
Nodal Expansion, Tumor Infiltration and Exhaustion of Neoepitope-Specific Th Cells After Prophylactic Peptide Vaccination and Anti-CTLA4 Therapy in Mouse Melanoma B16


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
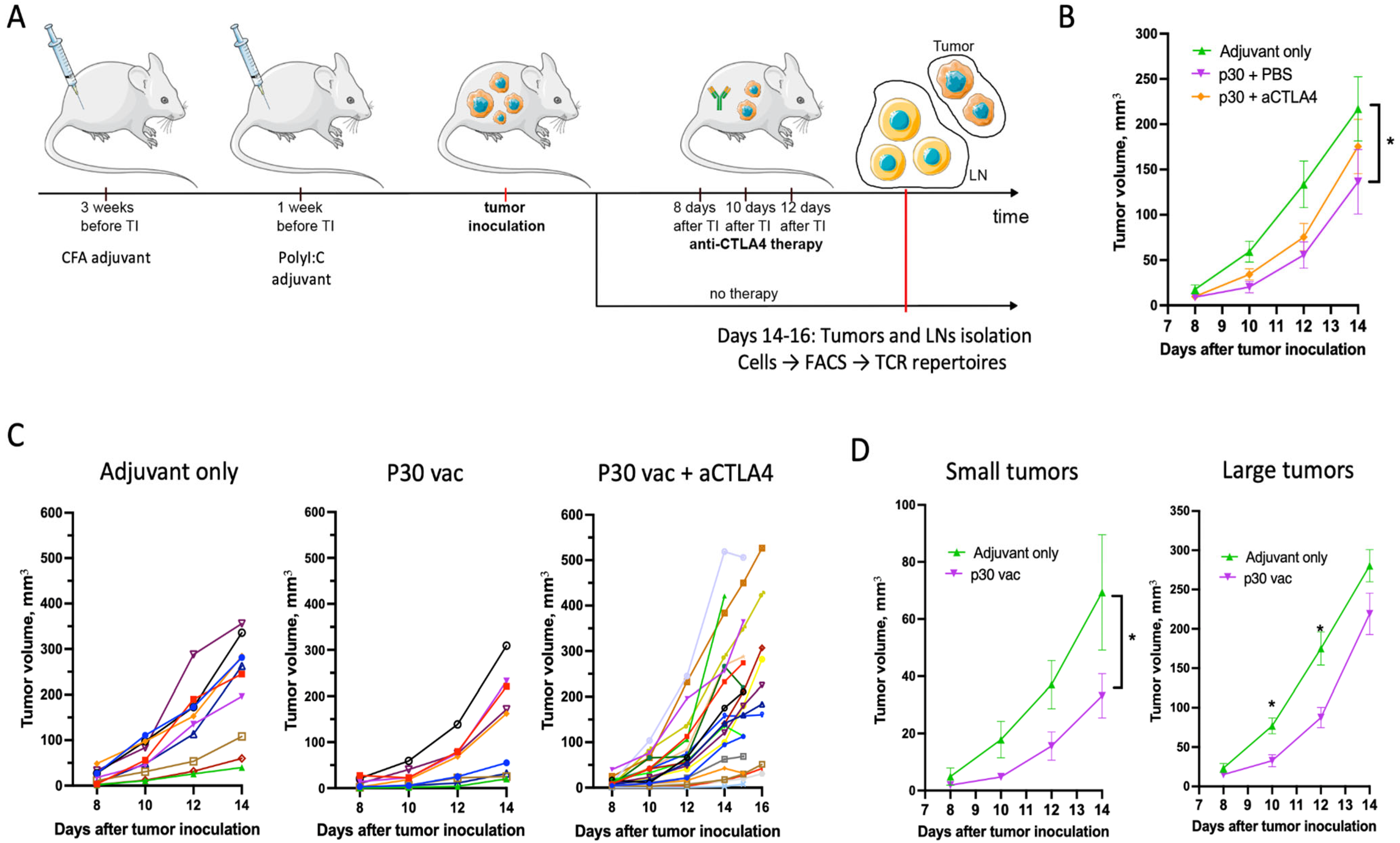
2.1. Peptide Neoantigen Vaccines Suppress Tumor Growth in Fraction of Mice
2.2. p30 Peptide and aCTLA4 Therapy Decrease Cell Sequestration and Immunosuppression of T- and B-Lymphocytes in CFA Plaques
2.3. aCTLA4 Therapy Stimulates Accumulation of Th Cells in Tumors
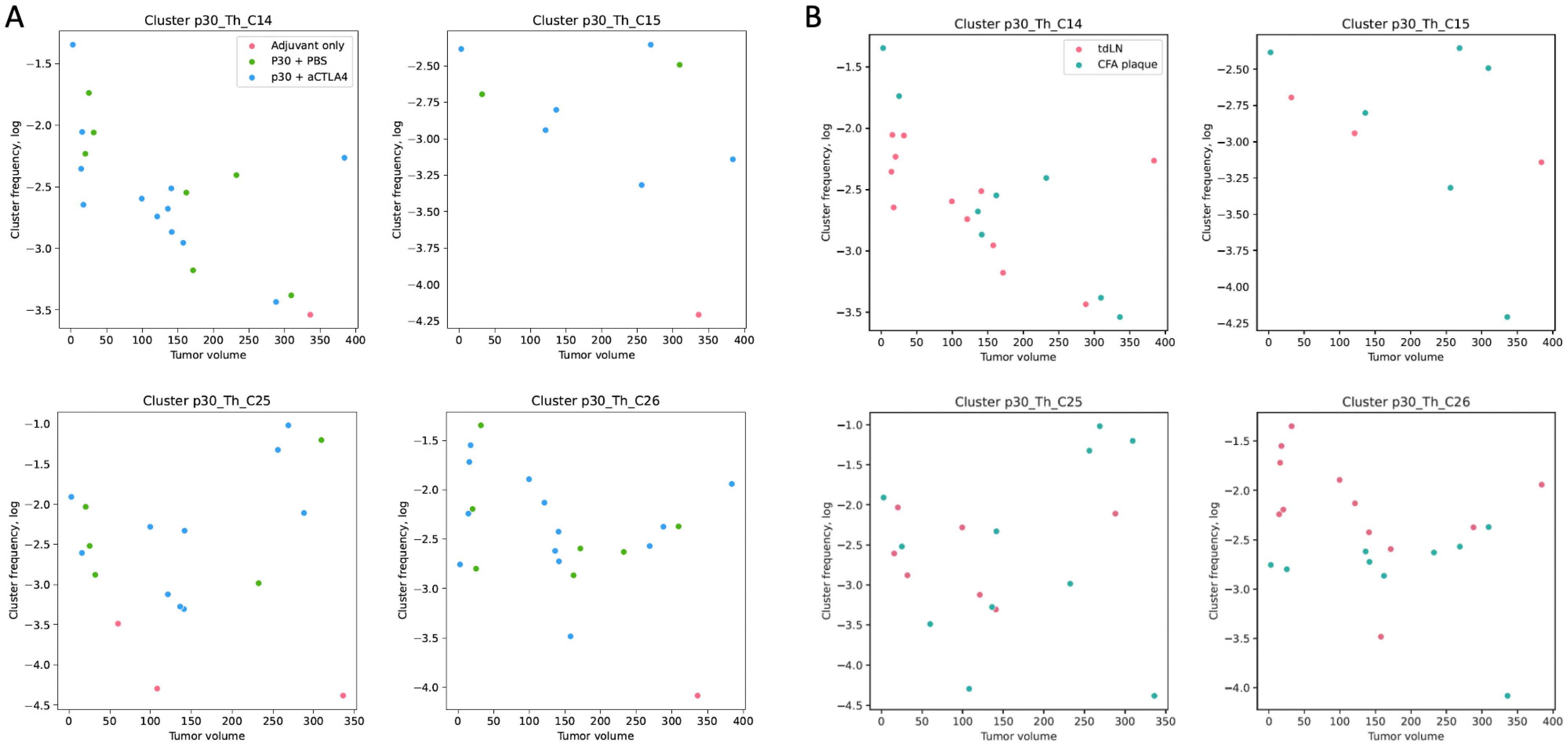
2.4. p30-Specific Th Cells Prevail in tdLNs and Have Trends Towards Negative Correlations with Tumor Size
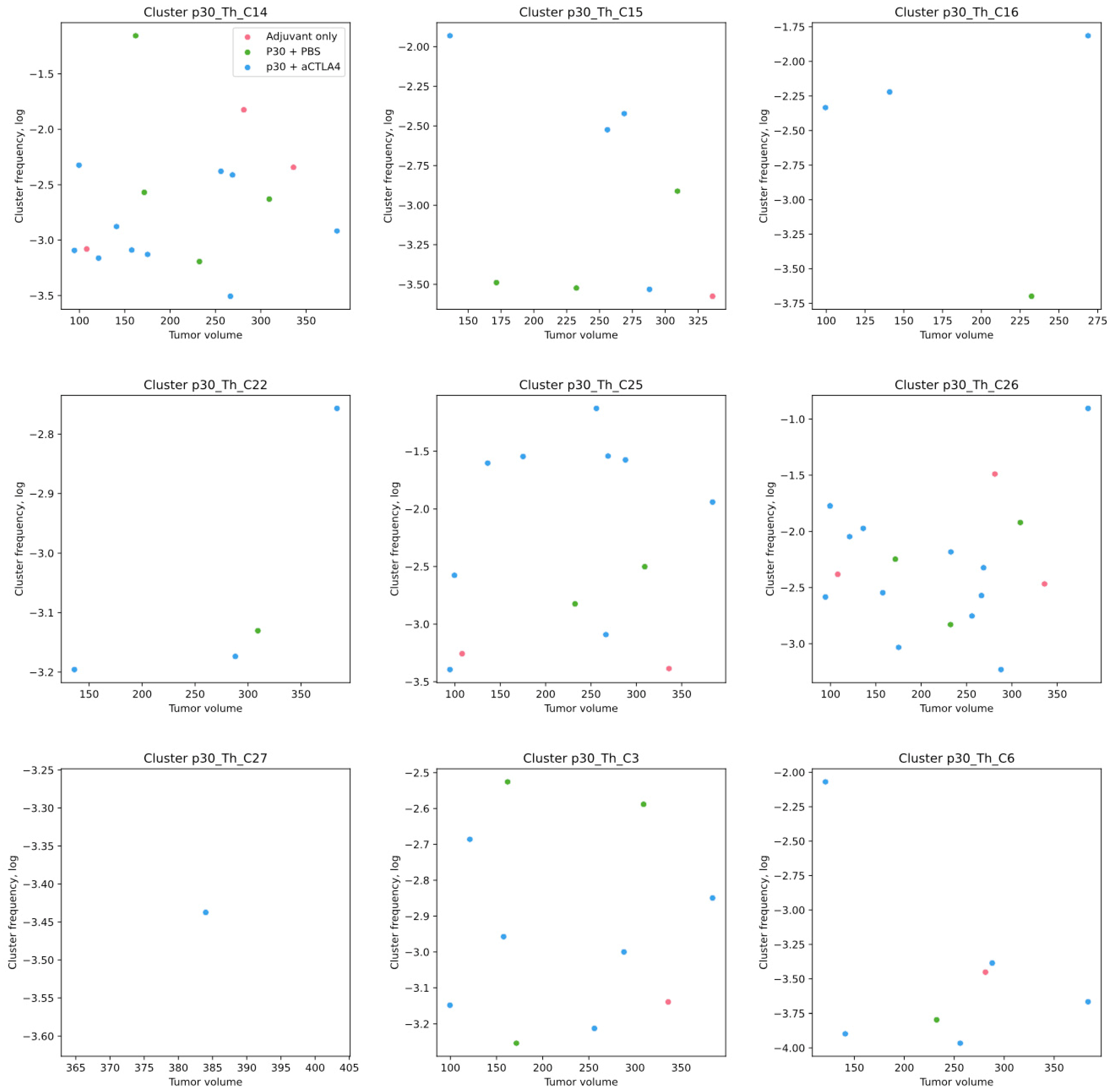
2.5. p30-Specific Th Clones Present in Tumors Without p30 Vaccination Are Not Correlated with Tumor Growth
3. Discussion
4. Materials and Methods
4.1. Mouse Vaccination
4.2. Tumor Engraftment, aCTLA4 Therapy and Tissue Sampling
4.3. Flow Cytometric Analysis and Sorting
4.4. TCR Library Preparation and Sequencing
4.5. TCR Repertoire Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Castle, J.C.; Kreiter, S.; Diekmann, J.; Löwer, M.; van de Roemer, N.; de Graaf, J.; Selmi, A.; Diken, M.; Boegel, S.; Paret, C.; et al. Exploiting the Mutanome for Tumor Vaccination. Cancer Res. 2012, 72, 1081–1091. [Google Scholar] [CrossRef]
- Kreiter, S.; Vormehr, M.; van de Roemer, N.; Diken, M.; Löwer, M.; Diekmann, J.; Boegel, S.; Schrörs, B.; Vascotto, F.; Castle, J.C.; et al. Mutant MHC Class II Epitopes Drive Therapeutic Immune Responses to Cancer. Nature 2015, 520, 692–696. [Google Scholar] [CrossRef]
- Hilf, N.; Kuttruff-Coqui, S.; Frenzel, K.; Bukur, V.; Stevanović, S.; Gouttefangeas, C.; Platten, M.; Tabatabai, G.; Dutoit, V.; van der Burg, S.H.; et al. Actively Personalized Vaccination Trial for Newly Diagnosed Glioblastoma. Nature 2018, 565, 240–245. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.-P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-Specific Therapeutic Immunity against Cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Purcell, A.W.; McCluskey, J.; Rossjohn, J. More than One Reason to Rethink the Use of Peptides in Vaccine Design. Nat. Rev. Drug Discov. 2007, 6, 404–414. [Google Scholar] [CrossRef]
- Serra, P.; Santamaria, P. Antigen-Specific Therapeutic Approaches for Autoimmunity. Nat. Biotechnol. 2019, 37, 238–251. [Google Scholar] [CrossRef]
- Sharonov, G.V.; Serebrovskaya, E.O.; Yuzhakova, D.V.; Britanova, O.V.; Chudakov, D.M. B Cells, Plasma Cells and Antibody Repertoires in the Tumour Microenvironment. Nat. Rev. Immunol. 2020, 20, 294–307. [Google Scholar] [CrossRef]
- Ben-Akiva, E.; Chapman, A.; Mao, T.; Irvine, D.J. Linking Vaccine Adjuvant Mechanisms of Action to Function. Sci. Immunol. 2025, 10, eado5937. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Wang, H.; Ding, X.-H.; Xiao, Y.-L.; Shao, Z.-M.; You, C.; Gu, Y.-J.; Jiang, Y.-Z. Bidirectional Crosstalk between Therapeutic Cancer Vaccines and the Tumor Microenvironment: Beyond Tumor Antigens. Fundam. Res. 2023, 3, 1005–1024. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards Personalized, Tumour-Specific, Therapeutic Vaccines for Cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Linnemann, C.; van Buuren, M.M.; Bies, L.; Verdegaal, E.M.E.; Schotte, R.; Calis, J.J.A.; Behjati, S.; Velds, A.; Hilkmann, H.; el Atmioui, D.; et al. High-Throughput Epitope Discovery Reveals Frequent Recognition of Neo-Antigens by CD4+ T Cells in Human Melanoma. Nat. Med. 2014, 21, 81–85. [Google Scholar] [CrossRef]
- Moravec, Z.; Zhao, Y.; Voogd, R.; Cook, D.R.; Kinrot, S.; Capra, B.; Yang, H.; Raud, B.; Ou, J.; Xuan, J.; et al. Discovery of Tumor-Reactive T Cell Receptors by Massively Parallel Library Synthesis and Screening. Nat. Biotechnol. 2024, 43, 214–222. [Google Scholar] [CrossRef]
- Genolet, R.; Bobisse, S.; Chiffelle, J.; Arnaud, M.; Petremand, R.; Queiroz, L.; Michel, A.; Reichenbach, P.; Cesbron, J.; Auger, A.; et al. TCR Sequencing and Cloning Methods for Repertoire Analysis and Isolation of Tumor-Reactive TCRs. Cell Rep. Methods 2023, 3, 100459. [Google Scholar] [CrossRef]
- Tan, C.L.; Lindner, K.; Boschert, T.; Meng, Z.; Ehrenfried, A.R.; Roia, A.D.; Haltenhof, G.; Faenza, A.; Imperatore, F.; Bunse, L.; et al. Prediction of Tumor-Reactive T Cell Receptors from ScRNA-Seq Data for Personalized T Cell Therapy. Nat. Biotechnol. 2024, 43, 134–142. [Google Scholar] [CrossRef]
- Lowery, F.J.; Krishna, S.; Yossef, R.; Parikh, N.B.; Chatani, P.D.; Zacharakis, N.; Parkhurst, M.R.; Levin, N.; Sindiri, S.; Sachs, A.; et al. Molecular Signatures of Antitumor Neoantigen-Reactive T Cells from Metastatic Human Cancers. Science 2022, 375, 877–884. [Google Scholar] [CrossRef]
- Liu, Z.; Li, J.P.; Chen, M.; Wu, M.; Shi, Y.; Li, W.; Teijaro, J.R.; Wu, P. Detecting Tumor Antigen-Specific T Cells via Interaction-Dependent Fucosyl-Biotinylation. Cell 2020, 183, 1117–1133.e19. [Google Scholar] [CrossRef]
- Li, G.; Bethune, M.T.; Wong, S.; Joglekar, A.V.; Leonard, M.T.; Wang, J.K.; Kim, J.T.; Cheng, D.; Peng, S.; Zaretsky, J.M.; et al. T Cell Antigen Discovery via Trogocytosis. Nat. Methods 2019, 16, 183–190. [Google Scholar] [CrossRef]
- Goncharov, M.M.; Bryushkova, E.A.; Sharaev, N.I.; Skatova, V.D.; Baryshnikova, A.M.; Sharonov, G.V.; Karnaukhov, V.; Vakhitova, M.T.; Samoylenko, I.V.; Demidov, L.V.; et al. Pinpointing the Tumor-Specific T Cells via TCR Clusters. eLife 2022, 11, e77274. [Google Scholar] [CrossRef]
- Shagina, I.A.; Nakonechanaya, T.O.; Izosimova, A.V.; Yuzhakova, D.V.; Skatova, V.D.; Lupyr, K.R.; Izraelson, M.; Davydov, A.N.; Shugay, M.; Britanova, O.V.; et al. Tracking Antigen-Specific T Cell Response to Cancer Immunotherapy. bioRxiv 2024, 54, 721–728. [Google Scholar] [CrossRef]
- Izosimova, A.V.; Shabalkina, A.V.; Myshkin, M.Y.; Shurganova, E.V.; Myalik, D.S.; Ryzhichenko, E.O.; Samitova, A.F.; Barsova, E.V.; Shagina, I.A.; Britanova, O.V.; et al. Local Enrichment with Convergence of Enriched T-Cell Clones Are Hallmarks of Effective Peptide Vaccination against B16 Melanoma. Vaccines 2024, 12, 345. [Google Scholar] [CrossRef]
- Speiser, D.E.; Chijioke, O.; Schaeuble, K.; Münz, C. CD4+ T Cells in Cancer. Nat. Cancer 2023, 4, 317–329. [Google Scholar] [CrossRef]
- Hirschhorn-Cymerman, D.; Budhu, S.; Kitano, S.; Liu, C.; Zhao, F.; Zhong, H.; Lesokhin, A.M.; Avogadri-Connors, F.; Yuan, J.; Li, Y.; et al. Induction of Tumoricidal Function in CD4+ T Cells Is Associated with Concomitant Memory and Terminally Differentiated Phenotype. J. Exp. Med. 2012, 209, 2113–2126. [Google Scholar] [CrossRef]
- Ott, P.A.; Hu-Lieskovan, S.; Chmielowski, B.; Govindan, R.; Naing, A.; Bhardwaj, N.; Margolin, K.; Awad, M.M.; Hellmann, M.D.; Lin, J.J.; et al. A Phase Ib Trial of Personalized Neoantigen Therapy Plus Anti-PD-1 in Patients with Advanced Melanoma, Non-Small Cell Lung Cancer, or Bladder Cancer. Cell 2020, 183, 347–362.e24. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Rasmussen, J.P.; Williams, L.M.; Dooley, J.L.; Farr, A.G.; Rudensky, A.Y. Regulatory T Cell Lineage Specification by the Forkhead Transcription Factor Foxp3. Immunity 2005, 22, 329–341. [Google Scholar] [CrossRef]
- Billiau, A.; Matthys, P. Modes of Action of Freund’s Adjuvants in Experimental Models of Autoimmune Diseases. J. Leukoc. Biol. 2001, 70, 849–860. [Google Scholar] [CrossRef]
- Meneveau, M.O.; Kumar, P.; Lynch, K.T.; Patel, S.P.; Slingluff, C.L. The vaccine-site microenvironment: Impacts of antigen, adjuvant, and same-site vaccination on antigen presentation and immune signaling. J. Immunother. Cancer 2022, 10, e003533. [Google Scholar] [CrossRef]
- Hailemichael, Y.; Dai, Z.; Jaffarzad, N.; Ye, Y.; Medina, M.A.; Huang, X.-F.; Dorta-Estremera, S.M.; Greeley, N.R.; Nitti, G.; Peng, W.; et al. Persistent Antigen at Vaccination Sites Induces Tumor-Specific CD8+ T Cell Sequestration, Dysfunction and Deletion. Nat. Med. 2013, 19, 465–472. [Google Scholar] [CrossRef]
- Riaz, B.; Islam, S.M.S.; Ryu, H.M.; Sohn, S. CD83 Regulates the Immune Responses in Inflammatory Disorders. Int. J. Mol. Sci. 2023, 24, 2831. [Google Scholar] [CrossRef]
- Zaunders, J.J.; Munier, M.L.; Seddiki, N.; Pett, S.; Ip, S.; Bailey, M.; Xu, Y.; Brown, K.; Dyer, W.B.; Kim, M.; et al. High Levels of Human Antigen-Specific CD4+ T Cells in Peripheral Blood Revealed by Stimulated Coexpression of CD25 and CD134 (OX40). J. Immunol. 2009, 183, 2827–2836. [Google Scholar] [CrossRef]
- Seddiki, N.; Cook, L.; Hsu, D.C.; Phetsouphanh, C.; Brown, K.; Xu, Y.; Kerr, S.J.; Cooper, D.A.; Munier, C.M.L.; Pett, S.; et al. Human Antigen-specific CD4+CD25+CD134+CD39+ T Cells Are Enriched for Regulatory T Cells and Comprise a Substantial Proportion of Recall Responses. Eur. J. Immunol. 2014, 44, 1644–1661. [Google Scholar] [CrossRef]
- Yadav, R.; Redmond, W.L. Current Clinical Trial Landscape of OX40 Agonists. Curr. Oncol. Rep. 2022, 24, 951–960. [Google Scholar] [CrossRef]
- Reinhardt, R.L.; Bullard, D.C.; Weaver, C.T.; Jenkins, M.K. Preferential Accumulation of Antigen-Specific Effector CD4 T Cells at an Antigen Injection Site Involves CD62E-Dependent Migration but Not Local Proliferation. J. Exp. Med. 2003, 197, 751–762. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, J.; Yue, S.; Li, Z.; Duan, X.; Lin, Y.; Yang, Y.; He, J.; Gao, L.; Pan, Z.; et al. An Oncolytic Virus Delivering Tumor-Irrelevant Bystander T Cell Epitopes Induces Anti-Tumor Immunity and Potentiates Cancer Immunotherapy. Nat. Cancer 2024, 5, 1063–1081. [Google Scholar] [CrossRef]
- Rizzuto, G.A.; Merghoub, T.; Hirschhorn-Cymerman, D.; Liu, C.; Lesokhin, A.M.; Sahawneh, D.; Zhong, H.; Panageas, K.S.; Perales, M.-A.; Altan-Bonnet, G.; et al. Self-Antigen–Specific CD8+ T Cell Precursor Frequency Determines the Quality of the Antitumor Immune Response. J. Exp. Med. 2009, 206, 849–866. [Google Scholar] [CrossRef]
- Grace, B.E.; Backlund, C.M.; Morgan, D.M.; Kang, B.H.; Singh, N.K.; Huisman, B.D.; Rappazzo, C.G.; Moynihan, K.D.; Maiorino, L.; Dobson, C.S.; et al. Identification of Highly Cross-Reactive Mimotopes for a Public T Cell Response in Murine Melanoma. Front. Immunol. 2022, 13, 886683. [Google Scholar] [CrossRef]
- Gordin, M.; Philip, H.; Zilberberg, A.; Gidoni, M.; Margalit, R.; Clouser, C.; Adams, K.; Vigneault, F.; Cohen, I.R.; Yaari, G.; et al. Breast Cancer Is Marked by Specific, Public T-Cell Receptor CDR3 Regions Shared by Mice and Humans. PLoS Comput. Biol. 2021, 17, e1008486. [Google Scholar] [CrossRef]
- Hu, Z.; Leet, D.E.; Allesøe, R.L.; Oliveira, G.; Li, S.; Luoma, A.M.; Liu, J.; Forman, J.; Huang, T.; Iorgulescu, J.B.; et al. Personal Neoantigen Vaccines Induce Persistent Memory T Cell Responses and Epitope Spreading in Patients with Melanoma. Nat. Med. 2021, 27, 515–525. [Google Scholar] [CrossRef]
- Hirschhorn, D.; Budhu, S.; Kraehenbuehl, L.; Gigoux, M.; Schröder, D.; Chow, A.; Ricca, J.M.; Gasmi, B.; Henau, O.D.; Mangarin, L.M.B.; et al. T Cell Immunotherapies Engage Neutrophils to Eliminate Tumor Antigen Escape Variants. Cell 2023, 186, 1432–1447.e17. [Google Scholar] [CrossRef]
- Kruse, B.; Buzzai, A.C.; Shridhar, N.; Braun, A.D.; Gellert, S.; Knauth, K.; Pozniak, J.; Peters, J.; Dittmann, P.; Mengoni, M.; et al. CD4+ T Cell-Induced Inflammatory Cell Death Controls Immune-Evasive Tumours. Nature 2023, 618, 1033–1040. [Google Scholar] [CrossRef]
- Gaylo-Moynihan, A.; Prizant, H.; Popović, M.; Fernandes, N.R.J.; Anderson, C.S.; Chiou, K.K.; Bell, H.; Schrock, D.C.; Schumacher, J.; Capece, T.; et al. Programming of Distinct Chemokine-Dependent and -Independent Search Strategies for Th1 and Th2 Cells Optimizes Function at Inflamed Sites. Immunity 2019, 51, 298–309.e6. [Google Scholar] [CrossRef]
- Dolina, J.S.; Lee, J.; Brightman, S.E.; McArdle, S.; Hall, S.M.; Thota, R.R.; Zavala, K.S.; Lanka, M.; Premlal, A.L.R.; Greenbaum, J.A.; et al. Linked CD4+/CD8+ T Cell Neoantigen Vaccination Overcomes Immune Checkpoint Blockade Resistance and Enables Tumor Regression. J. Clin. Investig. 2023, 133, e164258. [Google Scholar] [CrossRef] [PubMed]
- Ott, P.A.; Hu, Z.; Keskin, D.B.; Shukla, S.A.; Sun, J.; Bozym, D.J.; Zhang, W.; Luoma, A.; Giobbie-Hurder, A.; Peter, L.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients with Melanoma. Nature 2017, 547, 217–221. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shabalkina, A.V.; Izosimova, A.V.; Ryzhichenko, E.O.; Shurganova, E.V.; Myalik, D.S.; Maryanchik, S.V.; Ruppel, V.K.; Knyazev, D.I.; Khilal, N.R.; Barsova, E.V.; et al. Nodal Expansion, Tumor Infiltration and Exhaustion of Neoepitope-Specific Th Cells After Prophylactic Peptide Vaccination and Anti-CTLA4 Therapy in Mouse Melanoma B16. Int. J. Mol. Sci. 2025, 26, 6453. https://doi.org/10.3390/ijms26136453
Shabalkina AV, Izosimova AV, Ryzhichenko EO, Shurganova EV, Myalik DS, Maryanchik SV, Ruppel VK, Knyazev DI, Khilal NR, Barsova EV, et al. Nodal Expansion, Tumor Infiltration and Exhaustion of Neoepitope-Specific Th Cells After Prophylactic Peptide Vaccination and Anti-CTLA4 Therapy in Mouse Melanoma B16. International Journal of Molecular Sciences. 2025; 26(13):6453. https://doi.org/10.3390/ijms26136453
Chicago/Turabian StyleShabalkina, Alexandra V., Anna V. Izosimova, Ekaterina O. Ryzhichenko, Elizaveta V. Shurganova, Daria S. Myalik, Sofia V. Maryanchik, Valeria K. Ruppel, Dmitriy I. Knyazev, Nadezhda R. Khilal, Ekaterina V. Barsova, and et al. 2025. "Nodal Expansion, Tumor Infiltration and Exhaustion of Neoepitope-Specific Th Cells After Prophylactic Peptide Vaccination and Anti-CTLA4 Therapy in Mouse Melanoma B16" International Journal of Molecular Sciences 26, no. 13: 6453. https://doi.org/10.3390/ijms26136453
APA StyleShabalkina, A. V., Izosimova, A. V., Ryzhichenko, E. O., Shurganova, E. V., Myalik, D. S., Maryanchik, S. V., Ruppel, V. K., Knyazev, D. I., Khilal, N. R., Barsova, E. V., Shagina, I. A., & Sharonov, G. V. (2025). Nodal Expansion, Tumor Infiltration and Exhaustion of Neoepitope-Specific Th Cells After Prophylactic Peptide Vaccination and Anti-CTLA4 Therapy in Mouse Melanoma B16. International Journal of Molecular Sciences, 26(13), 6453. https://doi.org/10.3390/ijms26136453