
Parkinson Disease Signaling Pathways, Molecular Mechanisms, and Potential Therapeutic Strategies: A Comprehensive Review



Abstract
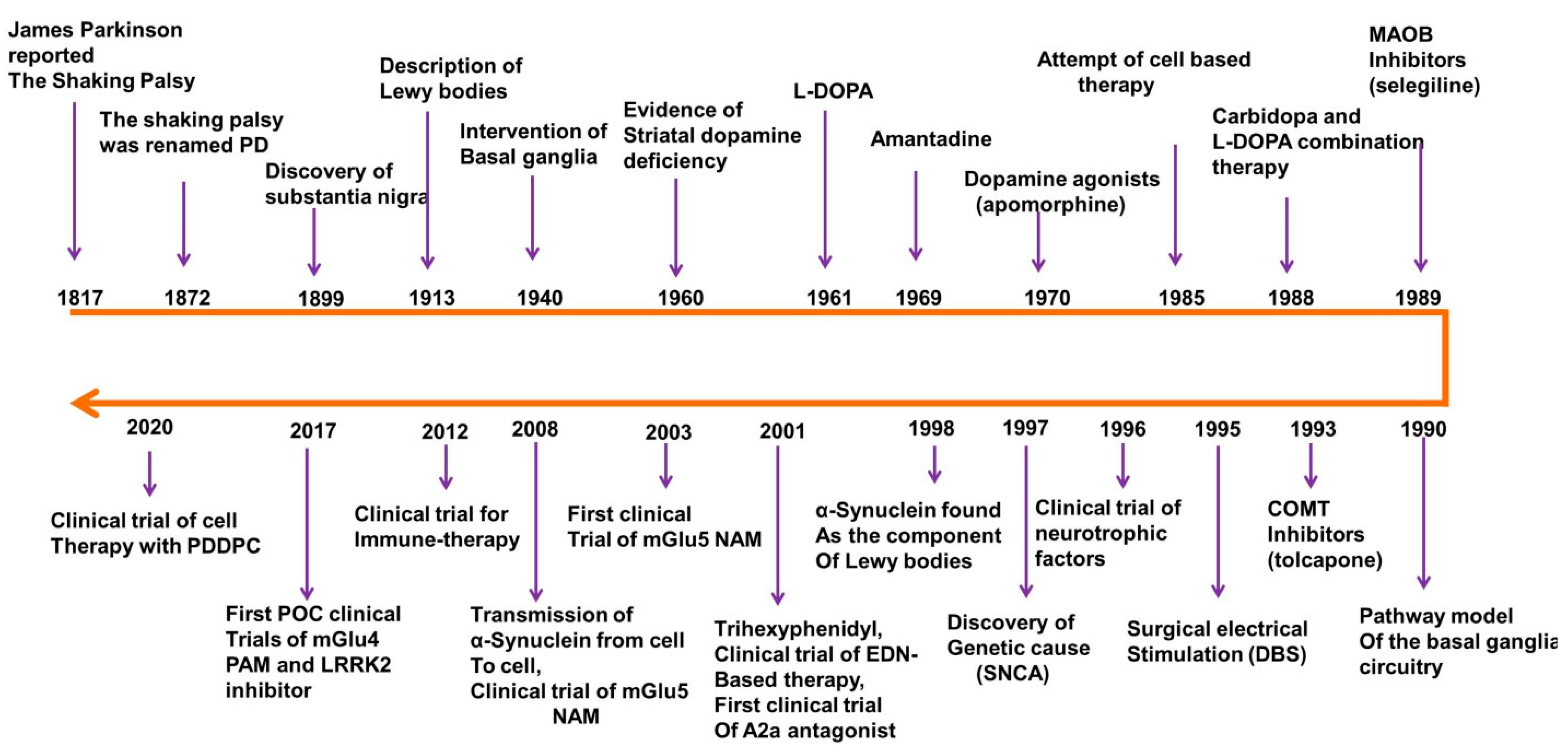
1. Introduction
2. Epidemiology, Clinical Features, and Diagnostic Criteria
3. Etiology and Pathogenesis of PD
3.1. Genetic and Environmental Factors and Pathogenesis of PD
3.2. Autosomal-Dominant Genes and PD Pathogenesis
3.3. Autosomal-Recessive Genes and PD Pathogenesis
4. Molecular Mechanisms of PD
4.1. Role of α-Synuclein Aggregation in PD Pathology
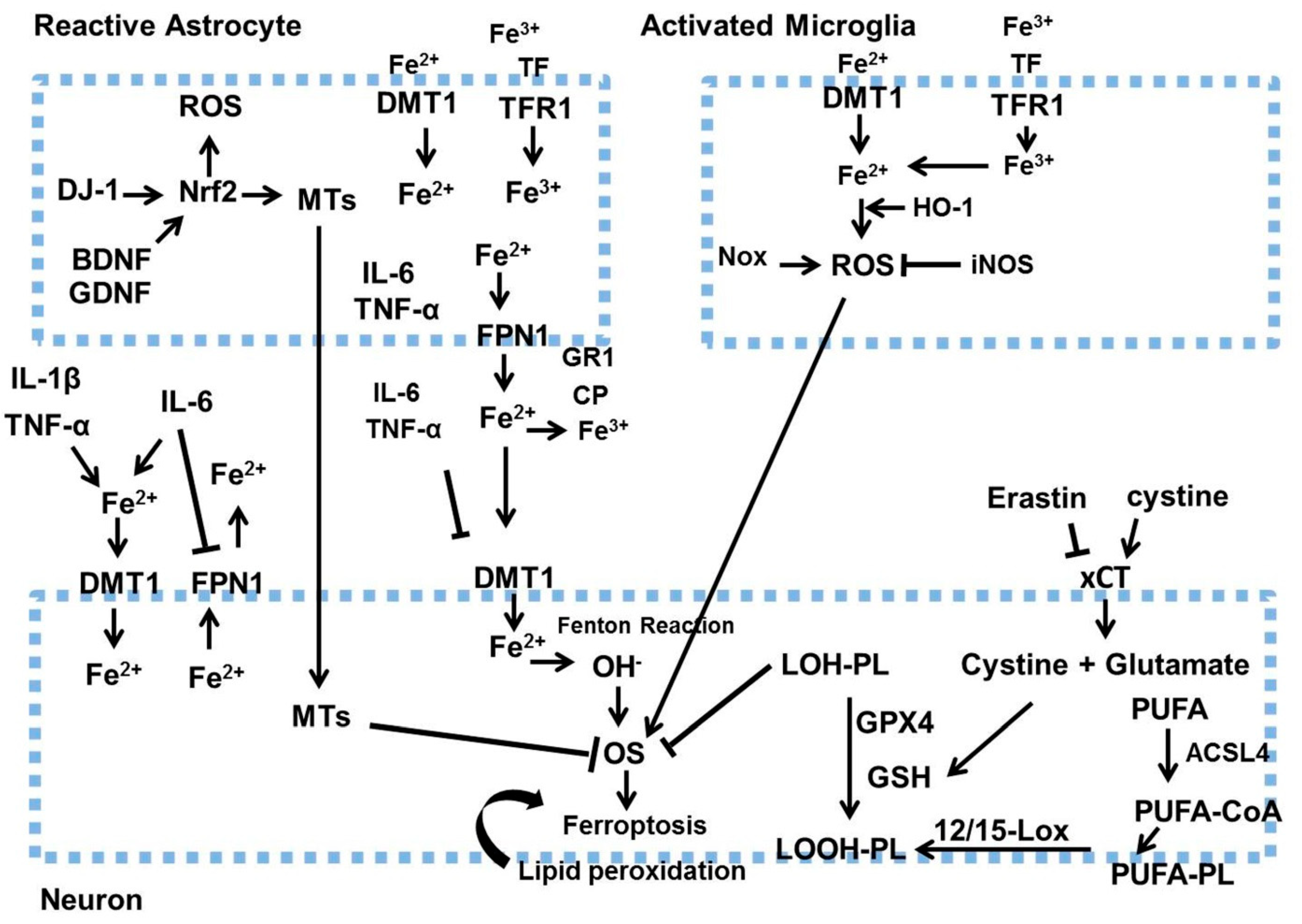
4.2. Role of Oxidative Stress (OS) in PD Pathology

4.3. Role of Ferroptosis in PD Pathology
4.4. Role of Mitochondrial Dysfunction in PD Pathology
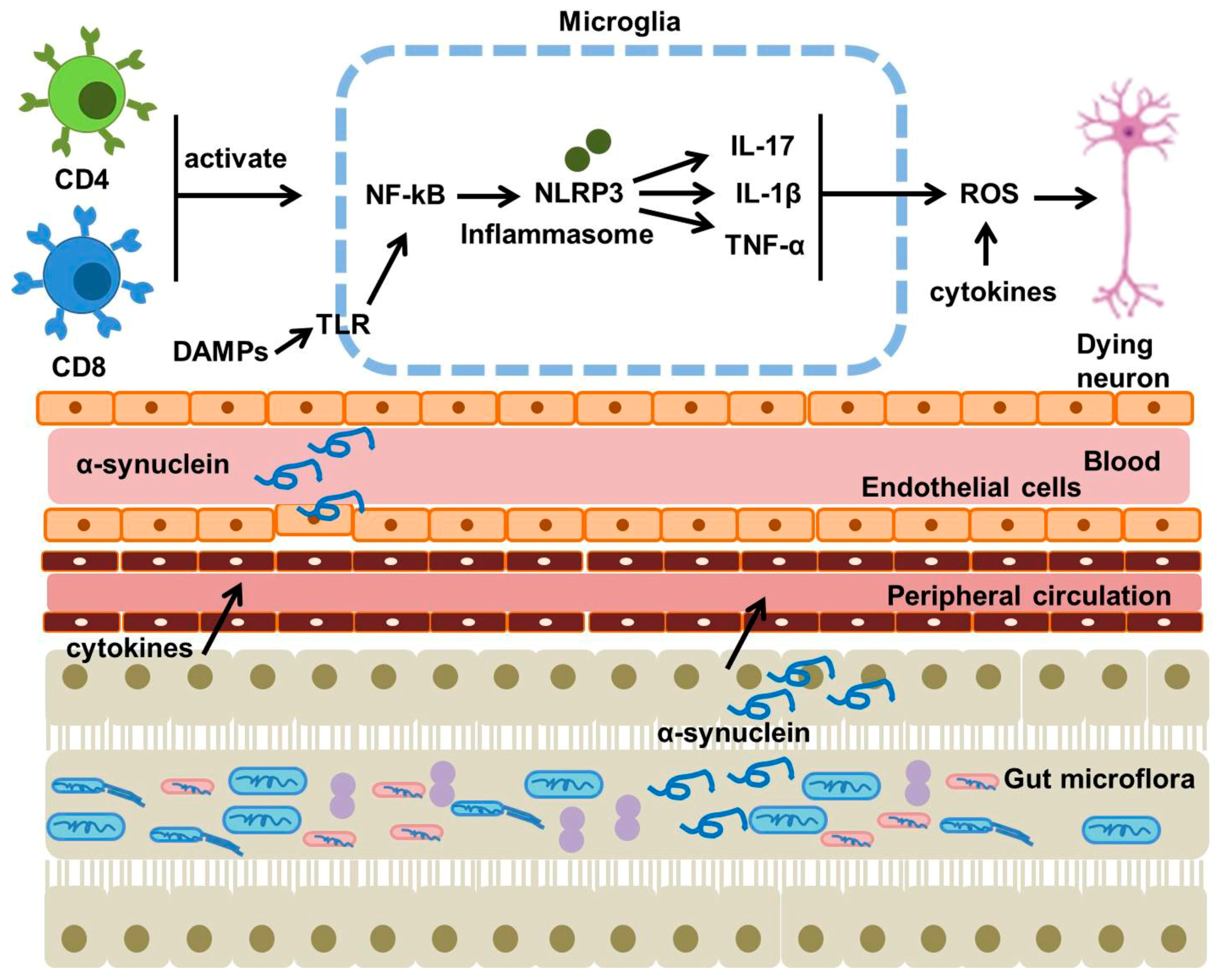
4.5. Role of Neuroinflammation in PD Pathology
4.6. Role of Gut Dysbiosis in PD Pathology
5. Research Models of PD
6. Therapeutic Strategies for PD
6.1. Commercially Available Drugs for PD
6.1.1. Levodopa
6.1.2. DA Agonists
6.1.3. Catechol-O-Methyltransferase and Monoamine Oxidase Type B Inhibitors
6.1.4. Non-Dopaminergic Targets
6.2. Drugs for PD Treatment Under Clinical Trials
6.3. Phytochemical-Based Therapeutic Strategy for PD Treatment
6.3.1. Phenol
6.3.2. Alkaloids
6.3.3. Flavonoids
6.3.4. Terpenoids
6.3.5. Saponins
7. Discussion and Perspectives
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Przedborski, S. The two-century journey of Parkinson disease research. Nat. Rev. Neurosci. 2017, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Charvin, D.; Medori, R.; Hauser, R.A.; Rascol, O. Therapeutic strategies for Parkinson disease: Beyond dopaminergic drugs. Nat. Rev. Drug Discov. 2018, 17, 804–822. [Google Scholar] [CrossRef]
- Twelves, D.; Perkins, K.S.; Counsell, C. Systematic review of incidence studies of Parkinson’s disease. Mov. Disord. 2003, 18, 19–31. [Google Scholar] [CrossRef]
- Savica, R.; Grossardt, B.R.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A. Incidence and pathology of synucleinopathies and tauopathies related to parkinsonism. JAMA Neurol. 2013, 70, 859–866. [Google Scholar] [CrossRef]
- Van Den Eeden, S.K.; Tanner, C.M.; Bernstein, A.L.; Fross, R.D.; Leimpeter, A.; Bloch, D.A.; Nelson, L.M. Incidence of Parkinson’s disease: Variation by age, gender, and race/ethnicity. Am. J. Epidemiol. 2003, 157, 1015–1022. [Google Scholar] [CrossRef]
- Pringsheim, T.; Jette, N.; Frolkis, A.; Steeves, T.D. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014, 29, 1583–1590. [Google Scholar] [CrossRef]
- Baldereschi, M.; Di Carlo, A.; Rocca, W.A.; Vanni, P.; Maggi, S.; Perissinotto, E.; Grigoletto, F.; Amaducci, L.; Inzitari, D. Parkinson’s disease and parkinsonism in a longitudinal study: Two-fold higher incidence in men. Neurology 2000, 55, 1358–1363. [Google Scholar] [CrossRef]
- Kusumi, M.; Nakashima, K.; Harada, H.; Nakayama, H.; Takahashi, K. Epidemiology of Parkinson’s disease in Yonago City, Japan: Comparison with a study carried out 12 years ago. Neuroepidemiology 1996, 15, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H.; Mehal, J.M.; Holman, R.C.; Rowland, A.S.; Cheek, J.E. Parkinson’s disease among American Indians and Alaska natives: A nationwide prevalence study. Mov. Disord. 2012, 27, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Davis, J.W.; Grandinetti, A.; Ross, G.W.; Popper, J.S.; White, L.R. Epidemiologic observations on Parkinson’s disease: Incidence and mortality in a prospective study of middle-aged men. Neurology 1996, 46, 1044–1050. [Google Scholar] [CrossRef]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Marras, C.; Lang, A. Parkinson’s disease subtypes: Lost in translation? J. Neurol. Neurosurg. Psychiatry 2013, 84, 409–415. [Google Scholar] [CrossRef]
- Jankovic, J.; McDermott, M.; Carter, J.; Gauthier, S.; Goetz, C.; Golbe, L.; Huber, S.; Koller, W.; Olanow, C.; Shoulson, I.; et al. Variable expression of Parkinson’s disease: A base-line analysis of the DATATOP cohort. Neurology 1990, 40, 1529–1534. [Google Scholar] [CrossRef]
- Khoo, T.K.; Yarnall, A.J.; Duncan, G.W.; Coleman, S.; O’Brien, J.T.; Brooks, D.J.; Barker, R.A.; Burn, D.J. The spectrum of nonmotor symptoms in early Parkinson disease. Neurology 2013, 80, 276–281. [Google Scholar] [CrossRef]
- Postuma, R.B.; Aarsland, D.; Barone, P.; Burn, D.J.; Hawkes, C.H.; Oertel, W.; Ziemssen, T. Identifying prodromal Parkinson’s disease: Pre-motor disorders in Parkinson’s disease. Mov. Disord. 2012, 27, 617–626. [Google Scholar] [CrossRef]
- Hely, M.A.; Morris, J.G.; Reid, W.G.; Trafficante, R. Sydney Multicenter Study of Parkinson’s disease: Non-L-dopa-responsive problems dominate at 15 years. Mov. Disord. 2005, 20, 190–199. [Google Scholar] [CrossRef]
- Fearnley, J.M.; Lees, A.J. Ageing and Parkinson’s disease: Substantia nigra regional selectivity. Brain 1991, 114 Pt 5, 2283–2301. [Google Scholar] [CrossRef] [PubMed]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Dijkstra, A.A.; Voorn, P.; Berendse, H.W.; Groenewegen, H.J.; Netherlands Brain Bank; Rozemuller, A.J.; van de Berg, W.D. Stage-dependent nigral neuronal loss in incidental Lewy body and Parkinson’s disease. Mov. Disord. 2014, 29, 1244–1251. [Google Scholar] [CrossRef] [PubMed]
- Iacono, D.; Geraci-Erck, M.; Rabin, M.L.; Adler, C.H.; Serrano, G.; Beach, T.G.; Kurlan, R. Parkinson disease and incidental Lewy body disease: Just a question of time? Neurology 2015, 85, 1670–1679. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Orieux, G.; Muriel, M.P.; Francois, C.; Feger, J. Nondopaminergic neurons in Parkinson’s disease. Adv. Neurol. 2003, 91, 29–37. [Google Scholar]
- Rizzo, G.; Copetti, M.; Arcuti, S.; Martino, D.; Fontana, A.; Logroscino, G. Accuracy of clinical diagnosis of Parkinson disease: A systematic review and meta-analysis. Neurology 2016, 86, 566–576. [Google Scholar] [CrossRef]
- Postuma, R.B.; Poewe, W.; Litvan, I.; Lewis, S.; Lang, A.E.; Halliday, G.; Goetz, C.G.; Chan, P.; Slow, E.; Seppi, K.; et al. Validation of the MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. 2018, 33, 1601–1608. [Google Scholar] [CrossRef]
- Krismer, F.; Pinter, B.; Mueller, C.; Mahlknecht, P.; Nocker, M.; Reiter, E.; Djamshidian-Tehrani, A.; Boesch, S.M.; Wenning, G.K.; Scherfler, C.; et al. Sniffing the diagnosis: Olfactory testing in neurodegenerative parkinsonism. Park. Relat. Disord. 2017, 35, 36–41. [Google Scholar] [CrossRef]
- Katzenschlager, R.; Lees, A.J. Olfaction and Parkinson’s syndromes: Its role in differential diagnosis. Curr. Opin. Neurol. 2004, 17, 417–423. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuka, C.; Sasaki, M.; Konno, K.; Koide, M.; Kato, K.; Takahashi, J.; Takahashi, S.; Kudo, K.; Yamashita, F.; Terayama, Y. Changes in substantia nigra and locus coeruleus in patients with early-stage Parkinson’s disease using neuromelanin-sensitive MR imaging. Neurosci. Lett. 2013, 541, 93–98. [Google Scholar] [CrossRef]
- Langkammer, C.; Schweser, F.; Krebs, N.; Deistung, A.; Goessler, W.; Scheurer, E.; Sommer, K.; Reishofer, G.; Yen, K.; Fazekas, F.; et al. Quantitative susceptibility mapping (QSM) as a means to measure brain iron? A post mortem validation study. Neuroimage 2012, 62, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Ehrminger, M.; Latimier, A.; Pyatigorskaya, N.; Garcia-Lorenzo, D.; Leu-Semenescu, S.; Vidailhet, M.; Lehericy, S.; Arnulf, I. The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain 2016, 139 Pt 4, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Stoessl, A.J.; Lehericy, S.; Strafella, A.P. Imaging insights into basal ganglia function, Parkinson’s disease, and dystonia. Lancet 2014, 384, 532–544. [Google Scholar] [CrossRef]
- Politis, M. Neuroimaging in Parkinson disease: From research setting to clinical practice. Nat. Rev. Neurol. 2014, 10, 708–722. [Google Scholar] [CrossRef]
- Kalinderi, K.; Bostantjopoulou, S.; Fidani, L. The genetic background of Parkinson’s disease: Current progress and future prospects. Acta Neurol. Scand. 2016, 134, 314–326. [Google Scholar] [CrossRef]
- Tambasco, N.; Nigro, P.; Romoli, M.; Prontera, P.; Simoni, S.; Calabresi, P. A53T in a parkinsonian family: A clinical update of the SNCA phenotypes. J. Neural Transm. 2016, 123, 1301–1307. [Google Scholar] [CrossRef]
- Marras, C.; Canning, C.G.; Goldman, S.M. Environment, lifestyle, and Parkinson’s disease: Implications for prevention in the next decade. Mov. Disord. 2019, 34, 801–811. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar] [CrossRef]
- Tanner, C.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1992, 10, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Ikeuchi, T.; Kakita, A.; Shiga, A.; Kasuga, K.; Kaneko, H.; Tan, C.F.; Idezuka, J.; Wakabayashi, K.; Onodera, O.; Iwatsubo, T.; et al. Patients homozygous and heterozygous for SNCA duplication in a family with parkinsonism and dementia. Arch. Neurol. 2008, 65, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, L.; Pan, H.; Liu, Z.; Jiang, L.; He, Y.; Zeng, Q.; Zhou, X.; Zhou, X.; Zhou, Y.; et al. The role of genetics in Parkinson’s disease: A large cohort study in Chinese mainland population. Brain 2020, 143, 2220–2234. [Google Scholar] [CrossRef]
- Monfrini, E.; Di Fonzo, A. Leucine-Rich Repeat Kinase (LRRK2) Genetics and Parkinson’s Disease. Adv. Neurobiol. 2017, 14, 3–30. [Google Scholar]
- Deng, H.; Wang, P.; Jankovic, J. The genetics of Parkinson disease. Ageing Res. Rev. 2018, 42, 72–85. [Google Scholar] [CrossRef]
- Hedrich, K.; Winkler, S.; Hagenah, J.; Kabakci, K.; Kasten, M.; Schwinger, E.; Volkmann, J.; Pramstaller, P.P.; Kostic, V.; Vieregge, P.; et al. Recurrent LRRK2 (Park8) mutations in early-onset Parkinson’s disease. Mov. Disord. 2006, 21, 1506–1510. [Google Scholar] [CrossRef]
- Martins, L.M.; Morrison, A.; Klupsch, K.; Fedele, V.; Moisoi, N.; Teismann, P.; Abuin, A.; Grau, E.; Geppert, M.; Livi, G.P.; et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol. Cell. Biol. 2004, 24, 9848–9862. [Google Scholar] [CrossRef]
- Plun-Favreau, H.; Klupsch, K.; Moisoi, N.; Gandhi, S.; Kjaer, S.; Frith, D.; Harvey, K.; Deas, E.; Harvey, R.J.; McDonald, N.; et al. The mitochondrial protease HtrA2 is regulated by Parkinson’s disease-associated kinase PINK1. Nat. Cell Biol. 2007, 9, 1243–1252. [Google Scholar] [CrossRef]
- Moisoi, N.; Klupsch, K.; Fedele, V.; East, P.; Sharma, S.; Renton, A.; Plun-Favreau, H.; Edwards, R.E.; Teismann, P.; Esposti, M.D.; et al. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ. 2009, 16, 449–464. [Google Scholar] [CrossRef]
- Fitzgerald, J.C.; Camprubi, M.D.; Dunn, L.; Wu, H.C.; Ip, N.Y.; Kruger, R.; Martins, L.M.; Wood, N.W.; Plun-Favreau, H. Phosphorylation of HtrA2 by cyclin-dependent kinase-5 is important for mitochondrial function. Cell Death Differ. 2012, 19, 257–266. [Google Scholar] [CrossRef]
- Basak, I.; Pal, R.; Patil, K.S.; Dunne, A.; Ho, H.P.; Lee, S.; Peiris, D.; Maple-Grodem, J.; Odell, M.; Chang, E.J.; et al. Arabidopsis AtPARK13, which confers thermotolerance, targets misfolded proteins. J. Biol. Chem. 2014, 289, 14458–14469. [Google Scholar] [CrossRef]
- Bonifacino, J.S.; Hurley, J.H. Retromer. Curr. Opin. Cell Biol. 2008, 20, 427–436. [Google Scholar] [CrossRef] [PubMed]
- Arighi, C.N.; Hartnell, L.M.; Aguilar, R.C.; Haft, C.R.; Bonifacino, J.S. Role of the mammalian retromer in sorting of the cation-independent mannose 6-phosphate receptor. J. Cell Biol. 2004, 165, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Chartier-Harlin, M.C.; Dachsel, J.C.; Vilarino-Guell, C.; Lincoln, S.J.; Lepretre, F.; Hulihan, M.M.; Kachergus, J.; Milnerwood, A.J.; Tapia, L.; Song, M.S.; et al. Translation initiator EIF4G1 mutations in familial Parkinson disease. Am. J. Hum. Genet. 2011, 89, 398–406. [Google Scholar] [CrossRef]
- Deng, H.; Wu, Y.; Jankovic, J. The EIF4G1 gene and Parkinson’s disease. Acta Neurol. Scand. 2015, 132, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Sonenberg, N.; Hinnebusch, A.G. Regulation of translation initiation in eukaryotes: Mechanisms and biological targets. Cell 2009, 136, 731–745. [Google Scholar] [CrossRef]
- Hattori, N.; Mizuno, Y. Twenty years since the discovery of the parkin gene. J. Neural Transm. 2017, 124, 1037–1054. [Google Scholar] [CrossRef]
- Klein, C.; Lohmann-Hedrich, K.; Rogaeva, E.; Schlossmacher, M.G.; Lang, A.E. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol. 2007, 6, 652–662. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef]
- Seirafi, M.; Kozlov, G.; Gehring, K. Parkin structure and function. FEBS J. 2015, 282, 2076–2088. [Google Scholar] [CrossRef]
- van der Merwe, C.; Jalali Sefid Dashti, Z.; Christoffels, A.; Loos, B.; Bardien, S. Evidence for a common biological pathway linking three Parkinson’s disease-causing genes: Parkin, PINK1 and DJ-1. Eur. J. Neurosci. 2015, 41, 1113–1125. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.; Nijland, P.G.; Drukarch, B.; de Vries, H.E.; van Horssen, J. Involvement and interplay of Parkin, PINK1, and DJ1 in neurodegenerative and neuroinflammatory disorders. Free. Radic. Biol. Med. 2012, 53, 983–992. [Google Scholar] [CrossRef]
- Zondler, L.; Miller-Fleming, L.; Repici, M.; Goncalves, S.; Tenreiro, S.; Rosado-Ramos, R.; Betzer, C.; Straatman, K.R.; Jensen, P.H.; Giorgini, F.; et al. DJ-1 interactions with alpha-synuclein attenuate aggregation and cellular toxicity in models of Parkinson’s disease. Cell Death Dis. 2014, 5, e1350. [Google Scholar] [CrossRef]
- Demirsoy, S.; Martin, S.; Motamedi, S.; van Veen, S.; Holemans, T.; Van den Haute, C.; Jordanova, A.; Baekelandt, V.; Vangheluwe, P.; Agostinis, P. ATP13A2/PARK9 regulates endo-/lysosomal cargo sorting and proteostasis through a novel PI(3, 5)P2-mediated scaffolding function. Hum. Mol. Genet. 2017, 26, 1656–1669. [Google Scholar] [CrossRef]
- Holemans, T.; Sorensen, D.M.; van Veen, S.; Martin, S.; Hermans, D.; Kemmer, G.C.; Van den Haute, C.; Baekelandt, V.; Gunther Pomorski, T.; Agostinis, P.; et al. A lipid switch unlocks Parkinson’s disease-associated ATP13A2. Proc. Natl. Acad. Sci. USA 2015, 112, 9040–9045. [Google Scholar] [CrossRef]
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815. [Google Scholar] [CrossRef]
- Tsunemi, T.; Krainc, D. Zn2+ dyshomeostasis caused by loss of ATP13A2/PARK9 leads to lysosomal dysfunction and alpha-synuclein accumulation. Hum. Mol. Genet. 2014, 23, 2791–2801. [Google Scholar] [CrossRef]
- Park, J.S.; Blair, N.F.; Sue, C.M. The role of ATP13A2 in Parkinson’s disease: Clinical phenotypes and molecular mechanisms. Mov. Disord. 2015, 30, 770–779. [Google Scholar] [CrossRef]
- Kong, S.M.; Chan, B.K.; Park, J.S.; Hill, K.J.; Aitken, J.B.; Cottle, L.; Farghaian, H.; Cole, A.R.; Lay, P.A.; Sue, C.M.; et al. Parkinson’s disease-linked human PARK9/ATP13A2 maintains zinc homeostasis and promotes alpha-Synuclein externalization via exosomes. Hum. Mol. Genet. 2014, 23, 2816–2833. [Google Scholar] [CrossRef]
- Teixeira, F.R.; Randle, S.J.; Patel, S.P.; Mevissen, T.E.; Zenkeviciute, G.; Koide, T.; Komander, D.; Laman, H. Gsk3beta and Tomm20 are substrates of the SCFFbxo7/PARK15 ubiquitin ligase associated with Parkinson’s disease. Biochem. J. 2016, 473, 3563–3580. [Google Scholar] [CrossRef]
- Ordureau, A.; Heo, J.M.; Duda, D.M.; Paulo, J.A.; Olszewski, J.L.; Yanishevski, D.; Rinehart, J.; Schulman, B.A.; Harper, J.W. Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc. Natl. Acad. Sci. USA 2015, 112, 6637–6642. [Google Scholar] [CrossRef]
- Duarte, F.V.; Amorim, J.A.; Varela, A.T.; Teodoro, J.S.; Gomes, A.P.; Cunha, R.A.; Palmeira, C.M.; Rolo, A.P. Adenosine receptors: Regulatory players in the preservation of mitochondrial function induced by ischemic preconditioning of rat liver. Purinergic Signal. 2017, 13, 179–190. [Google Scholar] [CrossRef]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W.; et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef]
- Zhou, Z.D.; Lee, J.C.T.; Tan, E.K. Pathophysiological mechanisms linking F-box only protein 7 (FBXO7) and Parkinson’s disease (PD). Mutat. Res. Rev. Mutat. Res. 2018, 778, 72–78. [Google Scholar] [CrossRef]
- Delgado-Camprubi, M.; Esteras, N.; Soutar, M.P.; Plun-Favreau, H.; Abramov, A.Y. Deficiency of Parkinson’s disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2017, 24, 2210. [Google Scholar] [CrossRef]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of alpha-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar] [CrossRef]
- Vekrellis, K.; Xilouri, M.; Emmanouilidou, E.; Rideout, H.J.; Stefanis, L. Pathological roles of alpha-synuclein in neurological disorders. Lancet Neurol. 2011, 10, 1015–1025. [Google Scholar] [CrossRef]
- Wales, P.; Pinho, R.; Lazaro, D.F.; Outeiro, T.F. Limelight on alpha-synuclein: Pathological and mechanistic implications in neurodegeneration. J. Park. Dis. 2013, 3, 415–459. [Google Scholar] [CrossRef]
- Burre, J. The Synaptic Function of alpha-Synuclein. J. Park. Dis. 2015, 5, 699–713. [Google Scholar]
- Ruiperez, V.; Darios, F.; Davletov, B. Alpha-synuclein, lipids and Parkinson’s disease. Prog. Lipid Res. 2010, 49, 420–428. [Google Scholar] [CrossRef]
- Kim, C.; Lee, S.J. Controlling the mass action of alpha-synuclein in Parkinson’s disease. J. Neurochem. 2008, 107, 303–316. [Google Scholar] [CrossRef] [PubMed]
- Melki, R. Role of Different Alpha-Synuclein Strains in Synucleinopathies, Similarities with other Neurodegenerative Diseases. J. Park. Dis. 2015, 5, 217–227. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. Proteostasis and aging. Nat. Med. 2015, 21, 1406–1415. [Google Scholar] [CrossRef]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef]
- Brieger, K.; Schiavone, S.; Miller, F.J., Jr.; Krause, K.H. Reactive oxygen species: From health to disease. Swiss Med. Wkly. 2012, 142, w13659. [Google Scholar] [CrossRef]
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: Therapeutic modulation via fumaric acid esters. Int. J. Mol. Sci. 2012, 13, 11783–11803. [Google Scholar] [CrossRef]
- Shukla, V.; Mishra, S.K.; Pant, H.C. Oxidative stress in neurodegeneration. Adv. Pharmacol. Sci. 2011, 2011, 572634. [Google Scholar] [CrossRef]
- Kemppainen, S.; Lindholm, P.; Galli, E.; Lahtinen, H.M.; Koivisto, H.; Hamalainen, E.; Saarma, M.; Tanila, H. Cerebral dopamine neurotrophic factor improves long-term memory in APP/PS1 transgenic mice modeling Alzheimer’s disease as well as in wild-type mice. Behav. Brain Res. 2015, 291, 1–11. [Google Scholar] [CrossRef]
- Melief, E.J.; Cudaback, E.; Jorstad, N.L.; Sherfield, E.; Postupna, N.; Wilson, A.; Darvas, M.; Montine, K.S.; Keene, C.D.; Montine, T.J. Partial depletion of striatal dopamine enhances penetrance of cognitive deficits in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. Res. 2015, 93, 1413–1422. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef]
- Kovac, S.; Angelova, P.R.; Holmstrom, K.M.; Zhang, Y.; Dinkova-Kostova, A.T.; Abramov, A.Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochim. Biophys. Acta 2015, 1850, 794–801. [Google Scholar] [CrossRef]
- Subramaniam, S.R.; Chesselet, M.F. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog. Neurobiol. 2013, 106–107, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Dionisio, P.A.; Amaral, J.D.; Rodrigues, C.M.P. Oxidative stress and regulated cell death in Parkinson’s disease. Ageing Res. Rev. 2021, 67, 101263. [Google Scholar] [CrossRef]
- Guiney, S.J.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I.; Ayton, S. Ferroptosis and cell death mechanisms in Parkinson’s disease. Neurochem. Int. 2017, 104, 34–48. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef]
- Gallagher, E.; Hou, C.; Zhu, Y.; Hsieh, C.J.; Lee, H.; Li, S.; Xu, K.; Henderson, P.; Chroneos, R.; Sheldon, M.; et al. Positron Emission Tomography with [18F]ROStrace Reveals Progressive Elevations in Oxidative Stress in a Mouse Model of Alpha-Synucleinopathy. Int. J. Mol. Sci. 2024, 25, 4943. [Google Scholar] [CrossRef]
- Dehay, B.; Bove, J.; Rodriguez-Muela, N.; Perier, C.; Recasens, A.; Boya, P.; Vila, M. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 2010, 30, 12535–12544. [Google Scholar] [CrossRef]
- Hernandes, M.S.; Cafe-Mendes, C.C.; Britto, L.R. NADPH oxidase and the degeneration of dopaminergic neurons in parkinsonian mice. Oxidative Med. Cell. Longev. 2013, 2013, 157857. [Google Scholar] [CrossRef]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef]
- Zhu, Y.; Kohli, N.; Young, A.; Sheldon, M.; Coni, J.; Rajasekaran, M.; Robinson, L.; Chroneos, R.; Riley, S.; Guarnieri, J.W.; et al. PET Imaging with [18F]ROStrace Detects Oxidative Stress and Predicts Parkinson’s Disease Progression in Mice. Antioxidants 2024, 13, 1226. [Google Scholar] [CrossRef]
- Dong-Chen, X.; Yong, C.; Yang, X.; Chen-Yu, S.; Li-Hua, P. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 73. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Do Van, B.; Gouel, F.; Jonneaux, A.; Timmerman, K.; Gele, P.; Petrault, M.; Bastide, M.; Laloux, C.; Moreau, C.; Bordet, R.; et al. Ferroptosis, a newly characterized form of cell death in Parkinson’s disease that is regulated by PKC. Neurobiol. Dis. 2016, 94, 169–178. [Google Scholar] [CrossRef]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef]
- Skouta, R.; Dixon, S.J.; Wang, J.; Dunn, D.E.; Orman, M.; Shimada, K.; Rosenberg, P.A.; Lo, D.C.; Weinberg, J.M.; Linkermann, A.; et al. Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J. Am. Chem. Soc. 2014, 136, 4551–4556. [Google Scholar] [CrossRef]
- Cao, J.; Chen, X.; Jiang, L.; Lu, B.; Yuan, M.; Zhu, D.; Zhu, H.; He, Q.; Yang, B.; Ying, M. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat. Commun. 2020, 11, 1251. [Google Scholar] [CrossRef]
- Forcina, G.C.; Dixon, S.J. GPX4 at the Crossroads of Lipid Homeostasis and Ferroptosis. Proteomics 2019, 19, 1800311. [Google Scholar] [CrossRef]
- Cozza, G.; Rossetto, M.; Bosello-Travain, V.; Maiorino, M.; Roveri, A.; Toppo, S.; Zaccarin, M.; Zennaro, L.; Ursini, F. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: The polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic. Biol. Med. 2017, 112, 1–11. [Google Scholar] [CrossRef]
- Castellani, R.J.; Siedlak, S.L.; Perry, G.; Smith, M.A. Sequestration of iron by Lewy bodies in Parkinson’s disease. Acta Neuropathol. 2000, 100, 111–114. [Google Scholar] [CrossRef]
- Golts, N.; Snyder, H.; Frasier, M.; Theisler, C.; Choi, P.; Wolozin, B. Magnesium inhibits spontaneous and iron-induced aggregation of alpha-synuclein. J. Biol. Chem. 2002, 277, 16116–16123. [Google Scholar] [CrossRef]
- Guo, J.J.; Yue, F.; Song, D.Y.; Bousset, L.; Liang, X.; Tang, J.; Yuan, L.; Li, W.; Melki, R.; Tang, Y.; et al. Intranasal administration of alpha-synuclein preformed fibrils triggers microglial iron deposition in the substantia nigra of Macaca fascicularis. Cell Death Dis. 2021, 12, 81. [Google Scholar] [CrossRef]
- Kenkhuis, B.; Somarakis, A.; de Haan, L.; Dzyubachyk, O.; IJsselsteijn, M.E.; de Miranda, N.F.C.C.; Lelieveldt, B.P.F.; Dijkstra, J.; van Roon-Mom, W.M.C.; Hollt, T.; et al. Iron loading is a prominent feature of activated microglia in Alzheimer’s disease patients. Acta Neuropathol. Commun. 2021, 9, 27. [Google Scholar] [CrossRef]
- Friedrich, I.; Reimann, K.; Jankuhn, S.; Kirilina, E.; Stieler, J.; Sonntag, M.; Meijer, J.; Weiskopf, N.; Reinert, T.; Arendt, T.; et al. Cell specific quantitative iron mapping on brain slices by immuno-microPIXE in healthy elderly and Parkinson’s disease. Acta Neuropathol. Commun. 2021, 9, 47. [Google Scholar] [CrossRef]
- Healy, S.; McMahon, J.; Owens, P.; FitzGerald, U. Significant glial alterations in response to iron loading in a novel organotypic hippocampal slice culture model. Sci. Rep. 2016, 6, 36410. [Google Scholar] [CrossRef]
- Wang, J.; Song, N.; Jiang, H.; Wang, J.; Xie, J. Pro-inflammatory cytokines modulate iron regulatory protein 1 expression and iron transportation through reactive oxygen/nitrogen species production in ventral mesencephalic neurons. Biochim. Biophys. Acta 2013, 1832, 618–625. [Google Scholar] [CrossRef]
- Urrutia, P.; Aguirre, P.; Esparza, A.; Tapia, V.; Mena, N.P.; Arredondo, M.; Gonzalez-Billault, C.; Nunez, M.T. Inflammation alters the expression of DMT1, FPN1 and hepcidin, and it causes iron accumulation in central nervous system cells. J. Neurochem. 2013, 126, 541–549. [Google Scholar] [CrossRef]
- Sharma, N.; Nehru, B. Apocyanin, a Microglial NADPH Oxidase Inhibitor Prevents Dopaminergic Neuronal Degeneration in Lipopolysaccharide-Induced Parkinson’s Disease Model. Mol. Neurobiol. 2016, 53, 3326–3337. [Google Scholar] [CrossRef]
- Kapralov, A.A.; Yang, Q.; Dar, H.H.; Tyurina, Y.Y.; Anthonymuthu, T.S.; Kim, R.; St Croix, C.M.; Mikulska-Ruminska, K.; Liu, B.; Shrivastava, I.H.; et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat. Chem. Biol. 2020, 16, 278–290. [Google Scholar] [CrossRef]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef] [PubMed]
- Ayton, S.; Lei, P.; Duce, J.A.; Wong, B.X.; Sedjahtera, A.; Adlard, P.A.; Bush, A.I.; Finkelstein, D.I. Ceruloplasmin dysfunction and therapeutic potential for Parkinson disease. Ann. Neurol. 2013, 73, 554–559. [Google Scholar] [CrossRef]
- Wang, J.; Bi, M.; Xie, J. Ceruloplasmin is Involved in the Nigral Iron Accumulation of 6-OHDA-Lesioned Rats. Cell. Mol. Neurobiol. 2015, 35, 661–668. [Google Scholar] [CrossRef]
- Fernandez-Mendivil, C.; Luengo, E.; Trigo-Alonso, P.; Garcia-Magro, N.; Negredo, P.; Lopez, M.G. Protective role of microglial HO-1 blockade in aging: Implication of iron metabolism. Redox Biol. 2021, 38, 101789. [Google Scholar] [CrossRef]
- Song, W.; Cressatti, M.; Zukor, H.; Liberman, A.; Galindez, C.; Schipper, H.M. Parkinsonian features in aging GFAP.HMOX1 transgenic mice overexpressing human HO-1 in the astroglial compartment. Neurobiol. Aging 2017, 58, 163–179. [Google Scholar] [CrossRef]
- Wang, Z.L.; Yuan, L.; Li, W.; Li, J.Y. Ferroptosis in Parkinson’s disease: Glia-neuron crosstalk. Trends Mol. Med. 2022, 28, 258–269. [Google Scholar] [CrossRef]
- Burns, R.S.; LeWitt, P.A.; Ebert, M.H.; Pakkenberg, H.; Kopin, I.J. The clinical syndrome of striatal dopamine deficiency. Parkinsonism induced by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). N. Engl. J. Med. 1985, 312, 1418–1421. [Google Scholar] [CrossRef]
- Chaturvedi, R.K.; Beal, M.F. Mitochondrial approaches for neuroprotection. Ann. N. Y. Acad. Sci. 2008, 1147, 395–412. [Google Scholar] [CrossRef]
- Panov, A.; Dikalov, S.; Shalbuyeva, N.; Taylor, G.; Sherer, T.; Greenamyre, J.T. Rotenone model of Parkinson disease: Multiple brain mitochondria dysfunctions after short term systemic rotenone intoxication. J. Biol. Chem. 2005, 280, 42026–42035. [Google Scholar] [CrossRef]
- Borland, M.K.; Trimmer, P.A.; Rubinstein, J.D.; Keeney, P.M.; Mohanakumar, K.; Liu, L.; Bennett, J.P., Jr. Chronic, low-dose rotenone reproduces Lewy neurites found in early stages of Parkinson’s disease, reduces mitochondrial movement and slowly kills differentiated SH-SY5Y neural cells. Mol. Neurodegener. 2008, 3, 21. [Google Scholar] [CrossRef]
- Dauer, W.; Przedborski, S. Parkinson’s disease: Mechanisms and models. Neuron 2003, 39, 889–909. [Google Scholar] [CrossRef] [PubMed]
- Klivenyi, P.; Siwek, D.; Gardian, G.; Yang, L.; Starkov, A.; Cleren, C.; Ferrante, R.J.; Kowall, N.W.; Abeliovich, A.; Beal, M.F. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol. Dis. 2006, 21, 541–548. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, R183–R194. [Google Scholar] [CrossRef]
- Mudo, G.; Makela, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Malkia, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1alpha protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jager, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Casarejos, M.J.; Menendez, J.; Solano, R.M.; Rodriguez-Navarro, J.A.; Garcia de Yebenes, J.; Mena, M.A. Susceptibility to rotenone is increased in neurons from parkin null mice and is reduced by minocycline. J. Neurochem. 2006, 97, 934–946. [Google Scholar] [CrossRef]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, C.; Yin, J.; Li, X.; Cheng, F.; Li, Y.; Yang, H.; Ueda, K.; Chan, P.; Yu, S. α-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci. Lett. 2009, 454, 187–192. [Google Scholar] [CrossRef]
- Bertolin, G.; Ferrando-Miguel, R.; Jacoupy, M.; Traver, S.; Grenier, K.; Greene, A.W.; Dauphin, A.; Waharte, F.; Bayot, A.; Salamero, J.; et al. The TOMM machinery is a molecular switch in PINK1 and PARK2/PARKIN-dependent mitochondrial clearance. Autophagy 2013, 9, 1801–1817. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef]
- Geisler, S.; Holmstrom, K.M.; Skujat, D.; Fiesel, F.C.; Rothfuss, O.C.; Kahle, P.J.; Springer, W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat. Cell Biol. 2010, 12, 119–131. [Google Scholar] [CrossRef]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef]
- Vives-Bauza, C.; Zhou, C.; Huang, Y.; Cui, M.; de Vries, R.L.; Kim, J.; May, J.; Tocilescu, M.A.; Liu, W.; Ko, H.S.; et al. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc. Natl. Acad. Sci. USA 2010, 107, 378–383. [Google Scholar] [CrossRef]
- Liu, W.; Wang, M.; Shen, L.; Zhu, Y.; Ma, H.; Liu, B.; Ouyang, L.; Guo, W.; Xu, Q.; Sun, Y. SHP2-mediated mitophagy boosted by lovastatin in neuronal cells alleviates parkinsonism in mice. Signal Transduct. Target. Ther. 2021, 6, 34. [Google Scholar] [CrossRef]
- Lavara-Culebras, E.; Paricio, N. Drosophila DJ-1 mutants are sensitive to oxidative stress and show reduced lifespan and motor deficits. Gene 2007, 400, 158–165. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef]
- Saha, S.; Guillily, M.D.; Ferree, A.; Lanceta, J.; Chan, D.; Ghosh, J.; Hsu, C.H.; Segal, L.; Raghavan, K.; Matsumoto, K.; et al. LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J. Neurosci. 2009, 29, 9210–9218. [Google Scholar] [CrossRef]
- Yue, M.; Hinkle, K.M.; Davies, P.; Trushina, E.; Fiesel, F.C.; Christenson, T.A.; Schroeder, A.S.; Zhang, L.; Bowles, E.; Behrouz, B.; et al. Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 2015, 78, 172–195. [Google Scholar] [CrossRef]
- Wang, X.; Yan, M.H.; Fujioka, H.; Liu, J.; Wilson-Delfosse, A.; Chen, S.G.; Perry, G.; Casadesus, G.; Zhu, X. LRRK2 regulates mitochondrial dynamics and function through direct interaction with DLP1. Hum. Mol. Genet. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1 beta, interleukin-6, epidermal growth factor and transforming growth factor-alpha are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Riederer, P.; Narabayashi, H.; Fujita, K.; Nagatsu, T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci. Lett. 1994, 165, 208–210. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Narabayashi, H.; Riederer, P.; Nagatsu, T. Transforming growth factor-beta 1 levels are elevated in the striatum and in ventricular cerebrospinal fluid in Parkinson’s disease. Neurosci. Lett. 1995, 193, 129–132. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Nagatsu, T. Interleukin-2 but not basic fibroblast growth factor is elevated in parkinsonian brain. J. Neural Transm. 1996, 103, 1077–1081. [Google Scholar] [CrossRef]
- Sommer, A.; Fadler, T.; Dorfmeister, E.; Hoffmann, A.C.; Xiang, W.; Winner, B.; Prots, I. Infiltrating T lymphocytes reduce myeloid phagocytosis activity in synucleinopathy model. J. Neuroinflamm. 2016, 13, 174. [Google Scholar] [CrossRef]
- Brochard, V.; Combadiere, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Stone, D.K.; Reynolds, A.D.; Mosley, R.L.; Gendelman, H.E. Innate and adaptive immunity for the pathobiology of Parkinson’s disease. Antioxid. Redox Signal. 2009, 11, 2151–2166. [Google Scholar] [CrossRef]
- Kustrimovic, N.; Comi, C.; Magistrelli, L.; Rasini, E.; Legnaro, M.; Bombelli, R.; Aleksic, I.; Blandini, F.; Minafra, B.; Riboldazzi, G.; et al. Parkinson’s disease patients have a complex phenotypic and functional Th1 bias: Cross-sectional studies of CD4+ Th1/Th2/T17 and Treg in drug-naive and drug-treated patients. J. Neuroinflamm. 2018, 15, 205. [Google Scholar] [CrossRef]
- Panicker, N.; Sarkar, S.; Harischandra, D.S.; Neal, M.; Kam, T.I.; Jin, H.; Saminathan, H.; Langley, M.; Charli, A.; Samidurai, M.; et al. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J. Exp. Med. 2019, 216, 1411–1430. [Google Scholar] [CrossRef]
- Ferreira, S.A.; Romero-Ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell. Neurosci. 2018, 12, 247. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yoshikawa, E.; Sekine, Y.; Futatsubashi, M.; Kanno, T.; Ogusu, T.; Torizuka, T. Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann. Neurol. 2005, 57, 168–175. [Google Scholar] [CrossRef]
- Ouchi, Y.; Yagi, S.; Yokokura, M.; Sakamoto, M. Neuroinflammation in the living brain of Parkinson’s disease. Park. Relat. Disord. 2009, 15 (Suppl. 3), S200–S204. [Google Scholar] [CrossRef]
- Haque, M.E.; Akther, M.; Jakaria, M.; Kim, I.S.; Azam, S.; Choi, D.K. Targeting the microglial NLRP3 inflammasome and its role in Parkinson’s disease. Mov. Disord. 2020, 35, 20–33. [Google Scholar] [CrossRef]
- Labzin, L.I.; Heneka, M.T.; Latz, E. Innate Immunity and Neurodegeneration. Annu. Rev. Med. 2018, 69, 437–449. [Google Scholar] [CrossRef]
- Earls, R.H.; Menees, K.B.; Chung, J.; Barber, J.; Gutekunst, C.A.; Hazim, M.G.; Lee, J.K. Intrastriatal injection of preformed alpha-synuclein fibrils alters central and peripheral immune cell profiles in non-transgenic mice. J. Neuroinflamm. 2019, 16, 250. [Google Scholar] [CrossRef]
- Kuhbandner, K.; Hoffmann, A.; Gonzalez Alvarado, M.N.; Seyler, L.; Bauerle, T.; Winkler, J.; Linker, R.A. alpha-Synuclein: A Modulator During Inflammatory CNS Demyelination. J. Mol. Neurosci. 2020, 70, 1038–1049. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- McGeer, P.L.; McGeer, E.G. Inflammation and the degenerative diseases of aging. Ann. N. Y. Acad. Sci. 2004, 1035, 104–116. [Google Scholar] [CrossRef]
- Ambrosi, G.; Kustrimovic, N.; Siani, F.; Rasini, E.; Cerri, S.; Ghezzi, C.; Dicorato, G.; Caputo, S.; Marino, F.; Cosentino, M.; et al. Complex Changes in the Innate and Adaptive Immunity Accompany Progressive Degeneration of the Nigrostriatal Pathway Induced by Intrastriatal Injection of 6-Hydroxydopamine in the Rat. Neurotox. Res. 2017, 32, 71–81. [Google Scholar] [CrossRef]
- Singh, S.S.; Rai, S.N.; Birla, H.; Zahra, W.; Rathore, A.S.; Singh, S.P. NF-kappaB-Mediated Neuroinflammation in Parkinson’s Disease and Potential Therapeutic Effect of Polyphenols. Neurotox. Res. 2020, 37, 491–507. [Google Scholar] [CrossRef]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Hunot, S.; Boissiere, F.; Faucheux, B.; Brugg, B.; Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 1996, 72, 355–363. [Google Scholar] [CrossRef]
- Noelker, C.; Morel, L.; Lescot, T.; Osterloh, A.; Alvarez-Fischer, D.; Breloer, M.; Henze, C.; Depboylu, C.; Skrzydelski, D.; Michel, P.P.; et al. Toll like receptor 4 mediates cell death in a mouse MPTP model of Parkinson disease. Sci. Rep. 2013, 3, 1393. [Google Scholar] [CrossRef]
- Sommer, A.; Marxreiter, F.; Krach, F.; Fadler, T.; Grosch, J.; Maroni, M.; Graef, D.; Eberhardt, E.; Riemenschneider, M.J.; Yeo, G.W.; et al. Th17 Lymphocytes Induce Neuronal Cell Death in a Human iPSC-Based Model of Parkinson’s Disease. Cell Stem Cell 2018, 23, 123–131.e6. [Google Scholar] [CrossRef]
- Galiano-Landeira, J.; Torra, A.; Vila, M.; Bove, J. CD8 T cell nigral infiltration precedes synucleinopathy in early stages of Parkinson’s disease. Brain 2020, 143, 3717–3733. [Google Scholar] [CrossRef]
- Karikari, A.A.; McFleder, R.L.; Ribechini, E.; Blum, R.; Bruttel, V.; Knorr, S.; Gehmeyr, M.; Volkmann, J.; Brotchie, J.M.; Ahsan, F.; et al. Neurodegeneration by alpha-synuclein-specific T cells in AAV-A53T-alpha-synuclein Parkinson’s disease mice. Brain Behav. Immun. 2022, 101, 194–210. [Google Scholar] [CrossRef]
- Williams, G.P.; Schonhoff, A.M.; Jurkuvenaite, A.; Gallups, N.J.; Standaert, D.G.; Harms, A.S. CD4 T cells mediate brain inflammation and neurodegeneration in a mouse model of Parkinson’s disease. Brain 2021, 144, 2047–2059. [Google Scholar] [CrossRef]
- Heiss, C.N.; Olofsson, L.E. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J. Neuroendocrinol. 2019, 31, e12684. [Google Scholar] [CrossRef]
- Lahiri, S.; Kim, H.; Garcia-Perez, I.; Reza, M.M.; Martin, K.A.; Kundu, P.; Cox, L.M.; Selkrig, J.; Posma, J.M.; Zhang, H.; et al. The gut microbiota influences skeletal muscle mass and function in mice. Sci. Transl. Med. 2019, 11, eaan5662. [Google Scholar] [CrossRef]
- Schwiertz, A.; Spiegel, J.; Dillmann, U.; Grundmann, D.; Burmann, J.; Fassbender, K.; Schafer, K.H.; Unger, M.M. Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Park. Relat. Disord. 2018, 50, 104–107. [Google Scholar] [CrossRef]
- Patterson, A.M.; Mulder, I.E.; Travis, A.J.; Lan, A.; Cerf-Bensussan, N.; Gaboriau-Routhiau, V.; Garden, K.; Logan, E.; Delday, M.I.; Coutts, A.G.P.; et al. Human Gut Symbiont Roseburia hominis Promotes and Regulates Innate Immunity. Front. Immunol. 2017, 8, 1166. [Google Scholar] [CrossRef]
- Matheoud, D.; Cannon, T.; Voisin, A.; Penttinen, A.M.; Ramet, L.; Fahmy, A.M.; Ducrot, C.; Laplante, A.; Bourque, M.J.; Zhu, L.; et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1−/− mice. Nature 2019, 571, 565–569. [Google Scholar] [CrossRef]
- Kelly, L.P.; Carvey, P.M.; Keshavarzian, A.; Shannon, K.M.; Shaikh, M.; Bakay, R.A.; Kordower, J.H. Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov. Disord. 2014, 29, 999–1009. [Google Scholar] [CrossRef]
- Gorecki, A.M.; Preskey, L.; Bakeberg, M.C.; Kenna, J.E.; Gildenhuys, C.; MacDougall, G.; Dunlop, S.A.; Mastaglia, F.L.; Akkari, P.A.; Koengten, F.; et al. Altered Gut Microbiome in Parkinson’s Disease and the Influence of Lipopolysaccharide in a Human alpha-Synuclein Over-Expressing Mouse Model. Front. Neurosci. 2019, 13, 839. [Google Scholar] [CrossRef]
- Chen, S.G.; Stribinskis, V.; Rane, M.J.; Demuth, D.R.; Gozal, E.; Roberts, A.M.; Jagadapillai, R.; Liu, R.; Choe, K.; Shivakumar, B.; et al. Exposure to the Functional Bacterial Amyloid Protein Curli Enhances Alpha-Synuclein Aggregation in Aged Fischer 344 Rats and Caenorhabditis elegans. Sci. Rep. 2016, 6, 34477. [Google Scholar] [CrossRef]
- Challis, C.; Hori, A.; Sampson, T.R.; Yoo, B.B.; Challis, R.C.; Hamilton, A.M.; Mazmanian, S.K.; Volpicelli-Daley, L.A.; Gradinaru, V. Gut-seeded alpha-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat. Neurosci. 2020, 23, 327–336. [Google Scholar] [CrossRef]
- Kim, S.; Kwon, S.H.; Kam, T.I.; Panicker, N.; Karuppagounder, S.S.; Lee, S.; Lee, J.H.; Kim, W.R.; Kook, M.; Foss, C.A.; et al. Transneuronal Propagation of Pathologic alpha-Synuclein from the Gut to the Brain Models Parkinson’s Disease. Neuron 2019, 103, 627–641.e7. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamguney, G.; et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Arotcarena, M.L.; Dovero, S.; Prigent, A.; Bourdenx, M.; Camus, S.; Porras, G.; Thiolat, M.L.; Tasselli, M.; Aubert, P.; Kruse, N.; et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain 2020, 143, 1462–1475. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W. Neuropathology of Parkinson disease. Park. Relat. Disord. 2018, 46 (Suppl. 1), S30–S33. [Google Scholar] [CrossRef]
- Schober, A. Classic toxin-induced animal models of Parkinson’s disease: 6-OHDA and MPTP. Cell Tissue Res. 2004, 318, 215–224. [Google Scholar] [CrossRef]
- Vandamme, T.F. Use of rodents as models of human diseases. J. Pharm. Bioallied Sci. 2014, 6, 2–9. [Google Scholar] [CrossRef]
- Ding, F.; Luan, L.; Ai, Y.; Walton, A.; Gerhardt, G.A.; Gash, D.M.; Grondin, R.; Zhang, Z. Development of a stable, early stage unilateral model of Parkinson’s disease in middle-aged rhesus monkeys. Exp. Neurol. 2008, 212, 431–439. [Google Scholar] [CrossRef]
- Kuwahara, T.; Koyama, A.; Gengyo-Ando, K.; Masuda, M.; Kowa, H.; Tsunoda, M.; Mitani, S.; Iwatsubo, T. Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J. Biol. Chem. 2006, 281, 334–340. [Google Scholar] [CrossRef]
- Lakso, M.; Vartiainen, S.; Moilanen, A.M.; Sirvio, J.; Thomas, J.H.; Nass, R.; Blakely, R.D.; Wong, G. Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J. Neurochem. 2003, 86, 165–172. [Google Scholar] [CrossRef]
- Chesselet, M.F. In vivo alpha-synuclein overexpression in rodents: A useful model of Parkinson’s disease? Exp. Neurol. 2008, 209, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Dawson, T.; Mandir, A.; Lee, M. Animal models of PD: Pieces of the same puzzle? Neuron 2002, 35, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Quinn, S.M.; Zhang, B.; Trojanowski, J.Q.; Lee, V.M. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 2002, 34, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.K.; Stirling, W.; Xu, Y.; Xu, X.; Qui, D.; Mandir, A.S.; Dawson, T.M.; Copeland, N.G.; Jenkins, N.A.; Price, D.L. Human alpha-synuclein-harboring familial Parkinson’s disease-linked Ala-53 --> Thr mutation causes neurodegenerative disease with alpha-synuclein aggregation in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 8968–8973. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Falkenberg, M.; Rantanen, A.; Park, C.B.; Gaspari, M.; Hultenby, K.; Rustin, P.; Gustafsson, C.M.; Larsson, N.G. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 2004, 13, 935–944. [Google Scholar] [CrossRef]
- Grunewald, A.; Kumar, K.R.; Sue, C.M. New insights into the complex role of mitochondria in Parkinson’s disease. Prog. Neurobiol. 2019, 177, 73–93. [Google Scholar] [CrossRef]
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.M.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J. Exp. Med. 2012, 209, 975–986. [Google Scholar] [CrossRef]
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef]
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V.; et al. α-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98. [Google Scholar] [CrossRef] [PubMed]
- PD MED Collaborative Group. Long-term effectiveness of dopamine agonists and monoamine oxidase B inhibitors compared with levodopa as initial treatment for Parkinson’s disease (PD MED): A large, open-label, pragmatic randomised trial. Lancet 2014, 384, 1196–1205. [Google Scholar] [CrossRef] [PubMed]
- LeWitt, P.A.; Fahn, S. Levodopa therapy for Parkinson disease: A look backward and forward. Neurology 2016, 86 (Suppl. 1), S3–S12. [Google Scholar] [CrossRef]
- Olanow, C.W.; Obeso, J.A.; Stocchi, F. Continuous dopamine-receptor treatment of Parkinson’s disease: Scientific rationale and clinical implications. Lancet Neurol. 2006, 5, 677–687. [Google Scholar] [CrossRef]
- Cenci, M.A. Presynaptic Mechanisms of l-DOPA-Induced Dyskinesia: The Findings, the Debate, and the Therapeutic Implications. Front. Neurol. 2014, 5, 242. [Google Scholar] [CrossRef]
- Poewe, W.; Antonini, A. Novel formulations and modes of delivery of levodopa. Mov. Disord. 2015, 30, 114–120. [Google Scholar] [CrossRef]
- Fox, S.H.; Katzenschlager, R.; Lim, S.Y.; Barton, B.; de Bie, R.M.A.; Seppi, K.; Coelho, M.; Sampaio, C.; Movement Disorder Society Evidence-Based Medicine Committee. International Parkinson and movement disorder society evidence-based medicine review: Update on treatments for the motor symptoms of Parkinson’s disease. Mov. Disord. 2018, 33, 1248–1266. [Google Scholar] [CrossRef]
- Connolly, B.S.; Lang, A.E. Pharmacological treatment of Parkinson disease: A review. JAMA 2014, 311, 1670–1683. [Google Scholar] [CrossRef]
- Seppi, K.; Weintraub, D.; Coelho, M.; Perez-Lloret, S.; Fox, S.H.; Katzenschlager, R.; Hametner, E.M.; Poewe, W.; Rascol, O.; Goetz, C.G.; et al. The Movement Disorder Society Evidence-Based Medicine Review Update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov. Disord. 2011, 26 (Suppl. 3), S42–S80. [Google Scholar] [CrossRef]
- Jankovic, J.; Poewe, W. Therapies in Parkinson’s disease. Curr. Opin. Neurol. 2012, 25, 433–447. [Google Scholar] [CrossRef] [PubMed]
- Voon, V.; Mehta, A.R.; Hallett, M. Impulse control disorders in Parkinson’s disease: Recent advances. Curr. Opin. Neurol. 2011, 24, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Frankel, J.P.; Lees, A.J.; Kempster, P.A.; Stern, G.M. Subcutaneous apomorphine in the treatment of Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 1990, 53, 96–101. [Google Scholar] [CrossRef]
- Katzenschlager, R.; Hughes, A.; Evans, A.; Manson, A.J.; Hoffman, M.; Swinn, L.; Watt, H.; Bhatia, K.; Quinn, N.; Lees, A.J. Continuous subcutaneous apomorphine therapy improves dyskinesias in Parkinson’s disease: A prospective study using single-dose challenges. Mov. Disord. 2005, 20, 151–157. [Google Scholar] [CrossRef]
- Hauser, R.A.; Olanow, C.W.; Dzyngel, B.; Bilbault, T.; Shill, H.; Isaacson, S.; Dubow, J.; Agro, A. Sublingual apomorphine (APL-130277) for the acute conversion of OFF to ON in Parkinson’s disease. Mov. Disord. 2016, 31, 1366–1372. [Google Scholar] [CrossRef]
- Muller, T. Catechol-O-methyltransferase inhibitors in Parkinson’s disease. Drugs 2015, 75, 157–174. [Google Scholar] [CrossRef]
- Ferreira, J.J.; Lees, A.; Rocha, J.F.; Poewe, W.; Rascol, O.; Soares-da-Silva, P.; Bi-Park 1 Investigators. Opicapone as an adjunct to levodopa in patients with Parkinson’s disease and end-of-dose motor fluctuations: A randomised, double-blind, controlled trial. Lancet Neurol. 2016, 15, 154–165. [Google Scholar] [CrossRef]
- Schapira, A.H. Monoamine oxidase B inhibitors for the treatment of Parkinson’s disease: A review of symptomatic and potential disease-modifying effects. CNS Drugs 2011, 25, 1061–1071. [Google Scholar] [CrossRef]
- Birkmayer, W.; Riederer, P.; Ambrozi, L.; Youdim, M.B. Implications of combined treatment with ‘Madopar’ and L-deprenil in Parkinson’s disease. A long-term study. Lancet 1977, 1, 439–443. [Google Scholar] [CrossRef]
- Schapira, A.H.; Fox, S.H.; Hauser, R.A.; Jankovic, J.; Jost, W.H.; Kenney, C.; Kulisevsky, J.; Pahwa, R.; Poewe, W.; Anand, R. Assessment of Safety and Efficacy of Safinamide as a Levodopa Adjunct in Patients with Parkinson Disease and Motor Fluctuations: A Randomized Clinical Trial. JAMA Neurol. 2017, 74, 216–224. [Google Scholar] [CrossRef]
- Chaudhuri, K.R.; Schapira, A.H. Non-motor symptoms of Parkinson’s disease: Dopaminergic pathophysiology and treatment. Lancet Neurol. 2009, 8, 464–474. [Google Scholar] [CrossRef] [PubMed]
- Connolly, B.; Fox, S.H. Treatment of cognitive, psychiatric, and affective disorders associated with Parkinson’s disease. Neurotherapeutics 2014, 11, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lloret, S.; Rey, M.V.; Pavy-Le Traon, A.; Rascol, O. Emerging drugs for autonomic dysfunction in Parkinson’s disease. Expert Opin. Emerg. Drugs 2013, 18, 39–53. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, R.; Werth, J.; Zhang, Y.; Duvvuri, S.; Gray, D. PF-06649751 efficacy and safety in early Parkinson’s disease: A randomized, placebo-controlled trial. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420911296. [Google Scholar] [CrossRef]
- Svenningsson, P.; Johansson, A.; Nyholm, D.; Tsitsi, P.; Hansson, F.; Sonesson, C.; Tedroff, J. Safety and tolerability of IRL790 in Parkinson’s disease with levodopa-induced dyskinesia-a phase 1b trial. npj Park. Dis. 2018, 4, 35. [Google Scholar] [CrossRef]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garcon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef]
- Hung, L.W.; Villemagne, V.L.; Cheng, L.; Sherratt, N.A.; Ayton, S.; White, A.R.; Crouch, P.J.; Lim, S.; Leong, S.L.; Wilkins, S.; et al. The hypoxia imaging agent CuII(atsm) is neuroprotective and improves motor and cognitive functions in multiple animal models of Parkinson’s disease. J. Exp. Med. 2012, 209, 837–854. [Google Scholar] [CrossRef]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Yang, J.; Song, S.; Li, J.; Liang, T. Neuroprotective effect of curcumin on hippocampal injury in 6-OHDA-induced Parkinson’s disease rat. Pathol. Res. Pract. 2014, 210, 357–362. [Google Scholar] [CrossRef]
- Baj, T.; Seth, R. Role of Curcumin in Regulation of TNF-alpha Mediated Brain Inflammatory Responses. Recent Pat. Inflamm. Allergy Drug Discov. 2018, 12, 69–77. [Google Scholar] [CrossRef] [PubMed]
- El Nebrisi, E.; Javed, H.; Ojha, S.K.; Oz, M.; Shehab, S. Neuroprotective Effect of Curcumin on the Nigrostriatal Pathway in a 6-Hydroxydopmine-Induced Rat Model of Parkinson’s Disease is Mediated by alpha7-Nicotinic Receptors. Int. J. Mol. Sci. 2020, 21, 7329. [Google Scholar] [CrossRef] [PubMed]
- Cretu, E.; Trifan, A.; Vasincu, A.; Miron, A. Plant-derived anticancer agents—Curcumin in cancer prevention and treatment. Rev. Med. Chir. Soc. Med. Nat. Iasi 2012, 116, 1223–1229. [Google Scholar] [PubMed]
- Ghasemi, F.; Bagheri, H.; Barreto, G.E.; Read, M.I.; Sahebkar, A. Effects of Curcumin on Microglial Cells. Neurotox. Res. 2019, 36, 12–26. [Google Scholar] [CrossRef]
- Fan, C.; Song, Q.; Wang, P.; Li, Y.; Yang, M.; Yu, S.Y. Neuroprotective Effects of Curcumin on IL-1beta-Induced Neuronal Apoptosis and Depression-like Behaviors Caused by Chronic Stress in Rats. Front. Cell. Neurosci. 2018, 12, 516. [Google Scholar]
- Ghosh, S.; Banerjee, S.; Sil, P.C. The beneficial role of curcumin on inflammation, diabetes and neurodegenerative disease: A recent update. Food Chem. Toxicol. 2015, 83, 111–124. [Google Scholar] [CrossRef]
- Gupta, S.C.; Patchva, S.; Koh, W.; Aggarwal, B.B. Discovery of curcumin, a component of golden spice, and its miraculous biological activities. Clin. Exp. Pharmacol. Physiol. 2012, 39, 283–299. [Google Scholar] [CrossRef]
- Sharma, N.; Nehru, B. Curcumin affords neuroprotection and inhibits alpha-synuclein aggregation in lipopolysaccharide-induced Parkinson’s disease model. Inflammopharmacology 2018, 26, 349–360. [Google Scholar] [CrossRef]
- Albani, D.; Polito, L.; Batelli, S.; De Mauro, S.; Fracasso, C.; Martelli, G.; Colombo, L.; Manzoni, C.; Salmona, M.; Caccia, S.; et al. The SIRT1 activator resveratrol protects SK-N-BE cells from oxidative stress and against toxicity caused by alpha-synuclein or amyloid-beta (1-42) peptide. J. Neurochem. 2009, 110, 1445–1456. [Google Scholar] [CrossRef]
- Zhang, J.; Fan, W.; Wang, H.; Bao, L.; Li, G.; Li, T.; Song, S.; Li, H.; Hao, J.; Sun, J. Resveratrol Protects PC12 Cell against 6-OHDA Damage via CXCR4 Signaling Pathway. Evid.-Based Complement. Altern. Med. 2015, 2015, 730121. [Google Scholar] [CrossRef]
- Zeng, W.; Zhang, W.; Lu, F.; Gao, L.; Gao, G. Resveratrol attenuates MPP+-induced mitochondrial dysfunction and cell apoptosis via AKT/GSK-3beta pathway in SN4741 cells. Neurosci. Lett. 2017, 637, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dong, X.; Liu, Z.; Zhu, S.; Liu, H.; Fan, W.; Hu, Y.; Hu, T.; Yu, Y.; Li, Y.; et al. Resveratrol Suppresses Rotenone-induced Neurotoxicity Through Activation of SIRT1/Akt1 Signaling Pathway. Anat. Rec. 2018, 301, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Zhang, Y.; Chen, M.; Jin, H.; Nie, J.; Luo, Y.; Zhou, S.; Shi, J.; Jin, F. Resveratrol delays 6-hydroxydopamine-induced apoptosis by activating the PI3K/Akt signaling pathway. Exp. Gerontol. 2019, 124, 110653. [Google Scholar] [CrossRef] [PubMed]
- Abolaji, A.O.; Adedara, A.O.; Adie, M.A.; Vicente-Crespo, M.; Farombi, E.O. Resveratrol prolongs lifespan and improves 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced oxidative damage and behavioural deficits in Drosophila melanogaster. Biochem. Biophys. Res. Commun. 2018, 503, 1042–1048. [Google Scholar] [CrossRef]
- Lin, K.L.; Lin, K.J.; Wang, P.W.; Chuang, J.H.; Lin, H.Y.; Chen, S.D.; Chuang, Y.C.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol provides neuroprotective effects through modulation of mitochondrial dynamics and ERK1/2 regulated autophagy. Free Radic. Res. 2018, 52, 1371–1386. [Google Scholar] [CrossRef]
- Palle, S.; Neerati, P. Improved neuroprotective effect of resveratrol nanoparticles as evinced by abrogation of rotenone-induced behavioral deficits and oxidative and mitochondrial dysfunctions in rat model of Parkinson’s disease. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 445–453. [Google Scholar] [CrossRef]
- Peng, K.; Tao, Y.; Zhang, J.; Wang, J.; Ye, F.; Dan, G.; Zhao, Y.; Cai, Y.; Zhao, J.; Wu, Q.; et al. Resveratrol Regulates Mitochondrial Biogenesis and Fission/Fusion to Attenuate Rotenone-Induced Neurotoxicity. Oxidative Med. Cell. Longev. 2016, 2016, 6705621. [Google Scholar] [CrossRef]
- Guo, Y.J.; Dong, S.Y.; Cui, X.X.; Feng, Y.; Liu, T.; Yin, M.; Kuo, S.H.; Tan, E.K.; Zhao, W.J.; Wu, Y.C. Resveratrol alleviates MPTP-induced motor impairments and pathological changes by autophagic degradation of alpha-synuclein via SIRT1-deacetylated LC3. Mol. Nutr. Food Res. 2016, 60, 2161–2175. [Google Scholar] [CrossRef]
- Liu, Q.; Zhu, D.; Jiang, P.; Tang, X.; Lang, Q.; Yu, Q.; Zhang, S.; Che, Y.; Feng, X. Resveratrol synergizes with low doses of L-DOPA to improve MPTP-induced Parkinson disease in mice. Behav. Brain Res. 2019, 367, 10–18. [Google Scholar] [CrossRef]
- Lin, T.K.; Chen, S.D.; Chuang, Y.C.; Lin, H.Y.; Huang, C.R.; Chuang, J.H.; Wang, P.W.; Huang, S.T.; Tiao, M.M.; Chen, J.B.; et al. Resveratrol partially prevents rotenone-induced neurotoxicity in dopaminergic SH-SY5Y cells through induction of heme oxygenase-1 dependent autophagy. Int. J. Mol. Sci. 2014, 15, 1625–1646. [Google Scholar] [CrossRef]
- Deng, H.; Ma, Z. Protective effects of berberine against MPTP-induced dopaminergic neuron injury through promoting autophagy in mice. Food Funct. 2021, 12, 8366–8375. [Google Scholar] [CrossRef] [PubMed]
- Dulovic, M.; Jovanovic, M.; Xilouri, M.; Stefanis, L.; Harhaji-Trajkovic, L.; Kravic-Stevovic, T.; Paunovic, V.; Ardah, M.T.; El-Agnaf, O.M.; Kostic, V.; et al. The protective role of AMP-activated protein kinase in alpha-synuclein neurotoxicity in vitro. Neurobiol. Dis. 2014, 63, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Liu, H.; Lin, Y.; Liu, M.; Li, Y.; Mao, H.; Zhang, Z.; Zhang, Y.; Ye, P.; Ding, L.; et al. Berberine Protects Against NLRP3 Inflammasome via Ameliorating Autophagic Impairment in MPTP-Induced Parkinson’s Disease Model. Front. Pharmacol. 2020, 11, 618787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tong, Q.; Ma, S.R.; Zhao, Z.X.; Pan, L.B.; Cong, L.; Han, P.; Peng, R.; Yu, H.; Lin, Y.; et al. Oral berberine improves brain dopa/dopamine levels to ameliorate Parkinson’s disease by regulating gut microbiota. Signal Transduct. Target. Ther. 2021, 6, 77. [Google Scholar] [CrossRef]
- Hou, Y.S.; Guan, J.J.; Xu, H.D.; Wu, F.; Sheng, R.; Qin, Z.H. Sestrin2 Protects Dopaminergic Cells against Rotenone Toxicity through AMPK-Dependent Autophagy Activation. Mol. Cell. Biol. 2015, 35, 2740–2751. [Google Scholar] [CrossRef]
- Lu, J.H.; Tan, J.Q.; Durairajan, S.S.; Liu, L.F.; Zhang, Z.H.; Ma, L.; Shen, H.M.; Chan, H.Y.; Li, M. Isorhynchophylline, a natural alkaloid, promotes the degradation of alpha-synuclein in neuronal cells via inducing autophagy. Autophagy 2012, 8, 98–108. [Google Scholar] [CrossRef]
- Li, X.M.; Zhang, X.J.; Dong, M.X. Isorhynchophylline Attenuates MPP+-Induced Apoptosis Through Endoplasmic Reticulum Stress- and Mitochondria-Dependent Pathways in PC12 Cells: Involvement of Antioxidant Activity. Neuromol. Med. 2017, 19, 480–492. [Google Scholar] [CrossRef]
- Hu, X.; Weng, Z.; Chu, C.T.; Zhang, L.; Cao, G.; Gao, Y.; Signore, A.; Zhu, J.; Hastings, T.; Greenamyre, J.T.; et al. Peroxiredoxin-2 protects against 6-hydroxydopamine-induced dopaminergic neurodegeneration via attenuation of the apoptosis signal-regulating kinase (ASK1) signaling cascade. J. Neurosci. 2011, 31, 247–261. [Google Scholar] [CrossRef]
- Kim, S.M.; Chung, M.J.; Ha, T.J.; Choi, H.N.; Jang, S.J.; Kim, S.O.; Chun, M.H.; Do, S.I.; Choo, Y.K.; Park, Y.I. Neuroprotective effects of black soybean anthocyanins via inactivation of ASK1-JNK/p38 pathways and mobilization of cellular sialic acids. Life Sci. 2012, 90, 874–882. [Google Scholar] [CrossRef]
- Lee, K.W.; Zhao, X.; Im, J.Y.; Grosso, H.; Jang, W.H.; Chan, T.W.; Sonsalla, P.K.; German, D.C.; Ichijo, H.; Junn, E.; et al. Apoptosis signal-regulating kinase 1 mediates MPTP toxicity and regulates glial activation. PLoS ONE 2012, 7, e29935. [Google Scholar] [CrossRef]
- Zhao, D.; Wu, Y.; Zhuang, J.; Xu, C.; Zhang, F. Activation of NLRP1 and NLRP3 inflammasomes contributed to cyclic stretch-induced pyroptosis and release of IL-1beta in human periodontal ligament cells. Oncotarget 2016, 7, 68292–68302. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.; Zhang, X.; Dong, M. Puerarin suppresses MPP+/MPTP-induced oxidative stress through an Nrf2-dependent mechanism. Food Chem. Toxicol. 2020, 144, 111644. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Qu, X.; Jin, G.; Nie, F.; Liu, F.; Chen, S.; Hu, F. Puerarin promoted proliferation and differentiation of dopamine-producing cells in Parkinson’s animal models. Biomed. Pharmacother. 2018, 106, 1236–1242. [Google Scholar]
- Zhao, Y.; Zhao, J.; Zhang, X.; Cheng, Y.; Luo, D.; Lee, S.M.; Lao, L.; Rong, J. Botanical Drug Puerarin Promotes Neuronal Survival and Neurite Outgrowth against MPTP/MPP+-Induced Toxicity via Progesterone Receptor Signaling. Oxidative Med. Cell. Longev. 2020, 2020, 7635291. [Google Scholar] [CrossRef]
- Ji, Y.; Wang, D.; Zhang, B.; Lu, H. Bergenin Ameliorates MPTP-Induced Parkinson’s Disease by Activating PI3K/Akt Signaling Pathway. J. Alzheimer’s Dis. 2019, 72, 823–833. [Google Scholar] [CrossRef]
- Luo, S.; Kang, S.S.; Wang, Z.H.; Liu, X.; Day, J.X.; Wu, Z.; Peng, J.; Xiang, D.; Springer, W.; Ye, K. Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing Its Antioxidative Activities in Parkinson’s Disease. J. Neurosci. 2019, 39, 7291–7305. [Google Scholar] [CrossRef]
- Yang, L.; Wang, H.; Liu, L.; Xie, A. The Role of Insulin/IGF-1/PI3K/Akt/GSK3beta Signaling in Parkinson’s Disease Dementia. Front. Neurosci. 2018, 12, 73. [Google Scholar] [CrossRef]
- Zhu, G.; Wang, X.; Wu, S.; Li, Q. Involvement of activation of PI3K/Akt pathway in the protective effects of puerarin against MPP+-induced human neuroblastoma SH-SY5Y cell death. Neurochem. Int. 2012, 60, 400–408. [Google Scholar] [CrossRef]
- Rui, W.; Li, S.; Xiao, H.; Xiao, M.; Shi, J. Baicalein Attenuates Neuroinflammation by Inhibiting NLRP3/caspase-1/GSDMD Pathway in MPTP Induced Mice Model of Parkinson’s Disease. Int. J. Neuropsychopharmacol. 2020, 23, 762–773. [Google Scholar] [CrossRef]
- Cookson, B.T.; Brennan, M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001, 9, 113–114. [Google Scholar] [CrossRef]
- Bergsbaken, T.; Fink, S.L.; Cookson, B.T. Pyroptosis: Host cell death and inflammation. Nat. Rev. Microbiol. 2009, 7, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Gordon, R.; Albornoz, E.A.; Christie, D.C.; Langley, M.R.; Kumar, V.; Mantovani, S.; Robertson, A.A.B.; Butler, M.S.; Rowe, D.B.; O’Neill, L.A.; et al. Inflammasome inhibition prevents alpha-synuclein pathology and dopaminergic neurodegeneration in mice. Sci. Transl. Med. 2018, 10, eaah4066. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Munoz-Planillo, R.; Nunez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; He, W.T.; Hu, L.; Li, J.; Fang, Y.; Wang, X.; Xu, X.; Wang, Z.; Huang, K.; Han, J. Pyroptosis is driven by non-selective gasdermin-D pore and its morphology is different from MLKL channel-mediated necroptosis. Cell Res. 2016, 26, 1007–1020. [Google Scholar] [CrossRef]
- Zhao, X.; Kong, D.; Zhou, Q.; Wei, G.; Song, J.; Liang, Y.; Du, G. Baicalein alleviates depression-like behavior in rotenone- induced Parkinson’s disease model in mice through activating the BDNF/TrkB/CREB pathway. Biomed. Pharmacother. 2021, 140, 111556. [Google Scholar] [CrossRef]
- Zheng, Z.V.; Cheung, C.Y.; Lyu, H.; Chan, H.Y.; Li, Y.; Bian, Z.X.; Wang, K.K.W.; Poon, W.S. Baicalein enhances the effect of low dose Levodopa on the gait deficits and protects dopaminergic neurons in experimental Parkinsonism. J. Clin. Neurosci. 2019, 64, 242–251. [Google Scholar] [CrossRef]
- Song, J.X.; Choi, M.Y.; Wong, K.C.; Chung, W.W.; Sze, S.C.; Ng, T.B.; Zhang, K.Y. Baicalein antagonizes rotenone-induced apoptosis in dopaminergic SH-SY5Y cells related to Parkinsonism. Chin. Med. 2012, 7, 1. [Google Scholar] [CrossRef]
- Zhang, C.; Zhao, M.; Wang, B.; Su, Z.; Guo, B.; Qin, L.; Zhang, W.; Zheng, R. The Nrf2-NLRP3-caspase-1 axis mediates the neuroprotective effects of Celastrol in Parkinson’s disease. Redox Biol. 2021, 47, 102134. [Google Scholar] [CrossRef]
- Lin, M.W.; Lin, C.C.; Chen, Y.H.; Yang, H.B.; Hung, S.Y. Celastrol Inhibits Dopaminergic Neuronal Death of Parkinson’s Disease through Activating Mitophagy. Antioxidants 2019, 9, 37. [Google Scholar] [CrossRef]
- Feng, Y.; Zheng, C.; Zhang, Y.; Xing, C.; Cai, W.; Li, R.; Chen, J.; Duan, Y. Triptolide Inhibits Preformed Fibril-Induced Microglial Activation by Targeting the MicroRNA155-5p/SHIP1 Pathway. Oxidative Med. Cell. Longev. 2019, 2019, 6527638. [Google Scholar] [CrossRef]
- Lu, S.; Liao, Q.S.; Tang, L. MiR-155 affects osteosarcoma cell proliferation and invasion through regulating NF-kappaB signaling pathway. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 7633–7639. [Google Scholar] [PubMed]
- Huang, Y.Y.; Zhang, Q.; Zhang, J.N.; Zhang, Y.N.; Gu, L.; Yang, H.M.; Xia, N.; Wang, X.M.; Zhang, H. Triptolide up-regulates metabotropic glutamate receptor 5 to inhibit microglia activation in the lipopolysaccharide-induced model of Parkinson’s disease. Brain Behav. Immun. 2018, 71, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gong, X.; Wang, L.; Liu, M.; Liu, Y.; Fu, X.; Wang, W.; Zhang, T.; Wang, X. Triptolide Promotes the Clearance of alpha-Synuclein by Enhancing Autophagy in Neuronal Cells. Mol. Neurobiol. 2017, 54, 2361–2372. [Google Scholar] [CrossRef] [PubMed]
- Qu, S.; Meng, X.; Liu, Y.; Zhang, X.; Zhang, Y. Ginsenoside Rb1 prevents MPTP-induced changes in hippocampal memory via regulation of the alpha-synuclein/PSD-95 pathway. Aging 2019, 11, 1934–1964. [Google Scholar] [CrossRef]
- Li, D.W.; Zhou, F.Z.; Sun, X.C.; Li, S.C.; Yang, J.B.; Sun, H.H.; Wang, A.H. Ginsenoside Rb1 protects dopaminergic neurons from inflammatory injury induced by intranigral lipopolysaccharide injection. Neural Regen. Res. 2019, 14, 1814–1822. [Google Scholar]
- Han, Y.; Wang, T.; Li, C.; Wang, Z.; Zhao, Y.; He, J.; Fu, L.; Han, B. Ginsenoside Rg3 exerts a neuroprotective effect in rotenone-induced Parkinson’s disease mice via its anti-oxidative properties. Eur. J. Pharmacol. 2021, 909, 174413. [Google Scholar] [CrossRef]
- Schweitzer, J.S.; Song, B.; Herrington, T.M.; Park, T.Y.; Lee, N.; Ko, S.; Jeon, J.; Cha, Y.; Kim, K.; Li, Q.; et al. Personalized iPSC-Derived Dopamine Progenitor Cells for Parkinson’s Disease. N. Engl. J. Med. 2020, 382, 1926–1932. [Google Scholar] [CrossRef]
- Zhu, Y.; Ouyang, Z.; Du, H.; Wang, M.; Wang, J.; Sun, H.; Kong, L.; Xu, Q.; Ma, H.; Sun, Y. New opportunities and challenges of natural products research: When target identification meets single-cell multiomics. Acta Pharm. Sin. B 2022, 12, 4011–4039. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanism | Role in PD Pathology | Effects |
|---|---|---|
| α-Synuclein Aggregation | Misfolded α-synuclein forms toxic oligomers and fibrils, accumulating as Lewy bodies in dopaminergic neurons | - Disrupts synaptic function - Impairs protein degradation - Spreads prion-like |
| Oxidative Stress (OS) | Excess ROS from mitochondrial impairment, dopamine metabolism, and inflammation | - Lipid peroxidation - DNA/protein damage - α-Synuclein misfolding |
| Ferroptosis | Iron-dependent, lipid-peroxide-driven cell death pathway observed in dopaminergic neuron degeneration | - Increased iron accumulation - GPX4 depletion - Membrane lipid peroxidation |
| Mitochondrial Dysfunction | Impaired complex I activity in the electron transport chain leads to energy failure and ROS generation | - ATP depletion - Increased oxidative stress - Cytochrome c release and apoptosis |
| Neuroinflammation | Activation of microglia and astrocytes promotes chronic inflammation and release of pro-inflammatory cytokines | - TNF-α, IL-1β release - Increased BBB permeability - Further neuron damage |
| Gut Dysbiosis | Altered microbiota composition affects gut-brain axis, promoting inflammation and α-synuclein aggregation | - Increased intestinal permeability - Elevated endotoxins (LPS) - Immune activation |
| Class | Drug | Therapeutic Applications |
|---|---|---|
| L-DOPA preparation | L-DOPA/benserazide tablet Carbidopa/L-DOPA tablet Carbidopa/L-DOPA controlled-release tablet | Parkinson’s syndrome Parkinson’s syndrome Parkinson’s syndrome, wearing-off, dyskinesia |
| DA agonists | Pramipexole tablet Ropinirole tablet Piribedil Transdermal rotigotine Injected apomorphine | Parkinson’s early syndrome, L-DOPA adjunct, wearing-off, dyskinesia Parkinson’s early syndrome, L-DOPA adjunct, wearing-off, dyskinesia Tremor, DA adjunct Parkinson’s early syndrome, L-DOPA adjunct, wearing-off, dyskinesia Wearing-off, L-DOPA-induced dyskinesias |
| N-methyl-D-aspartate receptor antagonist | Amantadine | Parkinson’s early syndrome, L-DOPA adjunct |
| Adenosine A2a receptor antagonists | Istradefylline | Wearing-off |
| Others | Clozapine | Dyskinesia |
| Anticholinergics | Benztropine Trihexyphenidy | Parkinson’s early syndrome, L-DOPA adjunct Parkinson’s early syndrome, L-DOPA adjunct |
| COMT inhibitors | Entacapone Opicapone Tolcapone | Wearing-off, dyskinesia Wearing-off, dyskinesia Wearing-off |
| MAO-B inhibitors | Selegiline Rasagiline Safinamide Zonisamide | Parkinson’s early syndrome, wearing-off, dyskinesia Parkinson’s early syndrome, L-DOPA adjunct, Wearing-off, Dyskinesia Wearing-off, Dyskinesia Wearing-off |
| Therapeutic Strategy | Name | Target and Classification |
|---|---|---|
| DA receptor agonists | PF-06649751/CVL 751/Tavapadon PF-06669571 PF-06412562 KDT-3594/AM-006 Lu-AF28996 | Small molecular DA D1/D5 agonist Small molecular DA D1 agonist Small molecular DA D1 agonist Small molecular DA agonist Small molecular DA D1/D2 agonist |
| Anti-α-synuclein aggregation therapy | Prasinezumab/PRX002/RO7046015 MEDI-1341/TAK-341 Lu AF82422 UCB7853 UCB 0599 Kenterin/Enterin-01 Ambroxol | Monoclonal antibody Monoclonal antibody Monoclonal antibody Monoclonal antibody Small molecular SNCA antagonists Small molecular SNCA antagonist Small molecular decrease in the cerebrospinal fluid α-synuclein level |
| Targeting ferroptosis | Cu(II)ATSM | Small molecular Peroxynitrite scavenger |
| Serotonin receptor agonists or antagonists | Landipirdine/SYN120/RO-5025181 SEP-363856 | Small molecular dual 5-HT6/5-HT2 antagonist Small molecular 5-HT1A agonist |
| Others | CNM-Au8 NLY01/NLY01-AD | Small molecular Small molecular GLP1R agonist |
| Gene therapy | AAV2-GDNF LY3884961/PR001A | AAV2-GDNF delivered to the putamen Glucocerebrosidase gene therapy by intra cisterna magna administration |
| Cell-based therapy | NTCELL ISC-hpNSC ANGE-S003 | Immunoprotected (alginate-encapsulated) porcine choroid pplexus cells Neural stem cells Neural stem cell |
| Botanical-based medication | DA 9805 Hypoestoxide WIN-1001X | Natural compounds Plant-based herbal dry powder Plant-based herbal extract |
| Kinase inhibitors | SUN-K706/Vodobatinib/SCC138/K0706 Nilotinib/Tasigna/AMN-107 Radotinib Dihydrochloride/IY-5511 BIIB-122/DNL151 DNL-201 | Small molecular Bcr-Abl antagonist Small molecular Bcr-Abl antagonist Small molecular Bcr-Abl antagonist Small molecular LRRK2 antagonist Small molecular LRRK2 antagonist |
| Muscarinic and nicotinic acetylcholine receptor agonists | Blarcamesine/AF710B/ANAVEX 2-73 | Small molecular Muscarinic acetylcholine receptor M1 agonist |
| Acetylcholinesterase antagonists | Buntanetap/ANVS-401 | Small molecular AchE antagonist/TAU antagonist |
| Adenosine A2a receptor antagonists | KW-6356 Caffeine | Small molecular adenosine A2A receptor antagonist Small molecular selective adenosine A2A antagonist |
| N-methyl-D-aspartate receptor (NMDAR) modulators | NBTX 001 NYX-458 DAAOI-P | Small molecular NMDAR modulato Small molecular NMDAR modulator Small molecular D-amino acid oxidase inhibitor |
| Class | Name | Targets |
|---|---|---|
| Phenol | Curcumin Resveratro | Trk/PI3K, JNK PI3K/Akt, SIRT 1, MAPK |
| Saponin | Ginsenoside Rb1 Ginsenoside Rg3 | NF-κB NF-κB |
| Terpenoid | Celastrol | MAPK, Nrf2-NLRP3-caspase-1 |
| Flavonoid | Puerarin Baicalein | Fyn/GSK-3β, PI3K/Akt NLRP3/caspase-1/gasdermin D, BDNF/TrkB/CREB |
| Alkaloid | Berberine Isorhynchophylline | AMPK ASK1/JNK |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.S.; Nasiripour, S.; Bopassa, J.C. Parkinson Disease Signaling Pathways, Molecular Mechanisms, and Potential Therapeutic Strategies: A Comprehensive Review. Int. J. Mol. Sci. 2025, 26, 6416. https://doi.org/10.3390/ijms26136416
Khan MS, Nasiripour S, Bopassa JC. Parkinson Disease Signaling Pathways, Molecular Mechanisms, and Potential Therapeutic Strategies: A Comprehensive Review. International Journal of Molecular Sciences. 2025; 26(13):6416. https://doi.org/10.3390/ijms26136416
Chicago/Turabian StyleKhan, Muhammad S., Somayyeh Nasiripour, and Jean C. Bopassa. 2025. "Parkinson Disease Signaling Pathways, Molecular Mechanisms, and Potential Therapeutic Strategies: A Comprehensive Review" International Journal of Molecular Sciences 26, no. 13: 6416. https://doi.org/10.3390/ijms26136416
APA StyleKhan, M. S., Nasiripour, S., & Bopassa, J. C. (2025). Parkinson Disease Signaling Pathways, Molecular Mechanisms, and Potential Therapeutic Strategies: A Comprehensive Review. International Journal of Molecular Sciences, 26(13), 6416. https://doi.org/10.3390/ijms26136416