in Solid Tumor Malignancies. Oncol. 2011, 16, 566–578. [Google Scholar] [CrossRef] [PubMed]
- Hennequin, L.F.; Allen, J.; Breed, J.; Curwen, J.; Fennell, M.; Green, T.P.; Lambert-van der Brempt, C.; Morgentin, R.; Norman, R.A.; Olivier, A.; et al. N-(5-Chloro-1,3-Benzodioxol-4-Yl)-7-[2-(4-Methylpiperazin-1-Yl)Ethoxy]-5-(Tetrahydro-2H-Pyran-4-Yloxy)Quinazolin-4-Amine, a Novel, Highly Selective, Orally Available, Dual-Specific c-Src/Abl Kinase Inhibitor. J. Med. Chem. 2006, 49, 6465–6488. [Google Scholar] [CrossRef] [PubMed]
- Ramos, R.; Vale, N. Dual Drug Repurposing: The Example of Saracatinib. Int. J. Mol. Sci. 2024, 25, 4565. [Google Scholar] [CrossRef]
- Lara, P.N.; Longmate, J.; Evans, C.P.; Quinn, D.I.; Twardowski, P.; Chatta, G.; Posadas, E.; Stadler, W.; Gandara, D.R. A Phase II Trial of the Src-Kinase Inhibitor AZD0530 in Patients with Advanced Castration-Resistant Prostate Cancer: A California Cancer Consortium Study. Anticancer. Drugs 2009, 20, 179–184. [Google Scholar] [CrossRef]
- Martellucci, S.; Clementi, L.; Sabetta, S.; Mattei, V.; Botta, L.; Angelucci, A. Src Family Kinases as Therapeutic Targets in Advanced Solid Tumors: What We Have Learned so Far. Cancers 2020, 12, 1448. [Google Scholar] [CrossRef]
- Lombardo, L.J.; Lee, F.Y.; Chen, P.; Norris, D.; Barrish, J.C.; Behnia, K.; Castaneda, S.; Cornelius, L.A.M.; Das, J.; Doweyko, A.M.; et al. Discovery of N-(2-Chloro-6-Methyl-Phenyl)-2-(6-(4-(2-Hydroxyethyl)-Piperazin-1-Yl)-2-Methylpyrimidin-4-Ylamino)Thiazole-5-Carboxamide (BMS-354825), a Dual Src/Abl Kinase Inhibitor with Potent Antitumor Activity in Preclinical Assays. J. Med. Chem. 2004, 47, 6658–6661. [Google Scholar] [CrossRef]
- Laurie, S.A.; Goss, G.D.; Shepherd, F.A.; Reaume, M.N.; Nicholas, G.; Philip, L.; Wang, L.; Schwock, J.; Hirsh, V.; Oza, A.; et al. A Phase II Trial of Saracatinib, an Inhibitor of Src Kinases, in Previously-Treated Advanced Non–Small-Cell Lung Cancer: The Princess Margaret Hospital Phase II Consortium. Clin. Lung Cancer 2014, 15, 52–57. [Google Scholar] [CrossRef]
- Chou, T.-C. Theoretical Basis, Experimental Design, and Computerized Simulation of Synergism and Antagonism in Drug Combination Studies. Pharmacol. Rev. 2006, 58, 621–681. [Google Scholar] [CrossRef]
- Dor, I.; Namba, M.; Sato, J. Establishment and Some Biological Characteristics of Human Hepatoma Cell Lines. Gan 1975, 66, 385–392. [Google Scholar]
- Arii, S.; Mise, M.; Harada, T.; Furutani, M.; Ishigami, S.; Niwano, M.; Mizumoto, M.; Fukumoto, M.; Imamura, M. Overexpression of Matrix Metalloproteinase 9 Gene in Hepatocellular Carcinoma with Invasive Potential. Hepatology 1996, 24, 316. [Google Scholar] [CrossRef] [PubMed]
- Pinto, E.; Pelizzaro, F.; Cardin, R.; Battistel, M.; Palano, G.; Bertellini, F.; Kitenge, M.P.; Peserico, G.; Farinati, F.; Russo, F.P. HIF-1α and VEGF as Prognostic Biomarkers in Hepatocellular Carcinoma Patients Treated with Transarterial Chemoembolization. Dig. Liver Dis. 2024, 56, 872–879. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Sinicrope, F.A. Sorafenib Inhibits STAT3 Activation to Enhance TRAIL-Mediated Apoptosis in Human Pancreatic Cancer Cells. Mol. Cancer Ther. 2010, 9, 742–750. [Google Scholar] [CrossRef]
- Kim, L.C.; Song, L.; Haura, E.B. Src Kinases as Therapeutic Targets for Cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Yu, H.; Pardoll, D.; Jove, R. STATs in Cancer Inflammation and Immunity: A Leading Role for STAT3. Nat. Rev. Cancer 2009, 9, 798–809. [Google Scholar] [CrossRef]
- Abu-Ghazaleh, R.; Kabir, J.; Jia, H.; Lobo, M.; Zachary, I. Src Mediates Stimulation by Vascular Endothelial Growth Factor of the Phosphorylation of Focal Adhesion Kinase at Tyrosine 861, and Migration and Anti-Apoptosis in Endothelial Cells. Biochem. J. 2001, 360, 255–264. [Google Scholar] [CrossRef]
- Eliceiri, B.P.; Paul, R.; Schwartzberg, P.L.; Hood, J.D.; Leng, J.; Cheresh, D.A. Selective Requirement for Src Kinases during VEGF-Induced Angiogenesis and Vascular Permeability. Mol. Cell 1999, 4, 915–924. [Google Scholar] [CrossRef]
- Mitra, S.K.; Hanson, D.A.; Schlaepfer, D.D. Focal Adhesion Kinase: In Command and Control of Cell Motility. Nat. Rev. Mol. Cell Biol. 2005, 6, 56–68. [Google Scholar] [CrossRef]
- Jeong, K.-Y.; Park, M.; Sim, J.-J.; Kim, H.M. Combination Antitumor Effect of Sorafenib via Calcium-Dependent Deactivation of Focal Adhesion Kinase Targeting Colorectal Cancer Cells. Molecules 2020, 25, 5299. [Google Scholar] [CrossRef]
- Zhou, Q.; Guo, X.; Choksi, R. Activation of Focal Adhesion Kinase and Src Mediates Acquired Sorafenib Resistance in A549 Human Lung Adenocarcinoma Xenografts. J. Pharmacol. Exp. Ther. 2017, 363, 428–443. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum Information for Publication of Quantitative Real-Time PCR Experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Cancer Genome Atlas Research Network Comprehensive and Integrative Genomic Characterization of Hepatocellular Carcinoma. Cell 2017, 169, 1327–1341.e23. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer F (5′→3′) | Primer R (5′→3′) |
|---|---|---|
| 18SRNA | TAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| GAPDH | CCCATGTTCGTCATGGGTGT | TGGTCATGAGTCCTTCCACGATA |
| SRC/ASV1 | CGCTGGCCGGTGGAGTG | CCAGCTTGCGGATCTTGTAGT |
| FGR | GGCCCGGCCTGCAT | TTGATGGCCTGAGAGGAGAAG |
| YES1 | ACAGCAAGACAAGGTGCAAA | GTAAACCGACCATACAGTGCAG |
| FYN | GGACATGGCAGCACAGGTG | TTTGCTGATCGCAGATCTCTATG |
| HIF1A | CCAGCAGACTCAAATACAAGAACC | TGTATGTGGGTAGGAGATGGAGAT |
| VEGFA | GTGAGGCGGCGGTGTG | GCAAGGCAAGGCTCCAATG |
| MMP2 | CCAAGAATAGATGCTGACTG | GGAGAAGAGCCTGAAGTG |
| MMP9 | CGGCAAGTCTTCCGAGTAGT | AGACCTGGGCAGATTCCAAAC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, L.K.; Disoma, C.; Tarchi, P.; El-Khobar, K.E.; Agustiningsih, A.; Dituri, F.; Tiribelli, C.; Sukowati, C. Dual Inhibition of SRC Family Kinases and Sorafenib Enhances Anti-Tumor Activity in Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2025, 26, 6506. https://doi.org/10.3390/ijms26136506
Cabral LK, Disoma C, Tarchi P, El-Khobar KE, Agustiningsih A, Dituri F, Tiribelli C, Sukowati C. Dual Inhibition of SRC Family Kinases and Sorafenib Enhances Anti-Tumor Activity in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences. 2025; 26(13):6506. https://doi.org/10.3390/ijms26136506
Chicago/Turabian StyleCabral, Loraine Kay, Cyrollah Disoma, Paola Tarchi, Korri Elvanita El-Khobar, Agustiningsih Agustiningsih, Francesco Dituri, Claudio Tiribelli, and Caecilia Sukowati. 2025. "Dual Inhibition of SRC Family Kinases and Sorafenib Enhances Anti-Tumor Activity in Hepatocellular Carcinoma Cells" International Journal of Molecular Sciences 26, no. 13: 6506. https://doi.org/10.3390/ijms26136506
APA StyleCabral, L. K., Disoma, C., Tarchi, P., El-Khobar, K. E., Agustiningsih, A., Dituri, F., Tiribelli, C., & Sukowati, C. (2025). Dual Inhibition of SRC Family Kinases and Sorafenib Enhances Anti-Tumor Activity in Hepatocellular Carcinoma Cells. International Journal of Molecular Sciences, 26(13), 6506. https://doi.org/10.3390/ijms26136506