
Heat Shock Protein and Disaggregase Influencing the Casein Structuralisation


Abstract
1. Introduction
2. Results
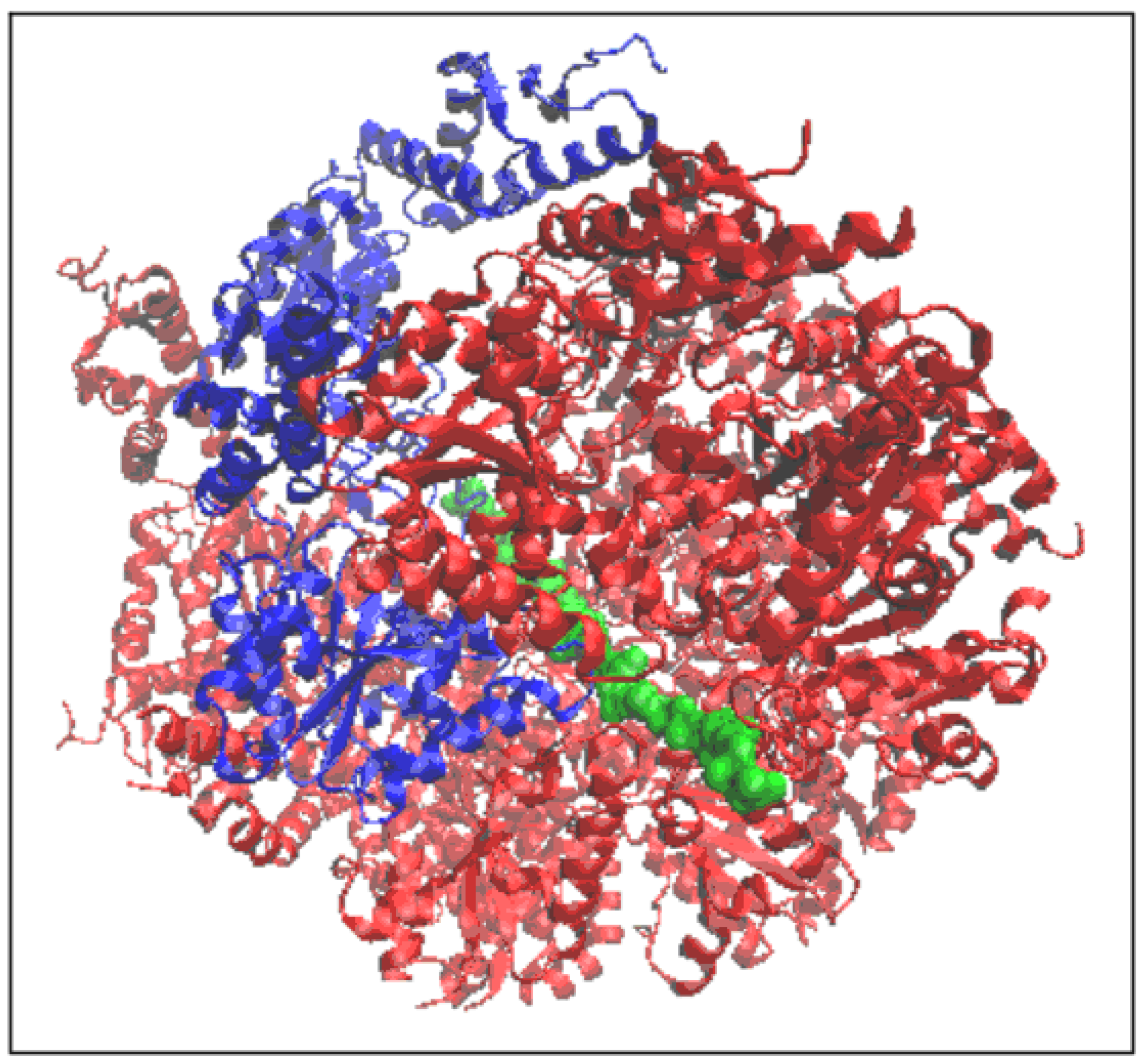
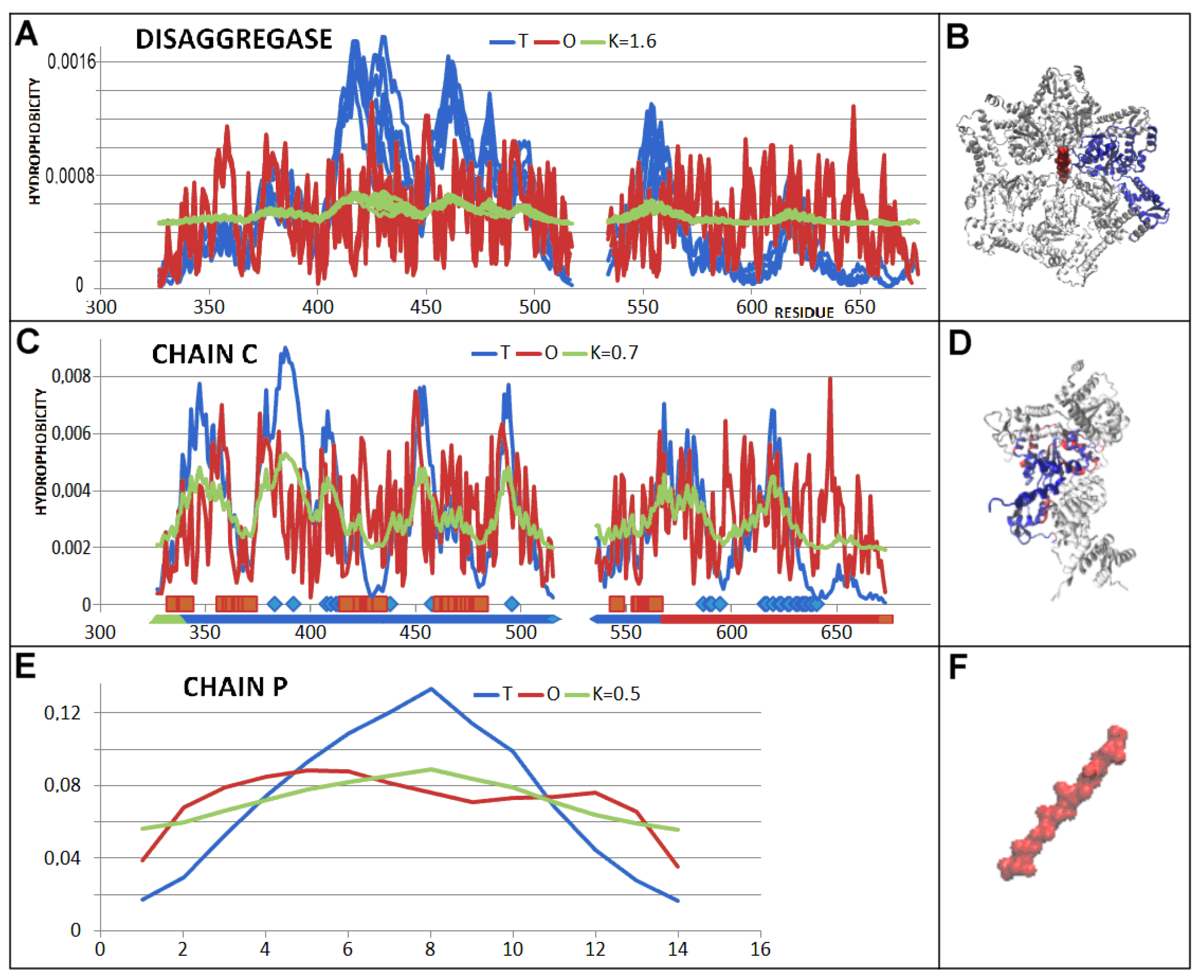
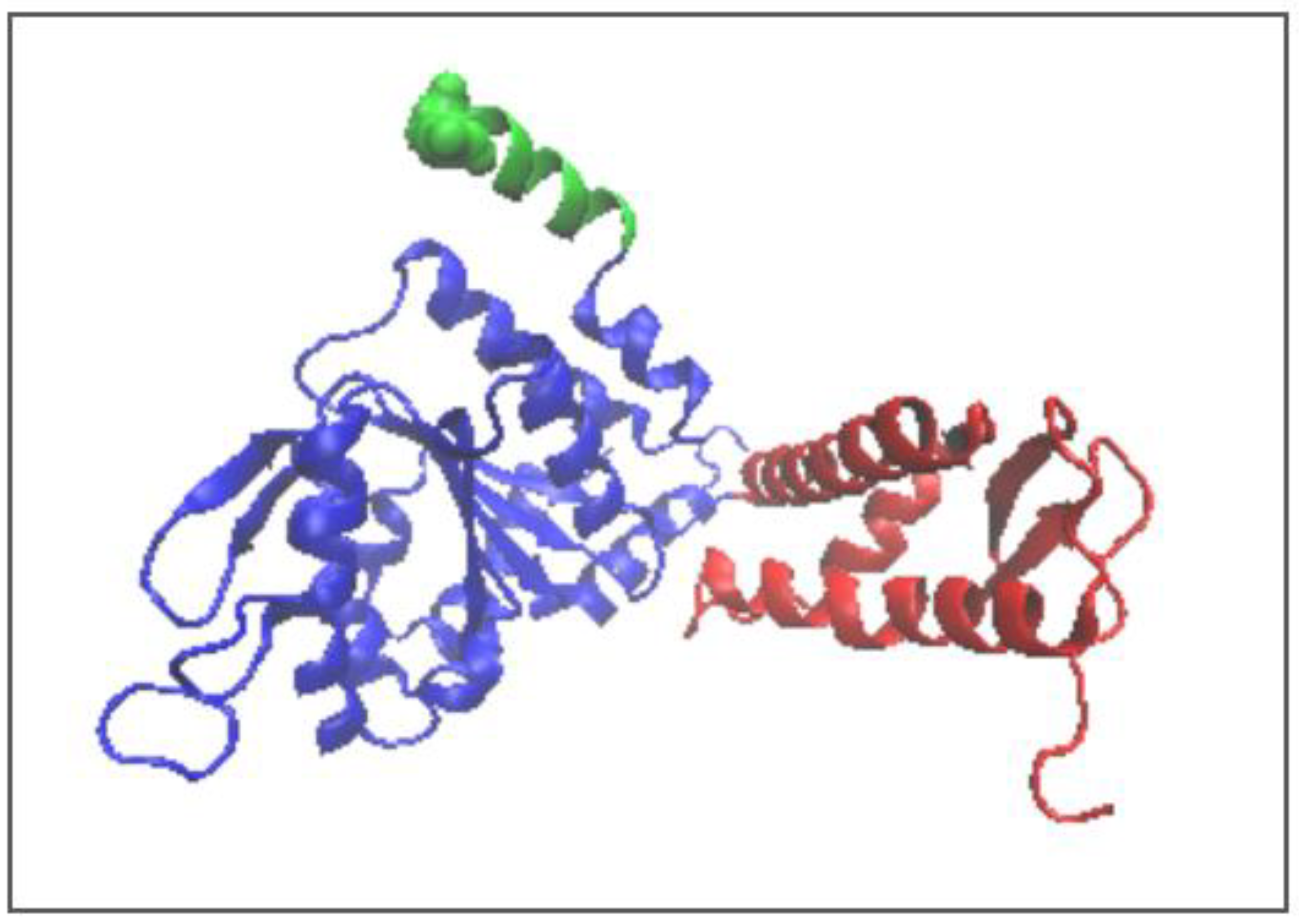
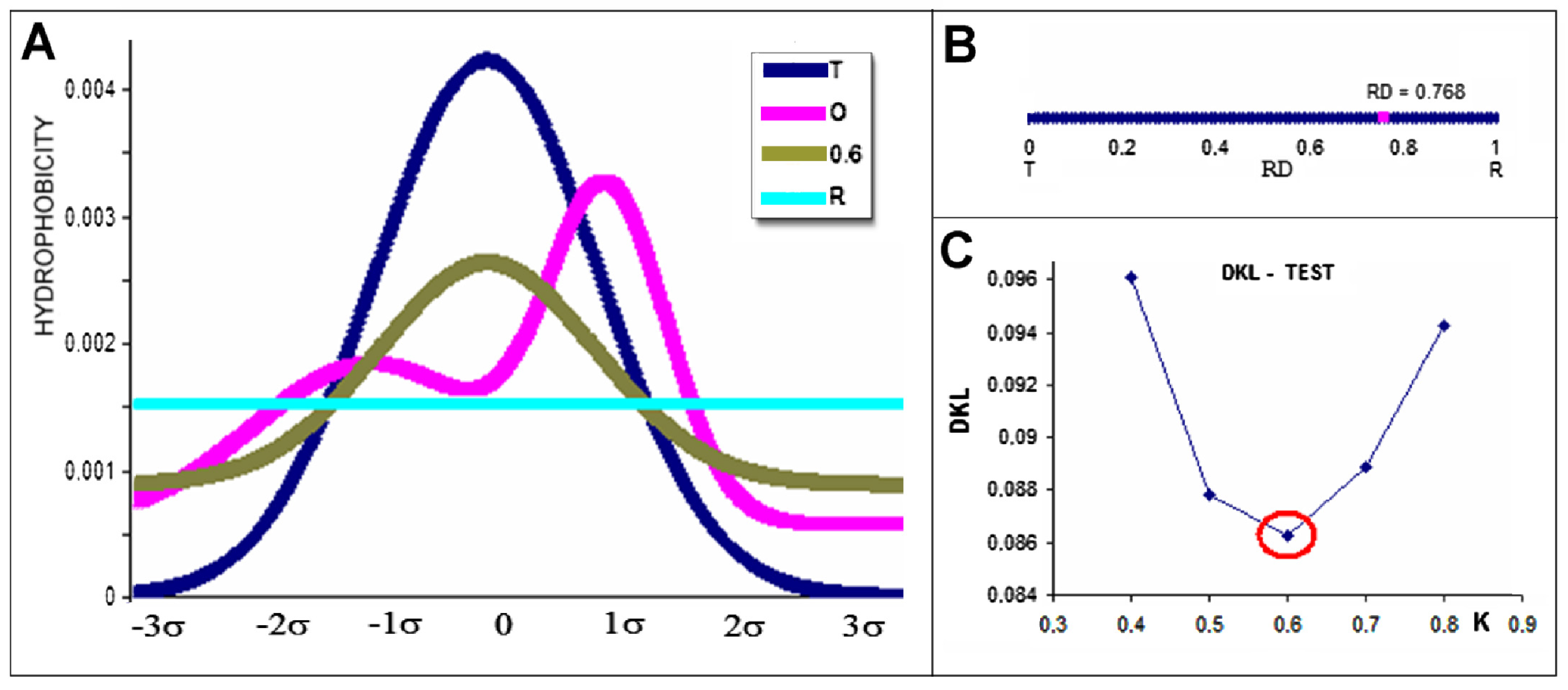
2.1. Structure of the Hsp104 Chaperonin Controlling the Casein Folding Process
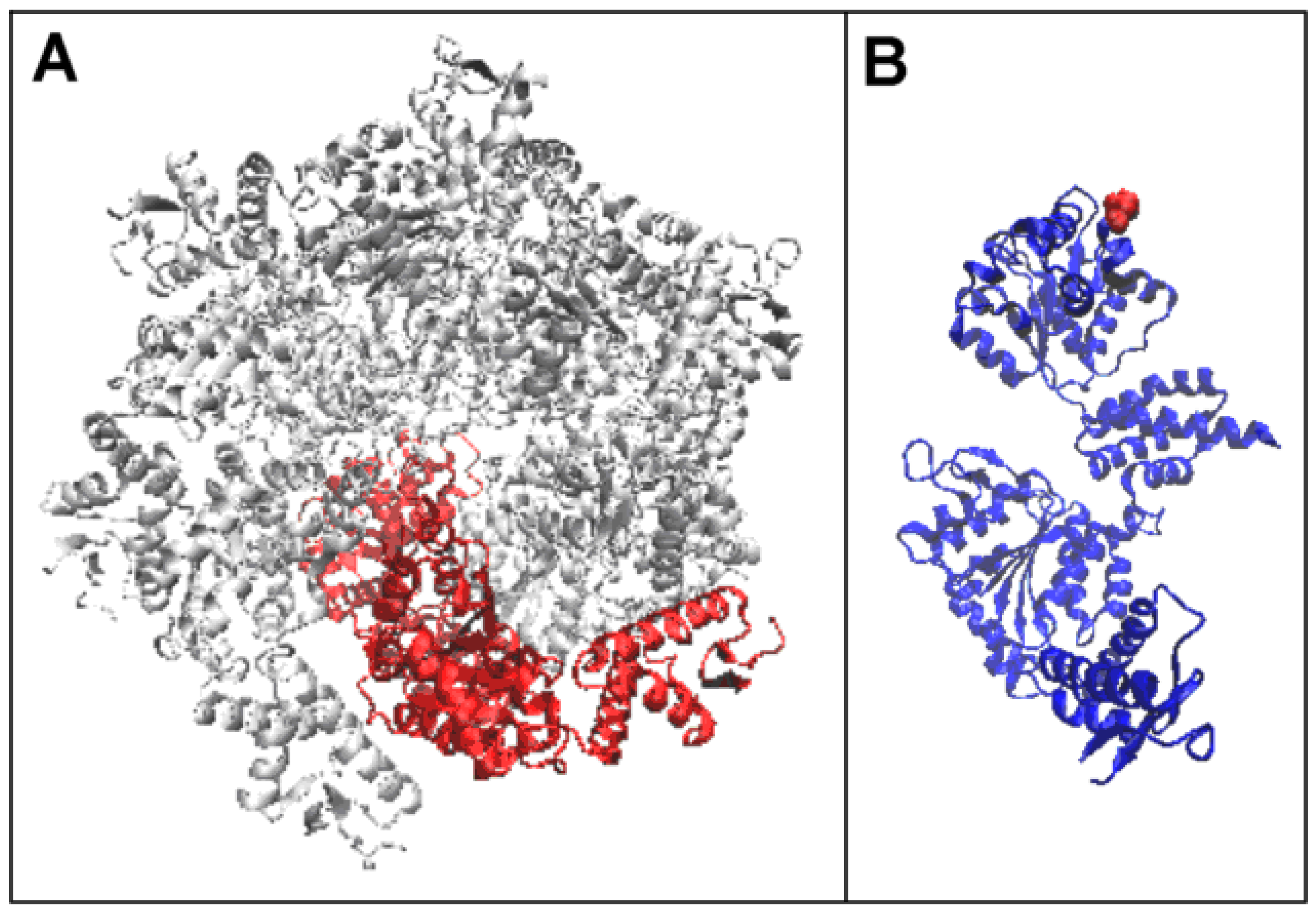
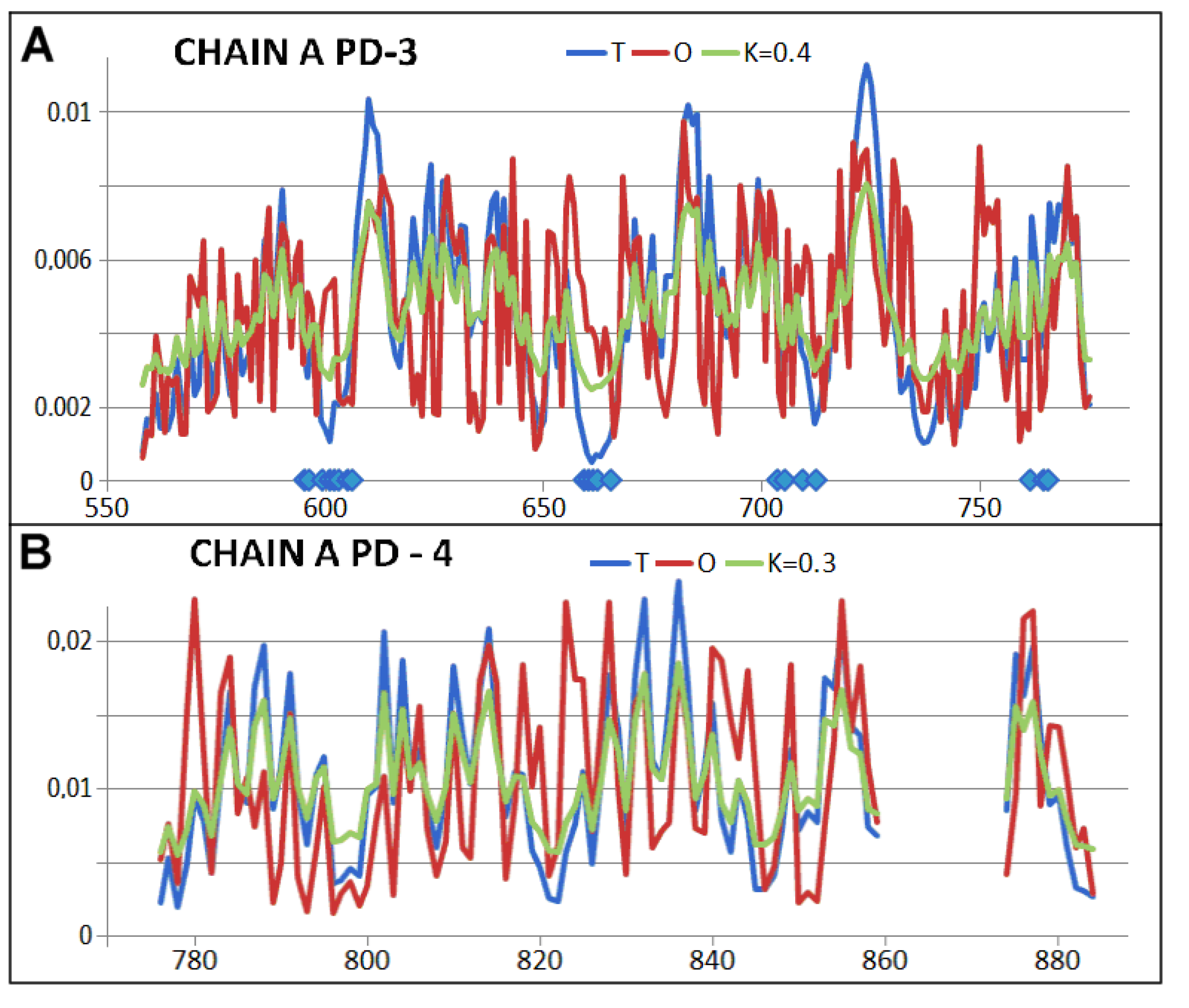
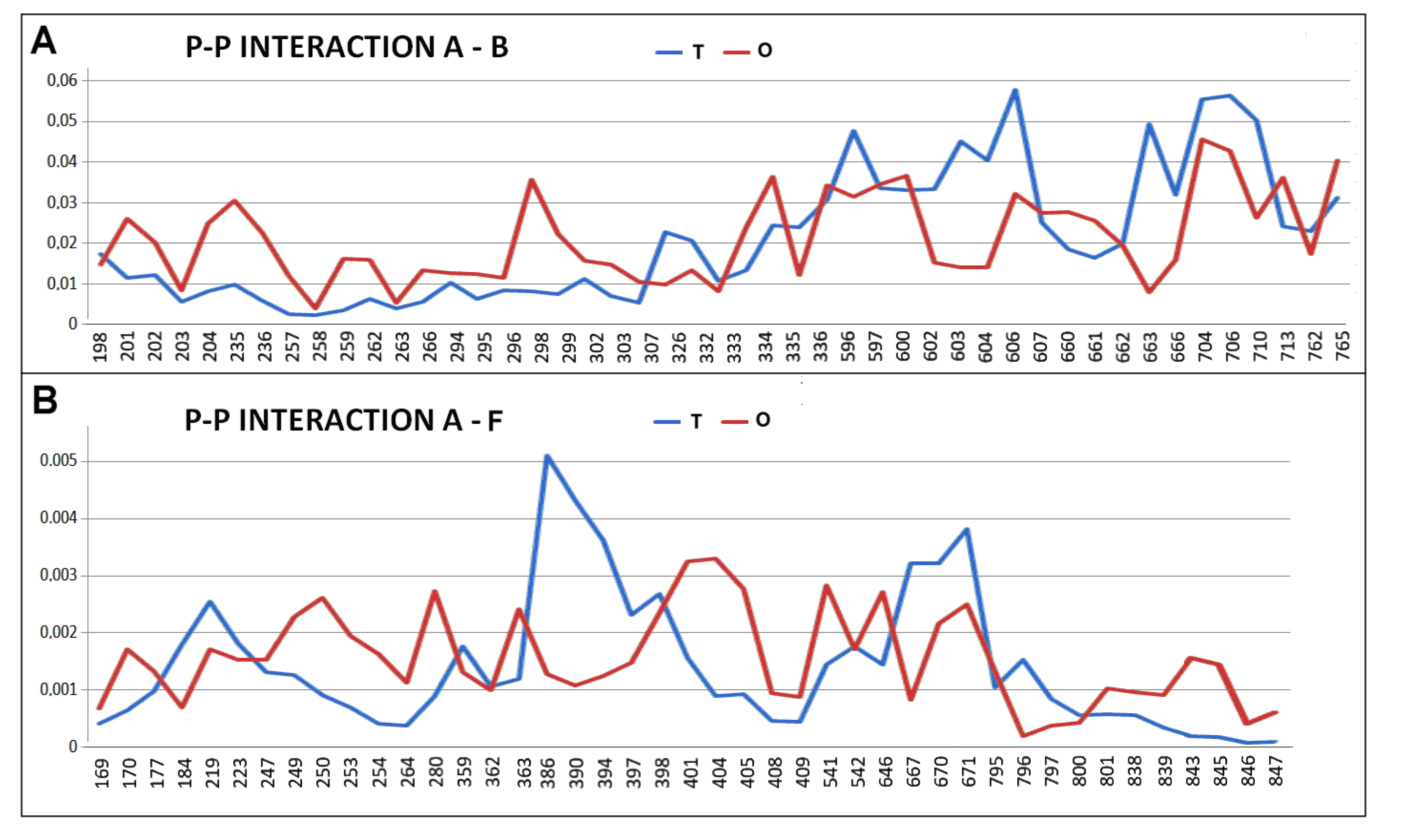
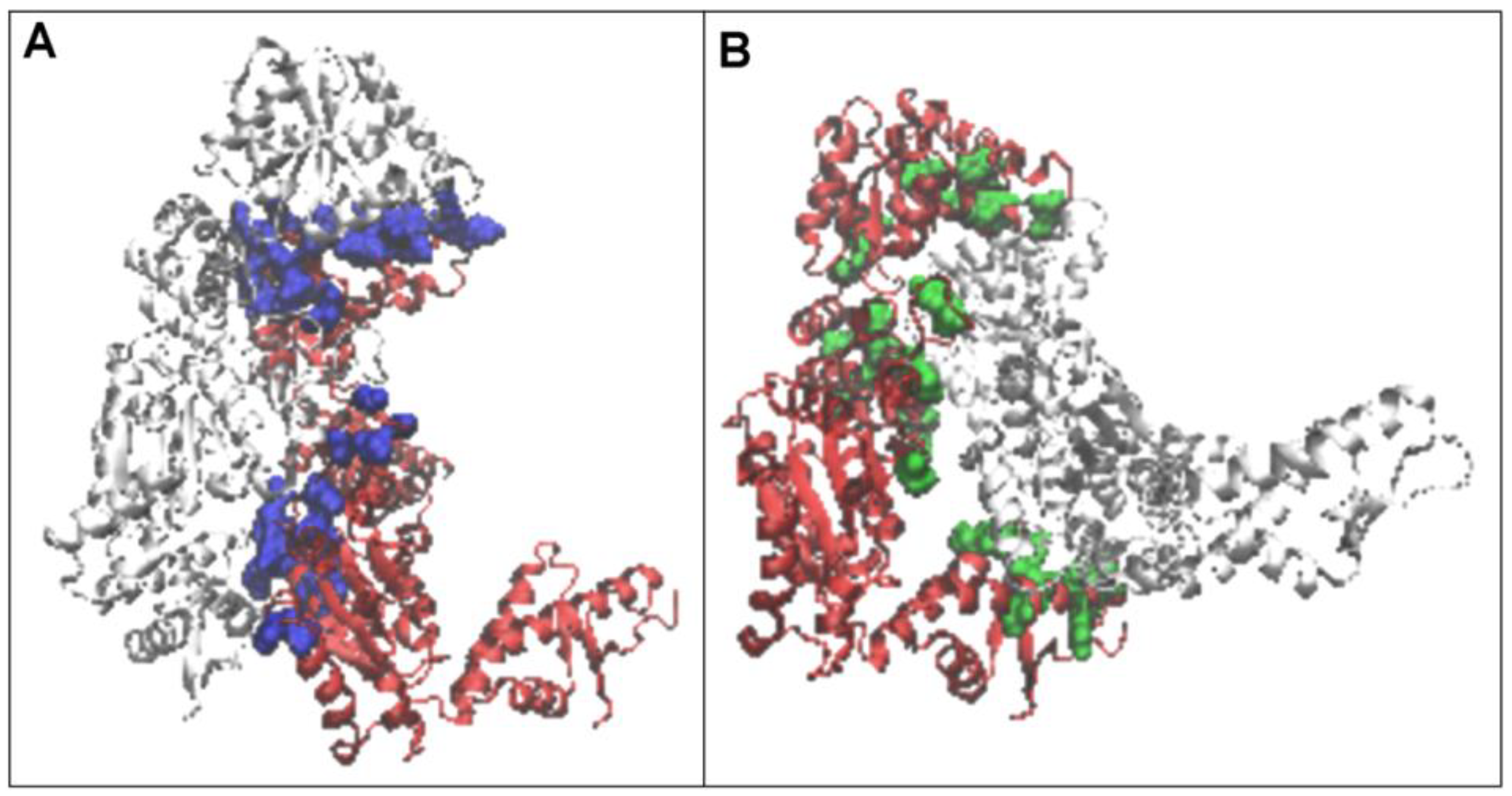
2.2. Mitochondrial Disaggregase from Homo Sapiens Preventing Incorrect Casein Complexation
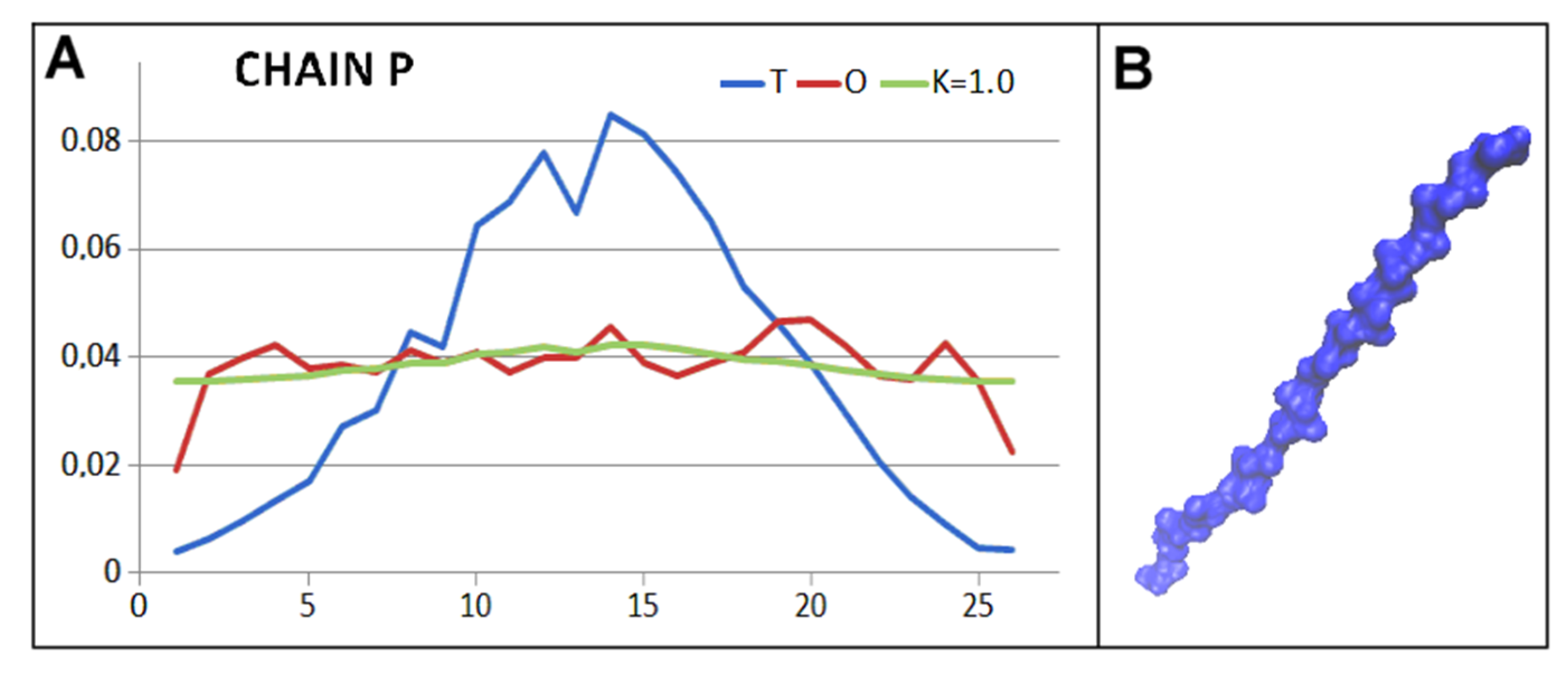
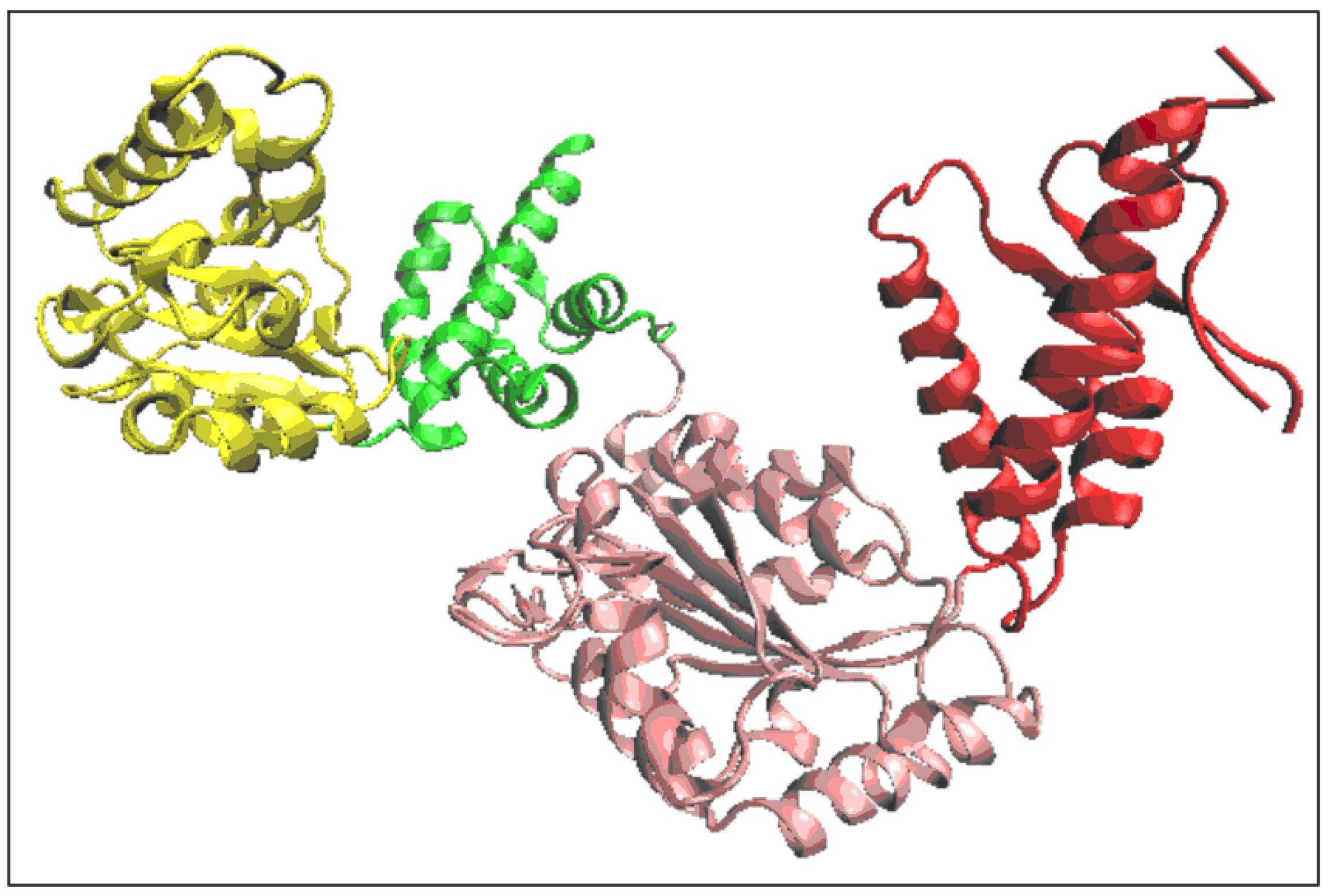
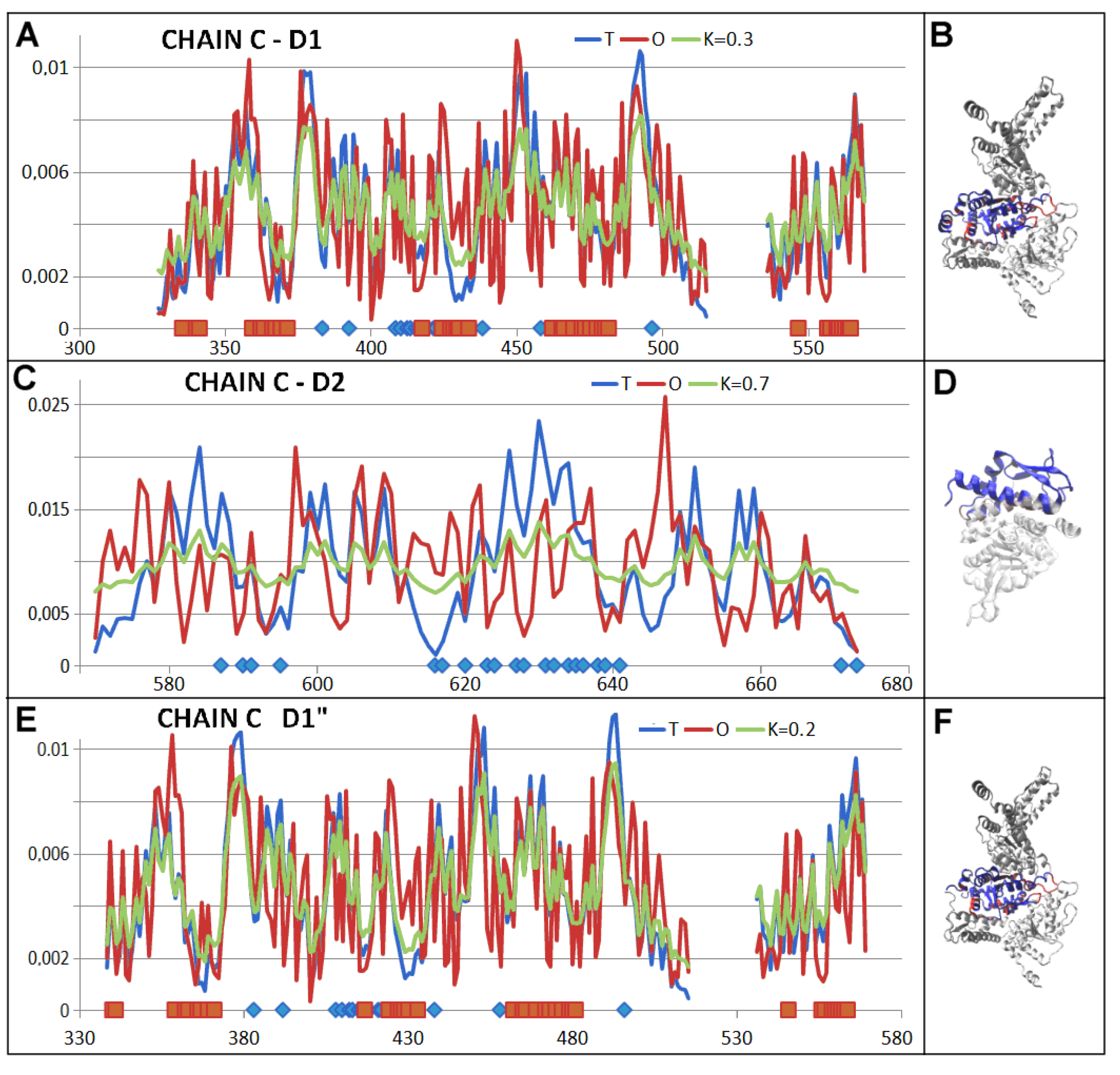
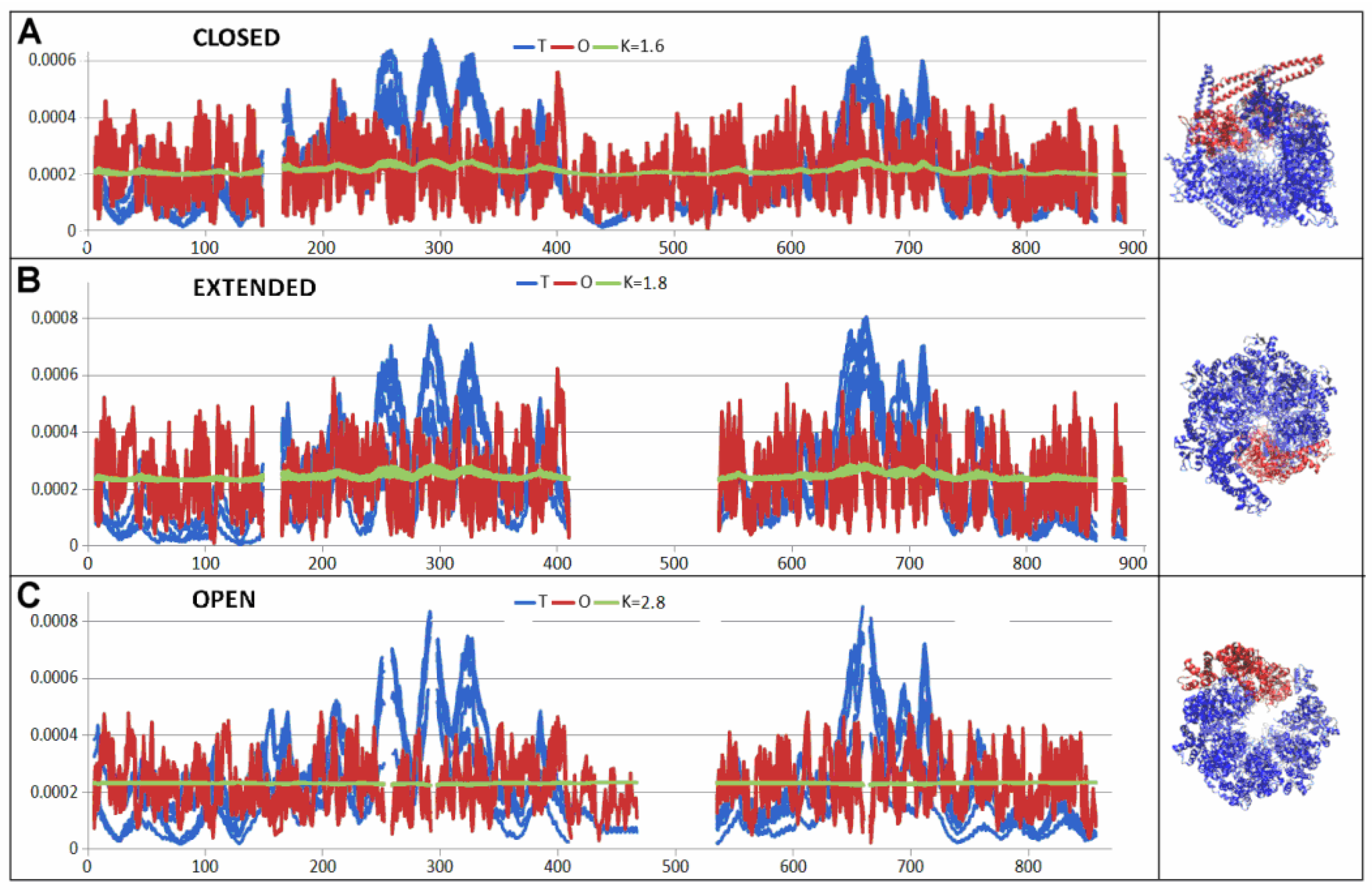
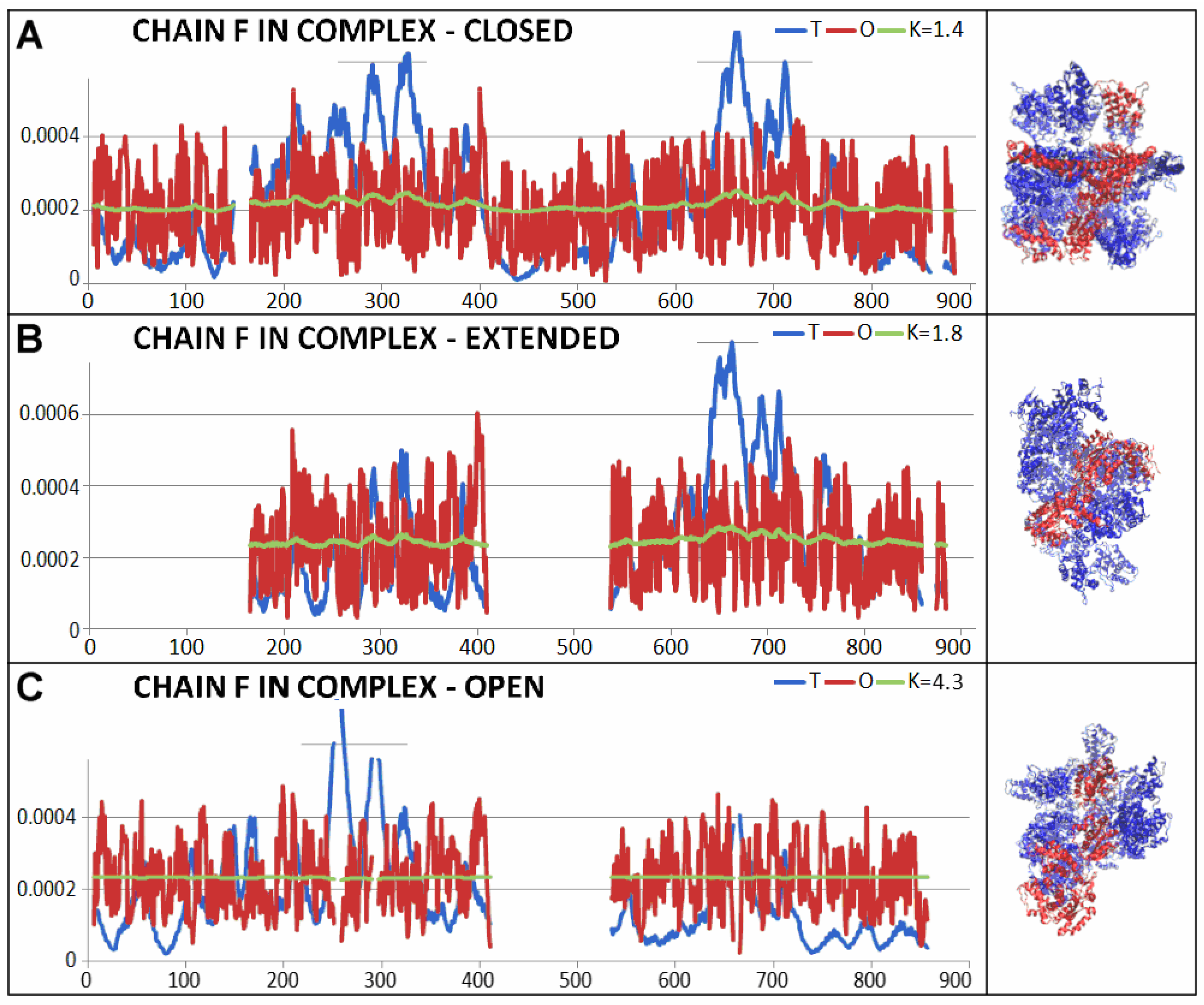
2.3. Function-Related Dynamic Structures of Disaggregase
3. Discussion
4. Materials and Methods
4.1. Data
4.2. Fuzzy Oil Drop Model—FOD-M
4.3. Programs Used
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritossa, F. A new puffing pattern induced by temperature shock and DNP in drosophila. Cell. Mol. Life Sci. 1962, 18, 571–573. [Google Scholar] [CrossRef]
- Hagymasi, A.T.; Dempsey, J.P.; Srivastava, P.K. Heat-Shock Proteins. Curr. Protoc. 2022, 2, e592. [Google Scholar] [CrossRef] [PubMed]
- Giovanella, B.C. Heat shock proteins. Adv. Exp. Med. Biol. 1990, 267, 95–98. [Google Scholar] [CrossRef] [PubMed]
- Gu, C.; Fan, X.; Yu, W. Functional Diversity of Mammalian Small Heat Shock Proteins: A Review. Cells 2023, 12, 1947. [Google Scholar] [CrossRef]
- Collier, M.P.; Benesch, J.L.P. Small heat-shock proteins and their role in mechanical stress. Cell Stress Chaperones 2020, 25, 601–613. [Google Scholar] [CrossRef]
- Zuo, W.F.; Pang, Q.; Zhu, X.; Yang, Q.Q.; Zhao, Q.; He, G.; Han, B.; Huang, W. Heat shock proteins as hallmarks of cancer: Insights from molecular mechanisms to therapeutic strategies. J. Hematol. Oncol. 2024, 17, 81. [Google Scholar] [CrossRef]
- Zhang, L.; Fok, J.H.; Davies, F.E. Heat shock proteins in multiple myeloma. Oncotarget 2014, 5, 1132–1148. [Google Scholar] [CrossRef]
- Milani, A.; Basirnejad, M.; Bolhassani, A. Heat-shock proteins in diagnosis and treatment: An overview of different biochemical and immunological functions. Immunotherapy 2019, 11, 215–239. [Google Scholar] [CrossRef]
- Youness, R.A.; Gohar, A.; Kiriacos, C.J.; El-Shazly, M. Heat Shock Proteins: Central Players in Oncological and Immuno-Oncological Tracks. Adv. Exp. Med. Biol. 2023, 1409, 193–203. [Google Scholar] [CrossRef]
- Nahleh, Z.; Tfayli, A.; Najm, A.; El Sayed, A.; Nahle, Z. Heat shock proteins in cancer: Targeting the ‘chaperones’. Future Med. Chem. 2012, 4, 927–935. [Google Scholar] [CrossRef]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.; Craig, E.A. Heat-shock proteins as molecular chaperones. Eur. J. Biochem. 1994, 219, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Tutar, L.; Tutar, Y. Heat shock proteins; an overview. Curr. Pharm. Biotechnol. 2010, 11, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Morrow, G.; Hightower, L.E.; Tanguay, R.M. Small heat shock proteins: Big folding machines. Cell Stress Chaperones 2015, 20, 207–212. [Google Scholar] [CrossRef]
- De Maio, A. Heat shock proteins: Facts, thoughts, and dreams. Shock 1999, 11, 1–12. [Google Scholar] [CrossRef]
- Fassler, J.S.; Skuodas, S.; Weeks, D.L.; Phillips, B.T. Protein Aggregation and Disaggregation in Cells and Development. J. Mol. Biol. 2021, 433, 167215. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Konieczny, L. Model of the external force field for the protein folding process-the role of prefoldin. Front. Chem. 2024, 12, 1342434. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Konieczny, L. Ab initio protein structure prediction: The necessary presence of external force field as it is delivered by Hsp40 chaperone. BMC Bioinform. 2023, 24, 418. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Dułak, D.; Konieczny, L. External Force Field for Protein Folding in Chaperonins-Potential Application in In Silico Protein Folding. ACS Omega 2024, 9, 18412–18428. [Google Scholar] [CrossRef]
- Roterman, I.; Konieczny, L. Protein Is an Intelligent Micelle. Entropy 2023, 25, 850. [Google Scholar] [CrossRef]
- Laskowski, R.A. PDBsum new things. Nucleic Acids Res. 2009, 37, D355–D359. [Google Scholar] [CrossRef] [PubMed]
- Orengo, C.A.; Michie, A.D.; Jones, S.; Jones, D.T.; Swindells, M.B.; Thornton, J.M. CATH—A hierarchic classification of protein domain structures. Structure 1997, 5, 1093–1109. [Google Scholar] [CrossRef] [PubMed]
- Cupo, R.R.; Rizo, A.N.; Braun, G.A.; Tse, E.; Chuang, E.; Gupta, K.; Southworth, D.R.; Shorter, J. Unique structural features govern the activity of a human mitochondrial AAA+ disaggregase, Skd3. Cell Rep. 2022, 40, 111408. [Google Scholar] [CrossRef]
- Lee, S.; Roh, S.H.; Lee, J.; Sung, N.; Liu, J.; Tsai, F.T.F. Cryo-EM Structures of the Hsp104 Protein Disaggregase Captured in the ATP Conformation. Cell Rep. 2019, 26, 29–36.e3. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Roterman, I.; Slupina, M.; Konieczny, L. Protein folding: Funnel model revised. Comput. Struct. Biotechnol. J. 2024, 23, 3827–3838. [Google Scholar] [CrossRef]
- Roterman, I.; Słupina, M.; Dułak, D.; Konieczny, L. Three scenarios for amyloid transformation in the context of the funnel model. Comput. Struct. Biotechnol. J. 2025, 27, 1648–1659. [Google Scholar] [CrossRef]
- Slupina, M.; Stapor, K.; Konieczny, L.; Gądek, K.; Nowakowski, P.; Roterman, I. Chameleon sequences-Structural effects. PLoS ONE 2025, 20, e0315901. [Google Scholar] [CrossRef]
- Roterman, I.; Slupina, M.; Stapor, K.; Konieczny, L.; Gądek, K.; Nowakowski, P. Chameleon Sequences-Structural Effects in Proteins Characterized by Hydrophobicity Disorder. ACS Omega 2024, 9, 38506–38522. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Konieczny, L. Role of environmental specificity in CASP results. BMC Bioinform. 2023, 24, 425. [Google Scholar] [CrossRef]
- Gadzała, M.; Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Determining protein similarity by comparing hydrophobic core structure. Heliyon 2017, 3, e00235. [Google Scholar] [CrossRef] [PubMed]
- Rehan, F.; Ahemad, N.; Gupta, M. Casein nanomicelle as an emerging biomaterial-A comprehensive review. Colloids Surf. B Biointerfaces 2019, 179, 280–292. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gates, S.N.; Yokom, A.L.; Lin, J.B.; Jackrel, M.E.; Rizo, A.N.; Kendsersky, N.M.; Buell, C.E.; Sweeny, E.A.; Mack, K.L.; Chuang, E.; et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 357, 273–279. [Google Scholar] [CrossRef]
- Yuan, R.; Zhang, J.; Kryshtafovych, A.; Schaeffer, R.D.; Zhou, J.; Cong, Q.; Grishin, N.V. CASP16 protein monomer structure prediction assessment. bioRxiv 2025. [Google Scholar] [CrossRef]
- Available online: https://predictioncenter.org/casp16/results.cgi?view=tables&target=T1266-D1& (accessed on 27 June 2025).
- Levitt, M.A. A simplified representation of protein conformations for rapid simulation of protein folding. J. Mol. Biol. 1976, 104, 59–107. [Google Scholar] [CrossRef]
- Kalinowska, B.; Banach, M.; Konieczny, L.; Roterman, I. Application of Divergence Entropy to Characterize the Structure of the Hydrophobic Core in DNA Interacting Proteins. Entropy 2015, 17, 1477–1507. [Google Scholar] [CrossRef]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Banach, M.; Stapor, K.; Konieczny, L.; Fabian, P.; Roterman, I. Downhill, Ultrafast and Fast Folding Proteins Revised. Int. J. Mol. Sci. 2020, 21, 7632. [Google Scholar] [CrossRef]
- Roterman, I.; Stapor, K.; Konieczny, L. Transmembrane proteins—Different anchoring systems. Proteins 2024, 92, 593–609. [Google Scholar] [CrossRef]
- Available online: https://www.ks.uiuc.edu/Research/vmd/ (accessed on 27 June 2025).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 5VJH | Fragment | Individual | In Complex | ||
|---|---|---|---|---|---|
| RD | K | RD | K | ||
| CHAINS A-F + P | 0.729 | 2.1 | |||
| CHAINS A-F | 0.729 | 2.1 | |||
| CHAIN A | 0.722 | 1.6 | 0.724 | ||
| CHAIN B | 0.702 | 1.3 | 0.732 | ||
| CHAIN C | 0.668 | 1.1 | 0.709 | ||
| CHAIN D | 0.722 | 1.1 | 0.710 | ||
| CHAIN E | 0.734 | 1.1 | 0.747 | ||
| CHAIN F | 0.755 | 1.5 | 0.737 | ||
| CHAIN P (casein) | 0.955 | 1.0 | 0.781 | ||
| PSEUDO-DOMAINS | 165–340 | 0.409 | 0.3 | ||
| 344–557 | 0.367 | 0.2 | |||
| 558–775 | 0.490 | 0.4 | |||
| 776–884 | 0.459 | 0.3 | |||
| Chain in Complex | Individual | ||||
|---|---|---|---|---|---|
| Chain | Fragment | RD | K | RD | K |
| A-F | 0.743 | 1.6 | |||
| A-F + P | 0.740 | ||||
| A | 0.609 | 0.6 | |||
| B | 0.637 | 0.7 | |||
| C | 0.737 | 1.6 | 0.613 | 0.7 | |
| D | 0.758 | 1.6 | 0.591 | 0.5 | |
| E | 0.749 | 1.3 | 0.544 | 0.4 | |
| F | 0.675 | 1.2 | 0.557 | 0.5 | |
| P | 0.745 | 1.7 | 0.802 | 0.5 | |
| C—PD 1 | (327–569) | 0.758 | 1.7 | 0.577 | 0.4 |
| C—PD 2 | (570–653) | 0.751 | 0.5 | 0.624 | 0.6 |
| C—PD 3 | (338–569) | 0.556 | 0.3 | ||
| Chain P-P | 0.786 | ||||
| Chain No P-P | 0.655 | ||||
| PDB ID | Complex | Chain in Complex | Chain Individual | |||
|---|---|---|---|---|---|---|
| Hsp104dwb | RD | K | RD | K | RD | K |
| 6N8T—closed conformation | 0.702 | 1.6 | ||||
| Chains: A | 0.711 | 1.6 | 0.748 | 1.4 | ||
| B | 0.683 | 1.4 | 0.748 | 1.4 | ||
| C | 0.705 | 1.5 | 0.739 | 1.4 | ||
| D | 0.689 | 1.5 | 0.776 | 1.6 | ||
| E | 0.716 | 1.7 | 0.744 | 1.4 | ||
| F | 0.706 | 1.4 | 0.735 | 1.2 | ||
| 6N8V—open conformation | 0.784 | 2.8 | ||||
| Chains: A—704 AA | 0.800 | 2.5 | 0.808 | 1.8 | ||
| B—704 AA | 0.800 | 2.2 | 0.845 | 2.1 | ||
| C—704 AA | 0.741 | 2.0 | 0.813 | 2.0 | ||
| D—769 AA | 0.769 | 2.2 | 0.782 | 1.6 | ||
| E—704 AA | 0.789 | 2.7 | 0.826 | 2.1 | ||
| F—704 AA | 0.786 | 4.3 | 0.824 | 1.2 | ||
| 6N8Z—extended conformation | 0.734 | 1.8 | ||||
| Chains: A—579 AA | 0.740 | 1.7 | 0.701 | 1.3 | ||
| B—723 AA | 0.738 | 1.8 | 0.760 | 1.5 | ||
| C—723 AA | 0.702 | 1.6 | 0.763 | 1.6 | ||
| D—723 AA | 0.751 | 1.7 | 0.737 | 1.4 | ||
| E—723 AA | 0.760 | 1.8 | 0.742 | 1.4 | ||
| F—579 AA | 0.692 | 1.7 | 0.703 | 1.2 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roterman, I.; Stapor, K.; Dułak, D.; Konieczny, L. Heat Shock Protein and Disaggregase Influencing the Casein Structuralisation. Int. J. Mol. Sci. 2025, 26, 6360. https://doi.org/10.3390/ijms26136360
Roterman I, Stapor K, Dułak D, Konieczny L. Heat Shock Protein and Disaggregase Influencing the Casein Structuralisation. International Journal of Molecular Sciences. 2025; 26(13):6360. https://doi.org/10.3390/ijms26136360
Chicago/Turabian StyleRoterman, Irena, Katarzyna Stapor, Dawid Dułak, and Leszek Konieczny. 2025. "Heat Shock Protein and Disaggregase Influencing the Casein Structuralisation" International Journal of Molecular Sciences 26, no. 13: 6360. https://doi.org/10.3390/ijms26136360
APA StyleRoterman, I., Stapor, K., Dułak, D., & Konieczny, L. (2025). Heat Shock Protein and Disaggregase Influencing the Casein Structuralisation. International Journal of Molecular Sciences, 26(13), 6360. https://doi.org/10.3390/ijms26136360