
Microfluidic Liquid Biopsy Minimally Invasive Cancer Diagnosis by Nano-Plasmonic Label-Free Detection of Extracellular Vesicles: Review



Abstract

1. Introduction
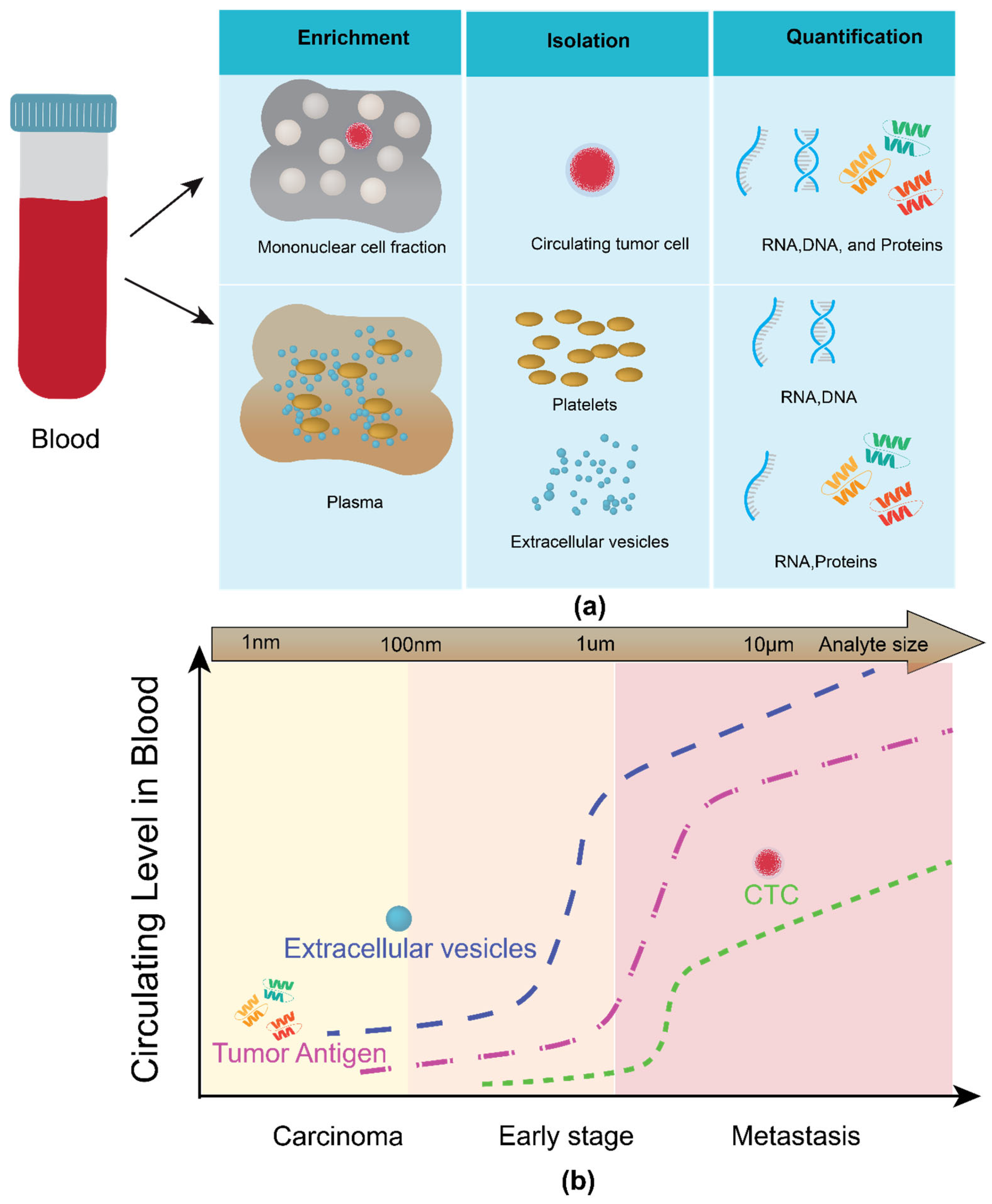
1.1. Classes of Analyte in Liquid Biopsy
1.1.1. Circulating Tumor Cells (CTC)
1.1.2. Circulating Cell-Free Nucleic Acids (Cf-NAs)
1.1.3. Tumor-Educated Platelets (TEPs)
1.1.4. Exosomes
1.2. Hurdles in Adapting Exosomes in Liquid Biopsy
1.3. Handling and Analysis of Exosomes: A Microfluidic Approach
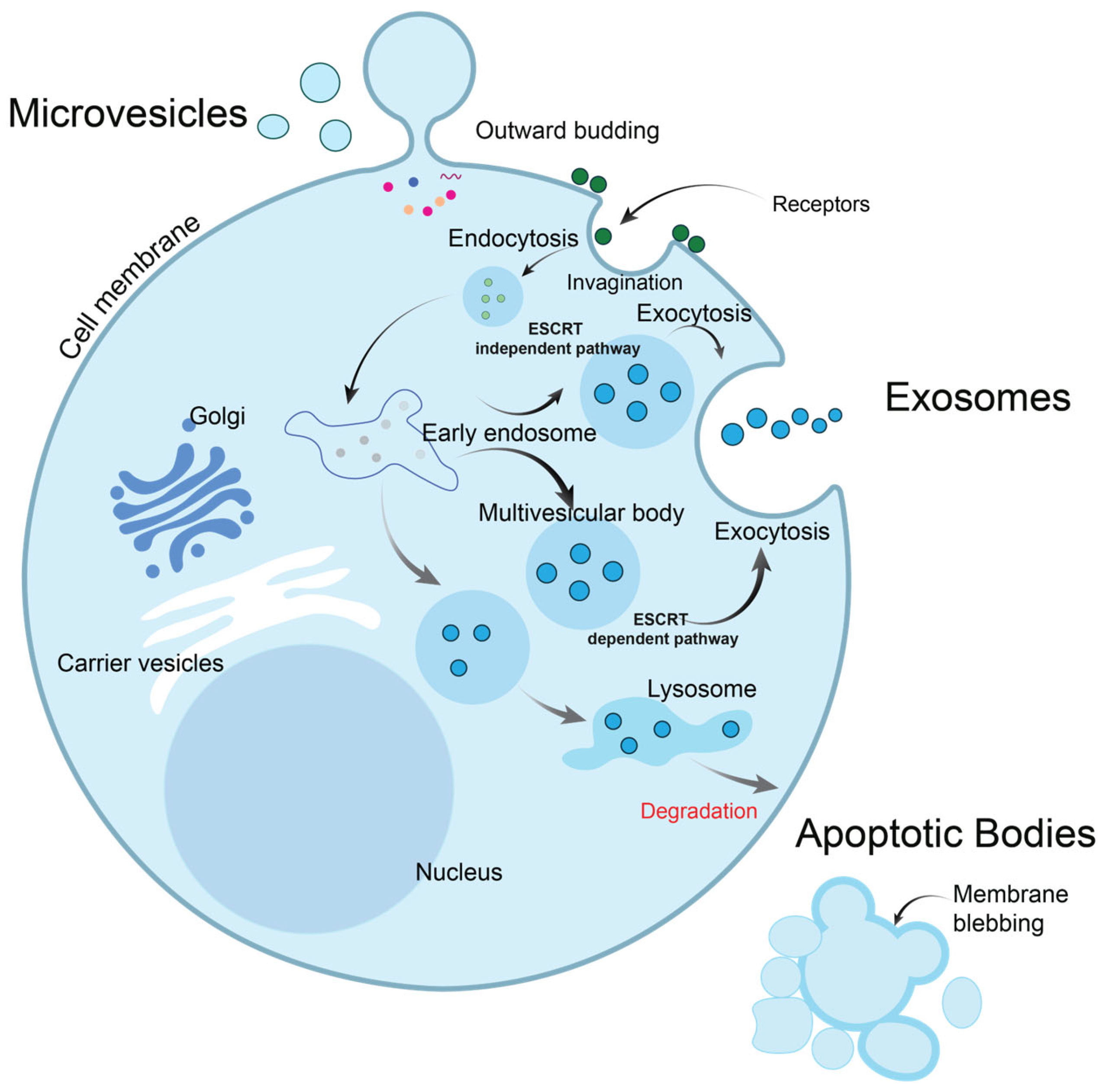
2. Extracellular Vesicles: Exosomes, Microvesicles, and Others
2.1. Exosomes
2.2. Microvesicles

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Exosomes | Microvesicles | Apoptotic Bodies | |
|---|---|---|---|
| Size | 20–100 nm | 50–1000 nm | 500–2000 nm |
| Density | 1.13–1.19 g/mL | 1.04–1.07 g/mL | 1.16–1.28 g/mL |
| Biogenesis | Formed through inward luminal budding of the membrane and fusion of multivesicular bodies with the cell membrane (endolysosomal pathway). | Directly shed from the cell’s plasma membrane through outward budding. | Formed through membrane blebbing from a cell undergoing apoptosis. |
| Composition |
|
|
|
| Biomarker | Tetraspanins family (such as TSPAN29 and TSPAN30, CD81, CD82, CD9, CD63), ESCRT proteins (Alix, TSG101), actin, flotillin, Hsc70, HSP 90, Hsp60 and Hsp20 clathrin, integrins (such as α3, α4, β1, β2) | Integrins, selectins, flotillin-2, CD40 ligand, metalloproteinase | Annexin V positivity, phosphatidyl serine |
2.3. Apoptotic Bodies
3. Microfluidic Platform for EV Isolation, Detection, and Characterization
3.1. EV Isolation
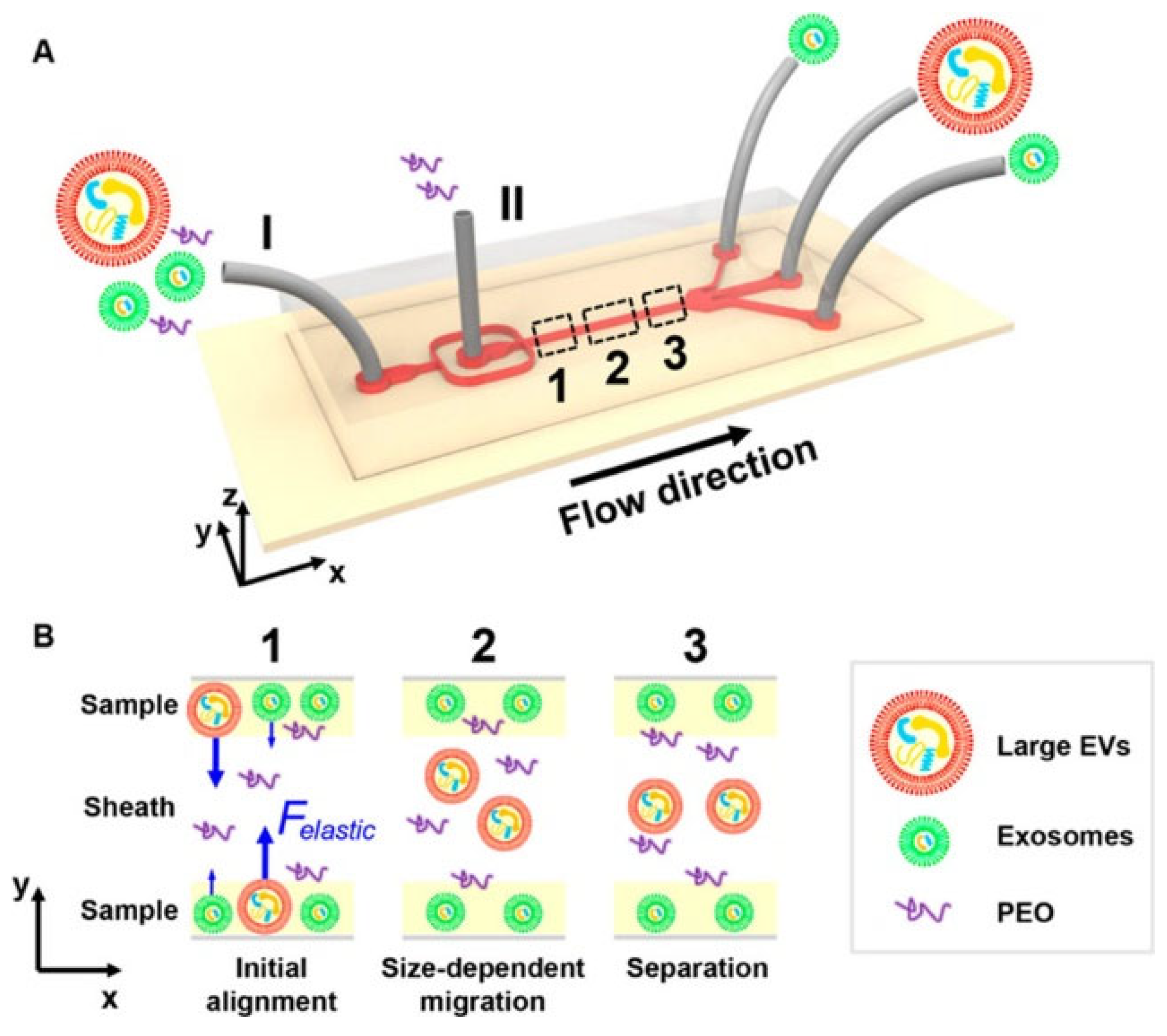
3.1.1. Physical Technique for EV Isolation in a Microfluidic Platform
3.1.2. Asymmetric Flow Field-Flow Fractionation (A4F)
3.1.3. Electrical Technique for EV Isolation in Microfluidic Platform
3.1.4. Immunoaffinity-Based Techniques for EV Isolation in Microfluidic Platform
3.2. EV Detection
4. Plasmonic Technologies for Exosome Analysis
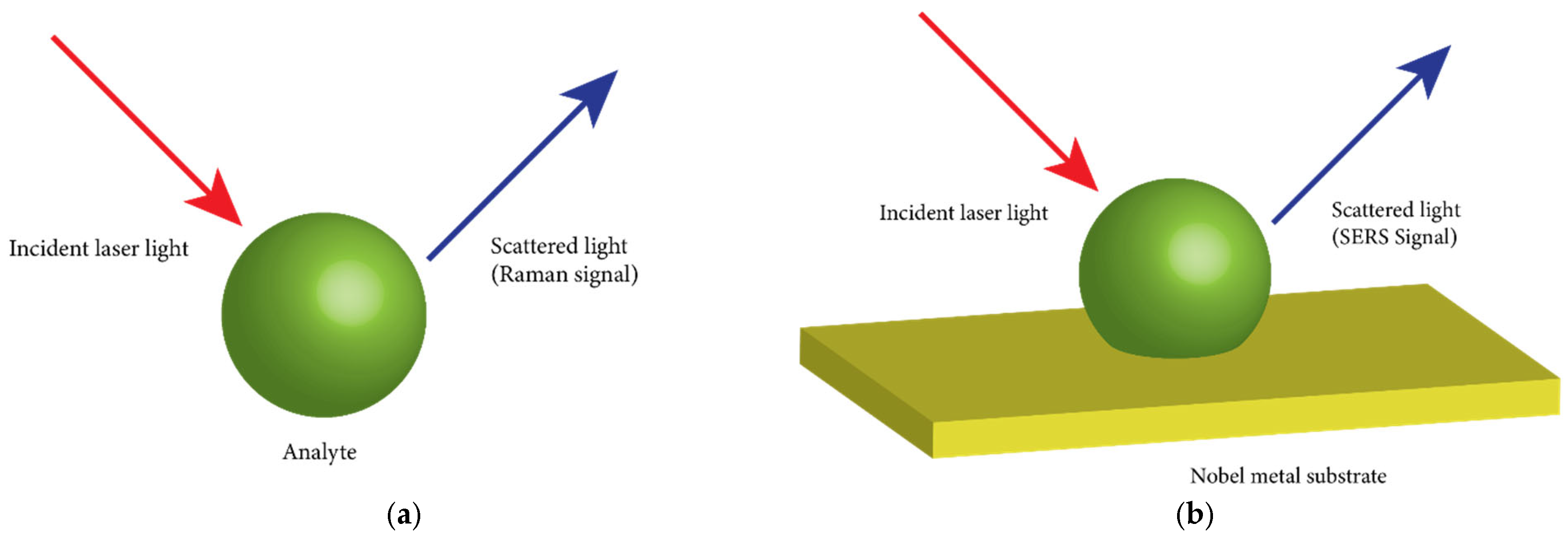
4.1. Raman Scattering
4.2. Surface-Enhanced Raman Scattering
4.2.1. Physical Modification of SERS Substrate
4.2.2. Chemical Modification of SERS Substrate
4.2.3. SERS Signal Detection Analysis
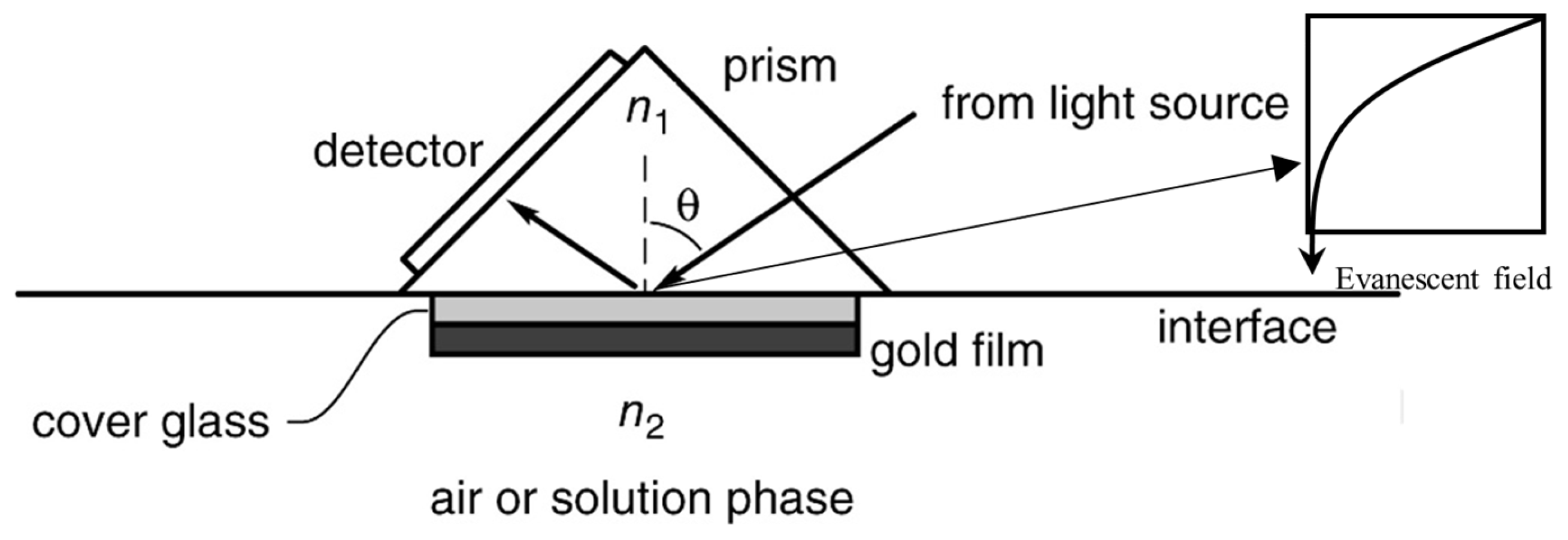
4.3. Surface Plasmon Resonance
4.4. Surface Plasmon Resonance Imaging
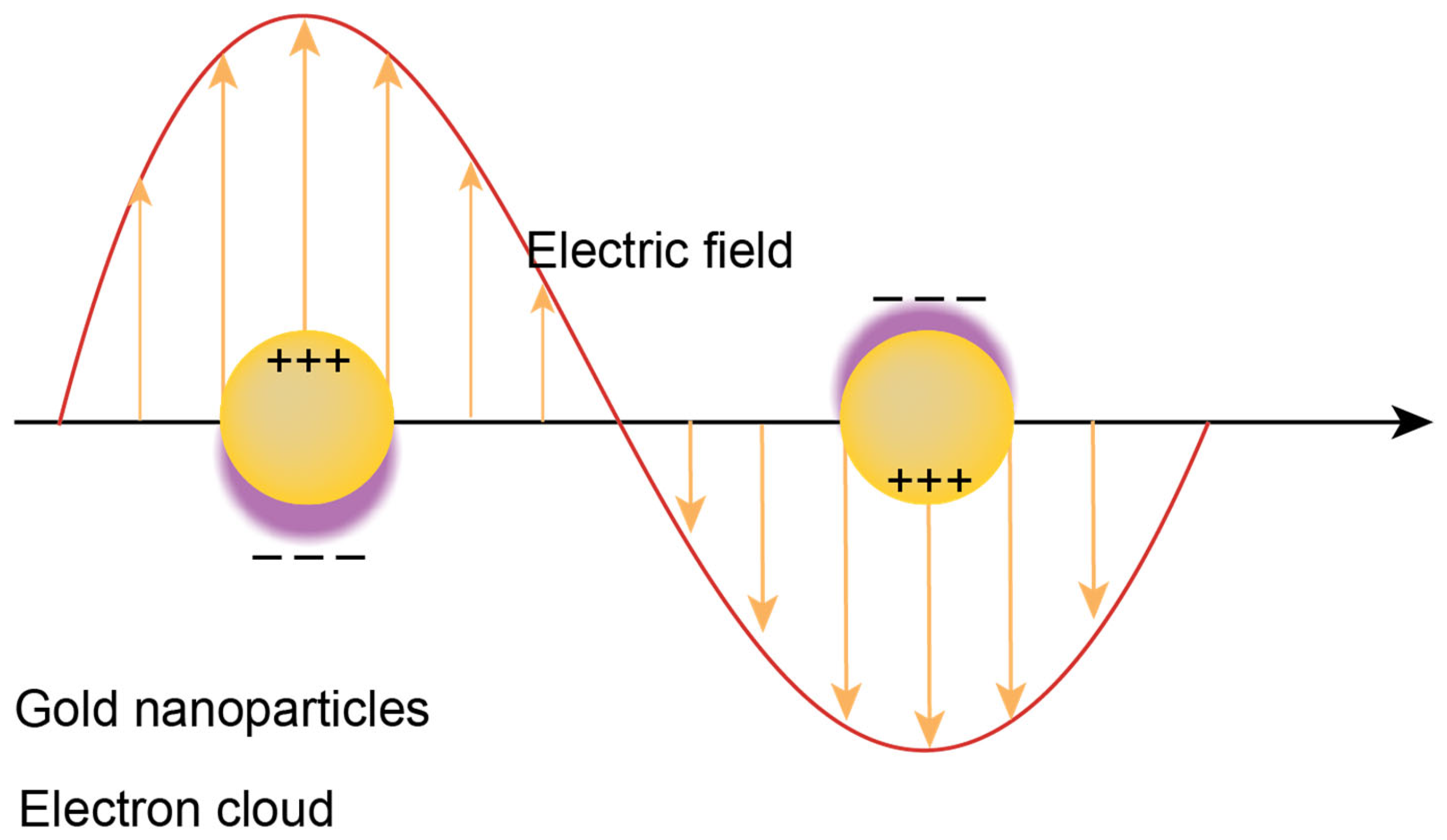
4.5. Localized Surface Plasmon Resonance (LSPR)
5. Research Outlook and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Li, S.; Yi, M.; Dong, B.; Tan, X.; Luo, S.; Wu, K. The role of exosomes in liquid biopsy for cancer diagnosis and prognosis prediction. Int. J. Cancer 2021, 148, 2640–2651. [Google Scholar] [CrossRef] [PubMed]
- Crowley, E.; Di Nicolantonio, F.; Loupakis, F.; Bardelli, A. Liquid biopsy: Monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol. 2013, 10, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Crosby, D.; Bhatia, S.; Brindle, K.M.; Coussens, L.M.; Dive, C.; Emberton, M.; Esener, S.; Fitzgerald, R.C.; Gambhir, S.S.; Kuhn, P.; et al. Early detection of cancer. Science 2022, 375, eaay9040. [Google Scholar] [CrossRef] [PubMed]
- Alix-Panabières, C.; Schwarzenbach, H.; Pantel, K. Circulating tumor cells and circulating tumor DNA. Annu. Rev. Med. 2012, 63, 199–215. [Google Scholar] [CrossRef]
- He, M.; Zeng, Y. Microfluidic Exosome Analysis toward Liquid Biopsy for Cancer. J. Lab. Autom. 2016, 21, 599–608. [Google Scholar] [CrossRef]
- Cowling, T.; Loshak, H. An Overview of Liquid Biopsy for Screening and Early Detection of Cancer. In CADTH Issues in Emerging Health Technologies; Canadian Agency for Drugs and Technologies in Health: Ottawa, ON, Canada, 2019; Chapter 179. Available online: https://www.ncbi.nlm.nih.gov/books/NBK555478/ (accessed on 14 October 2024).
- Vellanki, P.J.; Ghosh, S.; Pathak, A.; Fusco, M.J.; Bloomquist, E.W.; Tang, S.; Singh, H.; Philip, R.; Pazdur, R.; Beaver, J.A. Regulatory implications of ctDNA in immuno-oncology for solid tumors. J. Immunol. Ther. Cancer 2023, 11, e005344. [Google Scholar] [CrossRef]
- Guardant360® CDx—A Fast, FDA-Approved CGP Panel with Guideline-Recommended Biomarkers. Available online: https://www.guardantcomplete.com/products/guardant360-cdx (accessed on 14 October 2024).
- Bauml, J.M.; Li, B.T.; Velcheti, V.; Govindan, R.; Curioni-Fontecedro, A.; Dooms, C.; Takahashi, T.; Duda, A.W.; Odegaard, J.I.; Cruz-Guilloty, F.; et al. Clinical validation of Guardant360 CDx as a blood-based companion diagnostic for sotorasib. Lung Cancer 2022, 166, 270–278. [Google Scholar] [CrossRef]
- Gupta, R.; Othman, T.; Chen, C.; Sandhu, J.; Ouyang, C.; Fakih, M. Guardant360 Circulating Tumor DNA Assay Is Concordant with Foundation One Next-Generation Sequencing in Detecting Actionable Driver Mutations in Anti-EGFR Naive Metastatic Colorectal Cancer. Oncologist 2020, 25, 235–243. [Google Scholar] [CrossRef]
- FoundationOne®Liquid CDx. Available online: https://www.foundationmedicine.com/test/foundationone-liquid-cdx (accessed on 14 October 2024).
- Shirley, M. Epi proColon® for Colorectal Cancer Screening: A Profile of Its Use in the USA. Mol. Diagn. Ther. 2020, 24, 497–503. [Google Scholar] [CrossRef]
- Clebak, K.T.; Nickolich, S.; Mendez-Miller, M. Multitarget Stool DNA Testing (Cologuard) for Colorectal Cancer Screening. Am. Fam. Physician 2022, 105, 198–200. [Google Scholar]
- Riethdorf, S.; Fritsche, H.; Müller, V.; Rau, T.; Schindlbeck, C.; Rack, B.; Janni, W.; Coith, C.; Beck, K.; Jänicke, F.; et al. Detection of Circulating Tumor Cells in Peripheral Blood of Patients with Metastatic Breast Cancer: A Validation Study of the CellSearch System. Clin. Cancer Res. 2007, 13, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Pyzocha, N.J. Galleri test for the detection of cancer. Am. Fam. Physician 2022, 106, 459–460. [Google Scholar] [PubMed]
- García-Foncillas, J.; Tabernero, J.; Élez, E.; Aranda, E.; Benavides, M.; Camps, C.; Jantus-Lewintre, E.; López, R.; Muinelo-Romay, L.; Montagut, C.; et al. Prospective multicenter real-world RAS mutation comparison between OncoBEAM-based liquid biopsy and tissue analysis in metastatic colorectal cancer. Br. J. Cancer 2018, 119, 1464–1470. [Google Scholar] [CrossRef] [PubMed]
- Garcia, J.; Gauthier, A.; Lescuyer, G.; Barthelemy, D.; Geiguer, F.; Balandier, J.; Edelstein, D.L.; Jones, F.S.; Holtrup, F.; Duruisseau, M.; et al. Routine Molecular Screening of Patients with Advanced Non-SmallCell Lung Cancer in Circulating Cell-Free DNA at Diagnosis and During Progression Using OncoBEAMTM EGFR V2 and NGS Technologies. Mol. Diagn. Ther. 2021, 25, 239–250. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science (1979) 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Ulz, P.; Perakis, S.; Zhou, Q.; Moser, T.; Belic, J.; Lazzeri, I.; Wölfler, A.; Zebisch, A.; Gerger, A.; Pristauz, G.; et al. Inference of transcription factor binding from cell-free DNA enables tumor subtype prediction and early detection. Nat. Commun. 2019, 10, 4666. [Google Scholar] [CrossRef]
- Alix-Panabières, C.; Pantel, K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016, 6, 479–491. [Google Scholar] [CrossRef]
- Pös, O.; Biró, O.; Szemes, T.; Nagy, B. Circulating cell-free nucleic acids: Characteristics and applications. Eur. J. Human. Genet. 2018, 26, 937–945. [Google Scholar] [CrossRef]
- Tivey, A.; Church, M.; Rothwell, D.; Dive, C.; Cook, N. Circulating tumour DNA—Looking beyond the blood. Nat. Rev. Clin. Oncol. 2022, 19, 600–612. [Google Scholar] [CrossRef]
- Song, P.; Wu, L.R.; Yan, Y.H.; Zhang, J.X.; Chu, T.; Kwong, L.N.; Patel, A.A.; Zhang, D.Y. Limitations and opportunities of technologies for the analysis of cell-free DNA in cancer diagnostics. Nat. Biomed. Eng. 2022, 6, 232–245. [Google Scholar] [CrossRef]
- Cutts, R.; Ulrich, L.; Beaney, M.; Robert, M.; Coakley, M.; Bunce, C.; Crestani, G.W.; Hrebien, S.; Kalashnikova, E.; Wu, H.T.; et al. Association of post-operative ctDNA detection with outcomes of patients with early breast cancers. ESMO Open 2024, 9, 103687. [Google Scholar] [CrossRef] [PubMed]
- Best, M.G.; Sol, N.; Kooi, I.; Tannous, J.; Westerman, B.A.; Rustenburg, F.; Schellen, P.; Verschueren, H.; Post, E.; Koster, J.; et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell 2015, 28, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Joosse, S.A.; Pantel, K. Tumor-Educated Platelets as Liquid Biopsy in Cancer Patients. Cancer Cell 2015, 28, 552–554. [Google Scholar] [CrossRef] [PubMed]
- In ‘t Veld, S.G.J.G.; Wurdinger, T. Tumor-educated platelets. Blood 2019, 133, 2359–2364. [Google Scholar] [CrossRef]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef]
- Milane, L.; Singh, A.; Mattheolabakis, G.; Suresh, M.; Amiji, M.M. Exosome mediated communication within the tumor microenvironment. J. Control Release 2015, 219, 278–294. [Google Scholar] [CrossRef]
- Singh, R.; Pochampally, R.; Watabe, K.; Lu, Z.; Mo, Y.Y. Exosome-mediated transfer of miR-10b promotes cell invasion in breast cancer. Mol. Cancer 2014, 13, 256. [Google Scholar] [CrossRef]
- Ferguson, S.; Weissleder, R. Modeling EV Kinetics for Use in Early Cancer Detection. Adv. Biosyst. 2020, 4, 1900305. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.M.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Samanta, S.; Rajasingh, S.; Drosos, N.; Zhou, Z.; Dawn, B.; Rajasingh, J. Exosomes: New molecular targets of diseases. Acta Pharmacol. Sin. 2018, 39, 501–513. [Google Scholar] [CrossRef]
- Reátegui, E.; Van Der Vos, K.E.; Lai, C.P.; Zeinali, M.; Atai, N.A.; Aldikacti, B.; Floyd, F.P., Jr.; H. Khankhel, A.; Thapar, V.; Hochberg, F.H.; et al. Engineered nanointerfaces for microfluidic isolation and molecular profiling of tumor-specific extracellular vesicles. Nat. Commun. 2018, 9, 175. [Google Scholar] [CrossRef] [PubMed]
- Tamrin, S.H.; Nezhad, A.S.; Sen, A. Label-Free Isolation of Exosomes Using Microfluidic Technologies. ACS Nano 2021, 15, 17047–17079. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Geng, Z.; Su, Y.; Fan, Z.; Wang, S.; Fang, W.; Chen, H. Label-Free Exosome Detection Based on a Low-Cost Plasmonic Biosensor Array Integrated with Microfluidics. Langmuir 2019, 35, 9816–9824. [Google Scholar] [CrossRef] [PubMed]
- Sung, B.H.; von Lersner, A.; Guerrero, J.; Krystofiak, E.S.; Inman, D.; Pelletier, R.; Zijlstra, A.; Ponik, S.M.; Weaver, A.M. A live cell reporter of exosome secretion and uptake reveals pathfinding behavior of migrating cells. Nat. Commun. 2020, 11, 2092. [Google Scholar] [CrossRef]
- Dash, M.; Palaniyandi, K.; Ramalingam, S.; Sahabudeen, S.; Raja, N.S. Exosomes isolated from two different cell lines using three different isolation techniques show variation in physical and molecular characteristics. Biochim. Biophys. Acta Biomembr. 2021, 1863, 183490. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Pereira, F.; de Matos, A.P.A.; Fernandes, M.; Baptista, P.V.; Fernandes, A.R. Smuggling gold nanoparticles across cell types—A new role for exosomes in gene silencing. Nanomedicine 2017, 13, 1389–1398. [Google Scholar] [CrossRef]
- Simpson, R.J.; Lim, J.W.; Moritz, R.L.; Mathivanan, S. Exosomes: Proteomic insights and diagnostic potential. Expert. Rev. Proteom. 2009, 6, 267–283. [Google Scholar] [CrossRef]
- Cocucci, E.; Meldolesi, J. Ectosomes; exosomes: Shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015, 25, 364–372. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar] [CrossRef]
- Théry, C.; Zitvogel, L.; Amigorena, S. Exosomes: Composition, biogenesis and function. Nat. Rev. Immunol. 2002, 2, 569–579. [Google Scholar] [CrossRef]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Roma-Rodrigues, C.; Fernandes, A.R.; Baptista, P.V. Exosome in tumour microenvironment: Overview of the crosstalk between normal and cancer cells. BioMed Res. Int. 2014, 2014, 179486. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.J.; Nieto, M.A. Epithelial-Mesenchymal Transitions in Development and Disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharmacol. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef]
- Arisan, E.D.; Rencuzogullari, O.; Cieza-Borrella, C.; Miralles Arenas, F.; Dwek, M.; Lange, S.; Uysal-Onganer, P. MiR-21 Is Required for the Epithelial–Mesenchymal Transition in MDA-MB-231 Breast Cancer Cells. Int. J. Mol. Sci. 2021, 22, 1557. [Google Scholar] [CrossRef]
- Wolf, P. The Nature and Significance of Platelet Products in Human Plasma. Br. J. Haematol. 1967, 13, 269–288. [Google Scholar] [CrossRef]
- Karimi, N.; Cvjetkovic, A.; Jang, S.C.; Crescitelli, R.; Hosseinpour Feizi, M.A.; Nieuwland, R.; Lötvall, J.; Lässer, C. Detailed analysis of the plasma extracellular vesicle proteome after separation from lipoproteins. Cell. Mol. Life Sci. 2018, 75, 2873–2886. [Google Scholar] [CrossRef]
- Zhang, H.; Freitas, D.; Kim, H.S.; Fabijanic, K.; Li, Z.; Chen, H.; Mark, M.T.; Molina, H.; Martin, A.B.; Bojmar, L.; et al. Identification of distinct nanoparticles and subsets of extracellular vesicles by asymmetric flow field-flow fractionation. Nat. Cell Biol. 2018, 20, 332–343. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular Vesicles Released by Glioblastoma Cells Stimulate Normal Astrocytes to Acquire a Tumor-Supportive Phenotype Via p53 and MYC Signaling Pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef]
- Shao, H.; Im, H.; Castro, C.M.; Breakefield, X.; Weissleder, R.; Lee, H. New Technologies for Analysis of Extracellular Vesicles. Chem. Rev. 2018, 118, 1917–1950. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A Basic Biological Phenomenon with Wideranging Implications in Tissue Kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.; Ma, C.; Zhou, T.; Dong, X.; Luo, Q.; Geng, L.; Ding, L.; Zhang, Y.; Zhang, L.; Li, N.; et al. Exosomes derived from gemcitabine-resistant cells transfer malignant phenotypic traits via delivery of miRNA-222-3p. Mol. Cancer 2017, 16, 132. [Google Scholar] [CrossRef] [PubMed]
- Krug, A.K.; Enderle, D.; Karlovich, C.; Priewasser, T.; Bentink, S.; Spiel, A.; Brinkmann, K.; Emenegger, J.; Grimm, D.G.; Castellanos-Rizaldos, E.; et al. Improved EGFR mutation detection using combined exosomal RNA and circulating tumor DNA in NSCLC patient plasma. Ann. Oncol. 2018, 29, 700–706. [Google Scholar] [CrossRef]
- Kim, Y.; Shin, S.; Kim, B.; Lee, K.A. Selecting short length nucleic acids localized in exosomes improves plasma EGFR mutation detection in NSCLC patients. Cancer Cell Int. 2019, 19, 251. [Google Scholar] [CrossRef]
- Jin, X.; Chen, Y.; Chen, H.; Fei, S.; Chen, D.; Cai, X.; Liu, L.; Lin, B.; Su, H.; Zhao, L.; et al. Evaluation of tumor-derived exosomal miRNA as potential diagnostic biomarkers for early-stage non-small cell lung cancer using next-generation sequencing. Clin. Cancer Res. 2017, 23, 5311–5319. [Google Scholar] [CrossRef]
- Zhang, X.; Sai, B.; Wang, F.; Wang, L.; Wang, Y.; Zheng, L.; Li, G.; Tang, J.; Xiang, J. Hypoxic BMSC-derived exosomal miRNAs promote metastasis of lung cancer cells via STAT3-induced EMT. Mol. Cancer 2019, 18, 40. [Google Scholar] [CrossRef]
- Zhang, N.; Nan, A.; Chen, L.; Li, X.; Jia, Y.; Qiu, M.; Dai, X.; Zhou, H.; Zhu, J.; Zhang, H.; et al. Circular RNA circSATB2 promotes progression of non-small cell lung cancer cells. Mol. Cancer 2020, 19, 101. [Google Scholar] [CrossRef]
- Sandfeld-Paulsen, B.; Aggerholm-Pedersen, N.; Bæk, R.; Jakobsen, K.R.; Meldgaard, P.; Folkersen, B.H.; Rasmussen, T.R.; Varming, K.; Jørgensen, M.M.; Sorensen, B.S. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol. Oncol. 2016, 10, 1595. [Google Scholar] [CrossRef]
- Yang, Y.; Kannisto, E.; Yu, G.; Reid, M.E.; Patnaik, S.K.; Wu, Y. An Immuno-Biochip Selectively Captures Tumor-Derived Exosomes and Detects Exosomal RNAs for Cancer Diagnosis. ACS Appl. Mater. Interfaces 2018, 10, 43375–43386. [Google Scholar] [CrossRef]
- Zhai, L.Y.; Li, M.X.; Pan, W.L.; Chen, Y.; Li, M.M.; Pang, J.X.; Zheng, L.; Chen, J.X.; Duan, W.J. In Situ Detection of Plasma Exosomal MicroRNA-1246 for Breast Cancer Diagnostics by a Au Nanoflare Probe. ACS Appl. Mater. Interfaces 2018, 10, 39478–39486. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martínez, A.; de Miguel-Pérez, D.; Ortega, F.G.; García-Puche, J.L.; Robles-Fernández, I.; Exposito, J.; Martorell-Marugan, J.; Carmona-Sáez, P.; Garrido-Navas, M.D.C.; Rolfo, C.; et al. Exosomal miRNA profile as complementary tool in the diagnostic and prediction of treatment response in localized breast cancer under neoadjuvant chemotherapy. Breast Cancer Res. 2019, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Tao, J.; Li, Y.; Feng, Y.; Ju, H.; Wang, Z.; Ding, L. Quantitative Localized Analysis Reveals Distinct Exosomal Protein-Specific Glycosignatures: Implications in Cancer Cell Subtyping, Exosome Biogenesis, and Function. J. Am. Chem. Soc. 2020, 142, 7404–7412. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.H.; Xue, L.; Hsu, C.C.; Paez, J.S.P.; Pan, L.; Andaluz, H.; Wendt, M.K.; Iliuk, A.B.; Zhu, J.K.; Tao, W.A. Phosphoproteins in extracellular vesicles as candidate markers for breast cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 3175–3180. [Google Scholar] [CrossRef]
- Fang, S.; Tian, H.; Li, X.; Jin, D.; Li, X.; Kong, J.; Yang, C.; Yang, X.; Lu, Y.; Luo, Y.; et al. Clinical application of a microfluidic chip for immunocapture and quantification of circulating exosomes to assist breast cancer diagnosis and molecular classification. PLoS ONE 2017, 12, e0175050. [Google Scholar] [CrossRef]
- Wang, X.; Zhong, W.; Bu, J.; Li, Y.; Li, R.; Nie, R.; Xiao, C.; Ma, K.; Huang, X.; Li, Y. Exosomal protein CD82 as a diagnostic biomarker for precision medicine for breast cancer. Mol. Carcinog. 2019, 58, 674–685. [Google Scholar] [CrossRef]
- Zhao, J.; Liu, C.; Li, Y.; Ma, Y.; Deng, J.; Li, L.; Sun, J. Thermophoretic Detection of Exosomal microRNAs by Nanoflares. J. Am. Chem. Soc. 2020, 142, 4996–5001. [Google Scholar] [CrossRef]
- Xie, M.; Yu, T.; Jing, X.; Ma, L.; Fan, Y.; Yang, F.; Ma, P.; Jiang, H.; Wu, X.; Shu, Y.; et al. Exosomal circSHKBP1 promotes gastric cancer progression via regulating the miR-582-3p/HUR/VEGF axis and suppressing HSP90 degradation. Mol. Cancer 2020, 19, 112. [Google Scholar] [CrossRef]
- Zhao, R.; Zhang, Y.; Zhang, X.; Yang, Y.; Zheng, X.; Li, X.; Liu, Y.; Zhang, Y. Exosomal long noncoding RNA HOTTIP as potential novel diagnostic and prognostic biomarker test for gastric cancer. Mol. Cancer 2018, 17, 68. [Google Scholar] [CrossRef]
- Lin, L.Y.; Yang, L.; Zeng, Q.; Wang, L.; Chen, M.L.; Zhao, Z.H.; Ye, G.D.; Luo, Q.C.; Lv, P.Y.; Guo, Q.W.; et al. Tumor-originated exosomal lncUEGC1 as a circulating biomarker for early-stage gastric cancer. Mol. Cancer 2018, 17, 84. [Google Scholar] [CrossRef]
- Bjørnetrø, T.; Redalen, K.R.; Meltzer, S.; Thusyanthan, N.S.; Samiappan, R.; Jegerschöld, C.; Handeland, K.R.; Ree, A.H. An experimental strategy unveiling exosomal microRNAs 486-5p, 181a-5p and 30d-5p from hypoxic tumour cells as circulating indicators of high-risk rectal cancer. J. Extracell. Vesicles 2019, 8, 1567219. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Li, J.; Gong, Y.; Wu, Q.; Tan, S.; Sun, D.; Xu, X.; Zuo, Y.; Zhao, Y.; Wei, Y.Q.; et al. Exosomal tRNA-derived small RNA as a promising biomarker for cancer diagnosis. Mol. Cancer 2019, 18, 74. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.F.; Gao, C.; Huang, X.Y.; Lu, J.C.; Guo, X.J.; Shi, G.M.; Cai, J.B.; Ke, A.W. Cancer cell-derived exosomal circUHRF1 induces natural killer cell exhaustion and may cause resistance to anti-PD1 therapy in hepatocellular carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.P.; Wang, C.Y.; Jin, X.H.; Li, M.; Wang, F.W.; Huang, W.J.; Yun, J.P.; Xu, R.H.; Cai, Q.Q.; Xie, D. Acidic microenvironment up-regulates exosomal mir-21 and mir-10b in early-stage hepatocellular carcinoma to promote cancer cell proliferation and metastasis. Theranostics 2019, 9, 1965–1979. [Google Scholar] [CrossRef]
- Bernard, V.; Kim, D.U.; San Lucas, F.A.; Castillo, J.; Allenson, K.; Mulu, F.C.; Stephens, B.M.; Huang, J.; Semaan, A.; Guerrero, P.A.; et al. Circulating Nucleic Acids Are Associated with Outcomes of Patients with Pancreatic Cancer. Gastroenterology 2019, 156, 108–118.e4. [Google Scholar] [CrossRef]
- Allenson, K.; Castillo, J.; San Lucas, F.A.; Scelo, G.; Kim, D.U.; Bernard, V.; Davis, G.; Kumar, T.; Katz, M.; Overman, M.J.; et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann. Oncol. 2017, 28, 741–747. [Google Scholar] [CrossRef]
- Kimura, H.; Yamamoto, H.; Harada, T.; Fumoto, K.; Osugi, Y.; Sada, R.; Maehara, N.; Hikita, H.; Mori, S.; Eguchi, H.; et al. CKAP4, a DKK1 receptor, is a biomarker in exosomes derived from pancreatic cancer and a molecular target for therapy. Clin. Cancer Res. 2019, 25, 1936–1947. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N.; et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef]
- Del Re, M.; Biasco, E.; Crucitta, S.; Derosa, L.; Rofi, E.; Orlandini, C.; Miccoli, M.; Galli, L.; Falcone, A.; Jenster, G.W.; et al. The Detection of Androgen Receptor Splice Variant 7 in Plasma-derived Exosomal RNA Strongly Predicts Resistance to Hormonal Therapy in Metastatic Prostate Cancer Patients. Eur. Urol. 2017, 71, 680–687. [Google Scholar] [CrossRef]
- Rodríguez, M.; Bajo-Santos, C.; Hessvik, N.P.; Lorenz, S.; Fromm, B.; Berge, V.; Sandvig, K.; Linē, A.; Llorente, A. Identification of non-invasive miRNAs biomarkers for prostate cancer by deep sequencing analysis of urinary exosomes. Mol. Cancer 2017, 16, 156. [Google Scholar] [CrossRef]
- Huang, X.; Yuan, T.; Liang, M.; Du, M.; Xia, S.; Dittmar, R.; Wang, D.; See, W.; Costello, B.A.; Quevedo, F.; et al. Exosomal miR-1290 and miR-375 as prognostic markers in castration-resistant prostate cancer. Eur. Urol. 2015, 67, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Logozzi, M.; Angelini, D.F.; Iessi, E.; Mizzoni, D.; Di Raimo, R.; Federici, C.; Lugini, L.; Borsellino, G.; Gentilucci, A.; Pierella, F.; et al. Increased PSA expression on prostate cancer exosomes in in vitro condition and in cancer patients. Cancer Lett. 2017, 403, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Fujita, Y.; Matsuda, Y.; Arai, T.; Horie, K.; Kameyama, K.; Kato, T.; Masunaga, K.; Kasuya, Y.; Tanaka, M.; et al. Gamma-glutamyltransferase activity in exosomes as a potential marker for prostate cancer. BMC Cancer 2017, 17, 316. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Du, M.; Wang, X.; Xu, W.; Liang, J.; Wang, W.; Lv, Q.; Qin, C.; Chu, H.; Wang, M.; et al. Exosome-transmitted long non-coding RNA PTENP1 suppresses bladder cancer progression. Mol. Cancer 2018, 17, 143. [Google Scholar] [CrossRef]
- Chen, C.; Luo, Y.; He, W.; Zhao, Y.; Kong, Y.; Liu, H.; Zhong, G.; Li, Y.; Li, J.; Huang, J.; et al. Exosomal long noncoding RNA LNMAT2 promotes lymphatic metastasis in bladder cancer. J. Clin. Investig. 2020, 130, 404–421. [Google Scholar] [CrossRef]
- Zhan, Y.; Du, L.; Wang, L.; Jiang, X.; Zhang, S.; Li, J.; Yan, K.; Duan, W.; Zhao, Y.; Wang, L.; et al. Expression signatures of exosomal long non-coding RNAs in urine serve as novel non-invasive biomarkers for diagnosis and recurrence prediction of bladder cancer. Mol. Cancer 2018, 17, 142. [Google Scholar] [CrossRef]
- Li, J.; Chen, Y.; Guo, X.; Zhou, L.; Jia, Z.; Peng, Z.; Tang, Y.; Liu, W.; Zhu, B.; Wang, L.; et al. GPC1 exosome and its regulatory miRNAs are specific markers for the detection and target therapy of colorectal cancer. J. Cell Mol. Med. 2017, 21, 838–847. [Google Scholar] [CrossRef]
- Yokoyama, S.; Takeuchi, A.; Yamaguchi, S.; Mitani, Y.; Watanabe, T.; Matsuda, K.; Hotta, T.; Shively, J.E.; Yamaue, H. Clinical implications of carcinoembryonic antigen distribution in serum exosomal fraction—Measurement by ELISA. PLoS ONE 2017, 12, e0183337. [Google Scholar] [CrossRef]
- Arbelaiz, A.; Azkargorta, M.; Krawczyk, M.; Santos-Laso, A.; Lapitz, A.; Perugorria, M.J.; Erice, O.; Gonzalez, E.; Jimenez-Agüero, R.; Lacasta, A.; et al. Serum extracellular vesicles contain protein biomarkers for primary sclerosing cholangitis and cholangiocarcinoma. Hepatology 2017, 66, 1125–1143. [Google Scholar] [CrossRef]
- Tang, M.K.; Yue, P.Y.; Ip, P.P.; Huang, R.L.; Lai, H.C.; Cheung, A.N.; Tse, K.Y.; Ngan, H.Y.; Wong, A.S. Soluble E-cadherin promotes tumor angiogenesis and localizes to exosome surface. Nat. Commun. 2018, 9, 2270. [Google Scholar] [CrossRef]
- Meng, X.; Müller, V.; Milde-Langosch, K.; Trillsch, F.; Pantel, K.; Schwarzenbach, H. Diagnostic and prognostic relevance of circulating exosomal miR-373, miR-200a, miR-200b and miR-200c in patients with epithelial ovarian cancer. Oncotarget 2016, 7, 16923. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Yang, Y.; Zeng, Y.; He, M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip 2016, 16, 489–496. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Hou, L.; Ma, Y.; Zhou, L.; Wang, F.; Cheng, B.; Wang, W.; Lu, B.; Liu, P.; Lu, W.; et al. Exosomal let-7d-3p and miR-30d-5p as diagnostic biomarkers for non-invasive screening of cervical cancer and its precursors. Mol. Cancer 2019, 18, 76. [Google Scholar] [CrossRef] [PubMed]
- Iaccino, E.; Mimmi, S.; Dattilo, V.; Marino, F.; Candeloro, P.; Di Loria, A.; Marimpietri, D.; Pisano, A.; Albano, F.; Vecchio, E.; et al. Monitoring multiple myeloma by idiotype-specific peptide binders of tumor-derived exosomes. Mol. Cancer 2017, 16, 159. [Google Scholar] [CrossRef]
- Manier, S.; Liu, C.J.; Avet-Loiseau, H.; Park, J.; Shi, J.; Campigotto, F.; Salem, K.Z.; Huynh, D.; Glavey, S.V.; Rivotto, B.; et al. Prognostic role of circulating exosomal miRNAs in multiple myeloma. Blood J. Am. Soc. Hematol. 2017, 129, 2429–2436. [Google Scholar] [CrossRef]
- Xu, H.; Han, H.; Song, S.; Yi, N.; Qian, C.A.; Qiu, Y.; Zhou, W.; Hong, Y.; Zhuang, W.; Li, Z.; et al. Exosome-transmitted PSMA3 and PSMA3-AS1 promote proteasome inhibitor resistance in multiple myeloma. Clin. Cancer Res. 2019, 25, 1923–1935. [Google Scholar] [CrossRef]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef]
- Brinkman, K.; Meyer, L.; Bickel, A.; Enderle, D.; Berking, C.; Skog, J.; Noerholm, M. Extracellular vesicles from plasma have higher tumour RNA fraction than platelets. J. Extracell. Vesicles 2020, 9, 1741176. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; Van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry Jr, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Patel, G.K.; Khan, M.A.; Zubair, H.; Srivastava, S.K.; Khushman, M.D.; Singh, S.; Singh, A.P. Comparative analysis of exosome isolation methods using culture supernatant for optimum yield, purity and downstream applications. Sci. Rep. 2019, 9, 5335. [Google Scholar] [CrossRef]
- Liu, C.; Guo, J.; Tian, F.; Yang, N.; Yan, F.; Ding, Y.; Wei, J.; Hu, G.; Nie, G.; Sun, J. Field-Free Isolation of Exosomes from Extracellular Vesicles by Microfluidic Viscoelastic Flows. ACS Nano 2017, 11, 6968–6976. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zhao, J.; Tian, F.; Chang, J.; Zhang, W.; Sun, J. λ-DNA- and Aptamer-Mediated Sorting and Analysis of Extracellular Vesicles. J. Am. Chem. Soc. 2019, 141, 3817–3821. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Yang, X.; Yang, R.; Fatima, Z.; Xie, P.; Liu, F.; Cai, T.; Xu, X.; Zhou, C.; Jia, Z.; et al. Simultaneous Isolation and Preparation of Extracellular Vesicles by Circular Multicavity Electrophoresis. Anal. Chem. 2025, 97, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Liu, C.; Cheng, Y.; Li, Y.; Deng, J.; Bai, L.; Qin, L.; Mei, H.; Zeng, M.; Tian, F.; et al. Cascaded microfluidic circuits for pulsatile filtration of extracellular vesicles from whole blood for early cancer diagnosis. Sci. Adv. 2023, 9, eade2819. [Google Scholar] [CrossRef]
- Marassi, V.; Giordani, S.; Placci, A.; Punzo, A.; Caliceti, C.; Zattoni, A.; Reschiglian, P.; Roda, B.; Roda, A. Emerging Microfluidic Tools for Simultaneous Exosomes and Cargo Biosensing in Liquid Biopsy: New Integrated Miniaturized FFF-Assisted Approach for Colon Cancer Diagnosis. Sensors 2023, 23, 9432. [Google Scholar] [CrossRef]
- Midekessa, G.; Godakumara, K.; Ord, J.; Viil, J.; Lättekivi, F.; Dissanayake, K.; Kopanchuk, S.; Rinken, A.; Andronowska, A.; Bhattacharjee, S.; et al. Zeta Potential of Extracellular Vesicles: Toward Understanding the Attributes that Determine Colloidal Stability. ACS Omega 2020, 5, 16701–16710. [Google Scholar] [CrossRef]
- Thakur, A.; Qiu, G.; Siu-Pang, N.G.; Guan, J.; Yue, J.; Lee, Y.; Wu, C.M.L. Direct detection of two different tumor-derived extracellular vesicles by SAM-AuNIs LSPR biosensor. Biosens. Bioelectron. 2017, 94, 400–407. [Google Scholar] [CrossRef]
- Ibsen, S.D.; Wright, J.; Lewis, J.M.; Kim, S.; Ko, S.Y.; Ong, J.; Manouchehri, S.; Vyas, A.; Akers, J.; Chen, C.C.; et al. Rapid Isolation and Detection of Exosomes and Associated Biomarkers from Plasma. ACS Nano 2017, 11, 6641–6651. [Google Scholar] [CrossRef]
- Bu, Y.; Wang, J.; Ni, S.; Lu, Z.; Guo, Y.; Yobas, L. High-Performance Gel-Free and Label-Free Size Fractionation of Extracellular Vesicles with Two-Dimensional Electrophoresis in a Microfluidic Artificial Sieve. Anal. Chem. 2024, 96, 3508–3516. [Google Scholar] [CrossRef]
- Chen, Y.S.; Lai, C.P.K.; Chen, C.; Lee, G.B. Isolation and recovery of extracellular vesicles using optically-induced dielectrophoresis on an integrated microfluidic platform. Lab Chip 2021, 21, 1475–1483. [Google Scholar] [CrossRef]
- Gwak, H.; Park, S.; Yu, H.; Hyun, K.A.; Jung, H.I. A modular microfluidic platform for serial enrichment and harvest of pure extracellular vesicles. Analyst 2022, 147, 1117–1127. [Google Scholar] [CrossRef] [PubMed]
- Yaman, S.; Devoe, T.; Aygun, U.; Parlatan, U.; Bobbili, M.R.; Karim, A.H.; Grillari, J.; Durmus, N.G. EV-Lev: Extracellular vesicle isolation from human plasma using microfluidic magnetic levitation device. Lab Chip 2025, 25, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Van Der Pol, E.; Hoekstra, A.G.; Sturk, A.; Otto, C.; Van Leeuwen, T.G.; Nieuwland, R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J. Thromb. Haemost. 2010, 8, 2596–2607. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, N.; Chen, J.I.L. Sensing Biomarkers with Plasmonics. Anal. Chem. 2020, 92, 7373–7381. [Google Scholar] [CrossRef]
- Lee, T.H.; Hirst, D.J.; Kulkarni, K.; Del Borgo, M.P.; Aguilar, M.I. Exploring Molecular-Biomembrane Interactions with Surface Plasmon Resonance and Dual Polarization Interferometry Technology: Expanding the Spotlight onto Biomembrane Structure. Chem. Rev. 2018, 118, 5392–5487. [Google Scholar] [CrossRef]
- Liu, J.; Jalali, M.; Mahshid, S.; Wachsmann-Hogiu, S. Are plasmonic optical biosensors ready for use in point-of-need applications? Analyst 2020, 145, 364–384. [Google Scholar] [CrossRef]
- Krafft, C.; Wilhelm, K.; Eremin, A.; Nestel, S.; von Bubnoff, N.; Schultze-Seemann, W.; Popp, J.; Nazarenko, I. A specific spectral signature of serum and plasma-derived extracellular vesicles for cancer screening. Nanomedicine 2017, 13, 835–841. [Google Scholar] [CrossRef]
- Saleh, B.E.A.; Teich, M.C. Fundamentals of Photonics; Wiley: Hoboken, NJ, USA, 2007. [Google Scholar] [CrossRef]
- Smith, Z.J.; Lee, C.; Rojalin, T.; Carney, R.P.; Hazari, S.; Knudson, A.; Lam, K.; Saari, H.; Ibañez, E.L.; Viitala, T.; et al. Single exosome study reveals subpopulations distributed among cell lines with variability related to membrane content. J. Extracell. Vesicles 2015, 4, 28533. [Google Scholar] [CrossRef]
- Tatischeff, I.; Larquet, E.; Falcón-Pérez, J.M.; Turpin, P.Y.; Kruglik, S.G. Fast characterisation of cell-derived extracellular vesicles by nanoparticles tracking analysis, cryo-electron microscopy, and Raman tweezers microspectroscopy. J. Extracell. Vesicles 2012, 1, 19179. [Google Scholar] [CrossRef]
- Stremersch, S.; Marro, M.; Pinchasik, B.E.; Baatsen, P.; Hendrix, A.; De Smedt, S.C.; Loza-Alvarez, P.; Skirtach, A.G.; Raemdonck, K.; Braeckmans, K. Identification of individual exosome-like vesicles by surface enhanced raman spectroscopy. Small 2016, 12, 3292–3301. [Google Scholar] [CrossRef]
- Tirinato, L.; Gentile, F.; Di Mascolo, D.; Coluccio, M.L.; Das, G.; Liberale, C.; Pullano, S.A.; Perozziello, G.; Francardi, M.; Accardo, A.; et al. SERS analysis on exosomes using super-hydrophobic surfaces. Microelectron. Eng. 2012, 97, 337–340. [Google Scholar] [CrossRef]
- Yan, Z.; Dutta, S.; Liu, Z.; Yu, X.; Mesgarzadeh, N.; Ji, F.; Bitan, G.; Xie, Y.H. A Label-free platform for identification of exosomes from different sources. ACS Sens. 2019, 4, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Carney, R.P.; Hazari, S.; Smith, Z.J.; Knudson, A.; Robertson, C.S.; Lam, K.S.; Wachsmann-Hogiu, S. 3D plasmonic nanobowl platform for the study of exosomes in solution. Nanoscale 2015, 7, 9290–9297. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Carney, R.; Lam, K.; Chan, J.W. SERS analysis of selectively captured exosomes using an integrin-specific peptide ligand. J. Raman Spectrosc. 2017, 48, 1771–1776. [Google Scholar] [CrossRef]
- Shin, H.; Jeong, H.; Park, J.; Hong, S.; Choi, Y. Correlation between Cancerous Exosomes and Protein Markers Based on Surface-Enhanced Raman Spectroscopy (SERS) and Principal Component Analysis (PCA). ACS Sens. 2018, 3, 2637–2643. [Google Scholar] [CrossRef]
- Guerrini, L.; Garcia-Rico, E.; O’loghlen, A.; Giannini, V.; Alvarez-Puebla, R.A. Surface-enhanced raman scattering (Sers) spectroscopy for sensing and characterization of exosomes in cancer diagnosis. Cancers 2021, 13, 2179. [Google Scholar] [CrossRef]
- Rojalin, T.; Koster, H.J.; Liu, J.; Mizenko, R.R.; Tran, D.; Wachsmann-Hogiu, S.; Carney, R.P. Hybrid Nanoplasmonic Porous Biomaterial Scaffold for Liquid Biopsy Diagnostics Using Extracellular Vesicles. ACS Sens. 2020, 5, 2820–2833. [Google Scholar] [CrossRef]
- Kwizera, E.A.; O’Connor, R.; Vinduska, V.; Williams, M.; Butch, E.R.; Snyder, S.E.; Chen, X.; Huang, X. Molecular detection and analysis of exosomes using surface-enhanced Raman scattering gold nanorods and a miniaturized device. Theranostics 2018, 8, 2722–2738. [Google Scholar] [CrossRef]
- Li, G.; Zhu, N.; Zhou, J.; Kang, K.; Zhou, X.; Ying, B.; Yi, Q.; Wu, Y. A magnetic surface-enhanced Raman scattering platform for performing successive breast cancer exosome isolation and analysis. J. Mater. Chem. B 2021, 9, 2709–2716. [Google Scholar] [CrossRef]
- Park, J.; Hwang, M.; Choi, B.; Jeong, H.; Jung, J.H.; Kim, H.K.; Hong, S.; Park, J.H.; Choi, Y. Exosome Classification by Pattern Analysis of Surface-Enhanced Raman Spectroscopy Data for Lung Cancer Diagnosis. Anal. Chem. 2017, 89, 6695–6701. [Google Scholar] [CrossRef]
- Carmicheal, J.; Hayashi, C.; Huang, X.; Liu, L.; Lu, Y.; Krasnoslobodtsev, A.; Lushnikov, A.; Kshirsagar, P.G.; Patel, A.; Jain, M.; et al. Label-free characterization of exosome via surface enhanced Raman spectroscopy for the early detection of pancreatic cancer. Nanomedicine 2019, 16, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Seo, D.; Choi, Y. Extracellular Vesicle Identification Using Label-Free Surface-Enhanced Raman Spectroscopy: Detection and Signal Analysis Strategies. Molecules 2020, 25, 5209. [Google Scholar] [CrossRef] [PubMed]
- Jalali, M.; Hosseini, I.I.; AbdelFatah, T.; Montermini, L.; Hogiu, S.W.; Rak, J.; Mahshid, S. Plasmonic nanobowtiefluidic device for sensitive detection of glioma extracellular vesicles by Raman spectrometry. Lab Chip 2021, 21, 855–866. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Peng, X.; Yang, Y.; Yi, J.; Zhao, D.; Bao, Q.; Long, S.; Yu, S.X.; Xu, X.X.; Liu, B.; et al. Integrated microfluidic-SERS for exosome biomarker profiling and osteosarcoma diagnosis. Biosens. Bioelectron. 2022, 217, 114709. [Google Scholar] [CrossRef]
- Masson, J.F. Surface Plasmon Resonance Clinical Biosensors for Medical Diagnostics. ACS Sens. 2017, 2, 16–30. [Google Scholar] [CrossRef]
- Tang, Y.; Zeng, X.; Liang, J. Surface plasmon resonance: An introduction to a surface spectroscopy technique. J. Chem. Educ. 2010, 87, 742–746. [Google Scholar] [CrossRef]
- Li, T.D.; Zhang, R.; Chen, H.; Huang, Z.P.; Ye, X.; Wang, H.; Deng, A.M.; Kong, J.L. An ultrasensitive polydopamine bi-functionalized SERS immunoassay for exosome-based diagnosis and classification of pancreatic cancer. Chem. Sci. 2018, 9, 5372–5382. [Google Scholar] [CrossRef]
- Di Noto, G.; Bugatti, A.; Zendrini, A.; Mazzoldi, E.L.; Montanelli, A.; Caimi, L.; Rusnati, M.; Ricotta, D.; Bergese, P. Merging colloidal nanoplasmonics and surface plasmon resonance spectroscopy for enhanced profiling of multiple myeloma-derived exosomes. Biosens. Bioelectron. 2016, 77, 518–524. [Google Scholar] [CrossRef]
- Im, H.; Shao, H.; Park, Y.I.; Peterson, V.M.; Castro, C.M.; Weissleder, R.; Lee, H. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol. 2014, 32, 490–495. [Google Scholar] [CrossRef]
- Grasso, L.; Wyss, R.; Weidenauer, L.; Thampi, A.; Demurtas, D.; Prudent, M.; Lion, N.; Vogel, H. Molecular screening of cancer-derived exosomes by surface plasmon resonance spectroscopy. Anal. Bioanal. Chem. 2015, 407, 5425–5432. [Google Scholar] [CrossRef]
- Sina, A.A.I.; Vaidyanathan, R.; Dey, S.; Carrascosa, L.G.; Shiddiky, M.J.A.; Trau, M. Real time and label free profiling of clinically relevant exosomes. Sci. Rep. 2016, 6, 30460. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Zeng, X.; An, Z.; Yang, Y.; Eisenbaum, M.; Gu, X.; Jornet, J.M.; Dy, G.K.; Reid, M.E.; Gan, Q.; et al. Sensitive Detection of Exosomal Proteins via a Compact Surface Plasmon Resonance Biosensor for Cancer Diagnosis. ACS Sens. 2018, 3, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
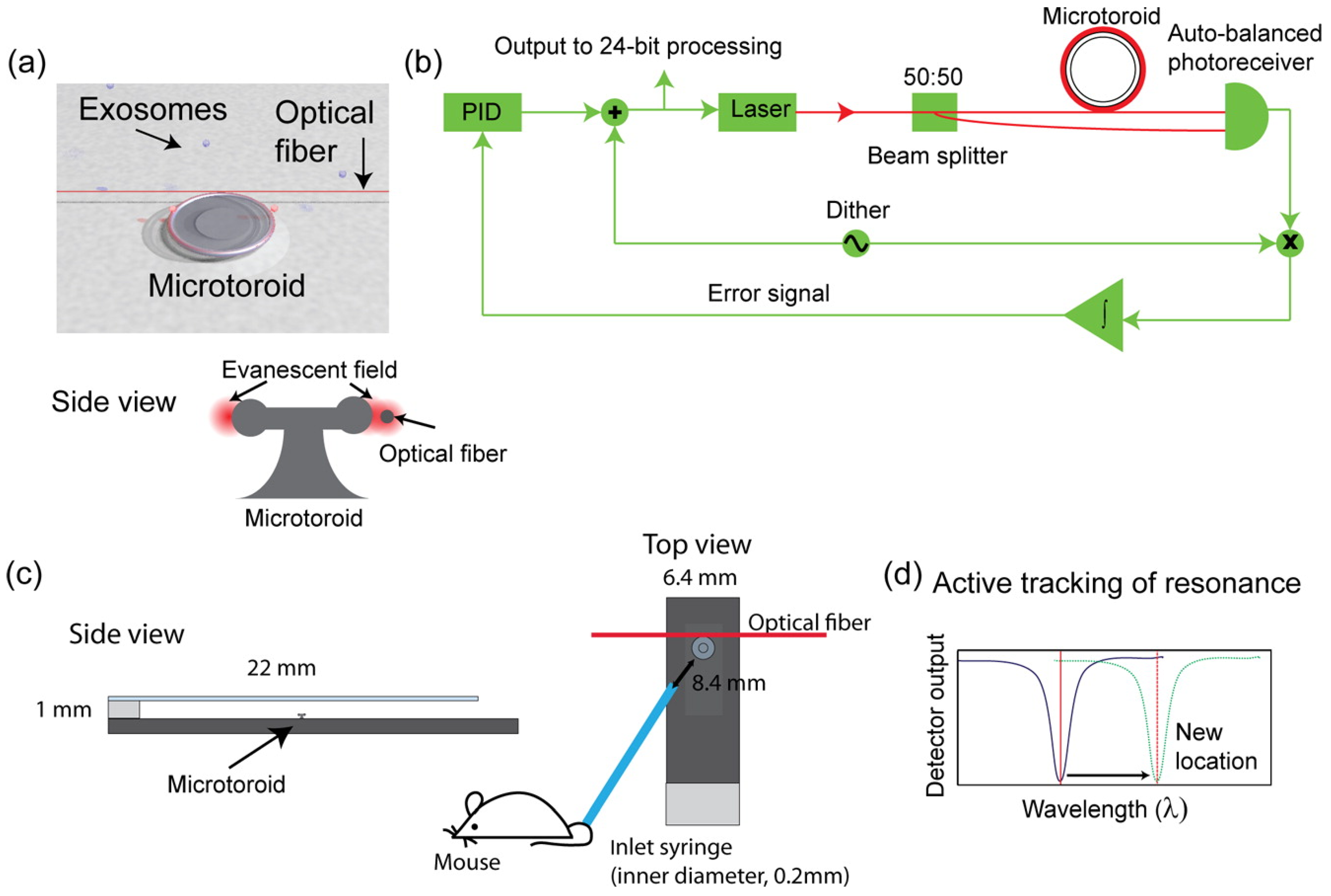
- Su, J. Label-Free Single Exosome Detection Using Frequency-Locked Microtoroid Optical Resonators. ACS Photonics 2015, 2, 1241–1245. [Google Scholar] [CrossRef]
- Rupert, D.L.; Lässer, C.; Eldh, M.; Block, S.; Zhdanov, V.P.; Lotvall, J.O.; Bally, M.; Höök, F. Determination of exosome concentration in solution using surface plasmon resonance spectroscopy. Anal. Chem. 2014, 86, 5929–5936. [Google Scholar] [CrossRef]
- Wu, W.; Yu, X.; Wu, J.; Wu, T.; Fan, Y.; Chen, W.; Zhao, M.; Wu, H.; Li, X.; Ding, S. Surface plasmon resonance imaging-based biosensor for multiplex and ultrasensitive detection of NSCLC-associated exosomal miRNAs using DNA programmed heterostructure of Au-on-Ag. Biosens. Bioelectron. 2021, 175, 112835. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, K.; Cui, J.; Liu, H.; Bu, X.; Ma, H.; Wang, W.; Gong, H.; Lausted, C.; Hood, L.; et al. Label-free quantitative detection of tumor-derived exosomes through surface plasmon resonance imaging. Anal. Chem. 2014, 86, 8857–8864. [Google Scholar] [CrossRef]
- Joshi, G.K.; Deitz-McElyea, S.; Liyanage, T.; Lawrence, K.; Mali, S.; Sardar, R.; Korc, M. Label-Free Nanoplasmonic-Based Short Noncoding RNA Sensing at Attomolar Concentrations Allows for Quantitative and Highly Specific Assay of MicroRNA-10b in Biological Fluids and Circulating Exosomes. ACS Nano 2015, 9, 11075–11089. [Google Scholar] [CrossRef]
- Bohren, C.F.; Huffman, D.R. Absorption and Scattering of Light by Small Particles; Wiley: Hoboken, NJ, USA, 2008. [Google Scholar] [CrossRef]
- Jung, L.S.; Campbell, C.T.; Chinowsky, T.M.; Mar, M.N.; Yee, S.S. Quantitative Interpretation of the Response of Surface Plasmon Resonance Sensors to Adsorbed Films. Langmuir 1998, 14, 5636–5648. [Google Scholar] [CrossRef]
- Jiang, Y.; Shi, M.; Liu, Y.; Wan, S.; Cui, C.; Zhang, L.; Tan, W. Aptamer/AuNP Biosensor for Colorimetric Profiling of Exosomal Proteins. Angew. Chem. 2017, 129, 12078–12082. [Google Scholar] [CrossRef]
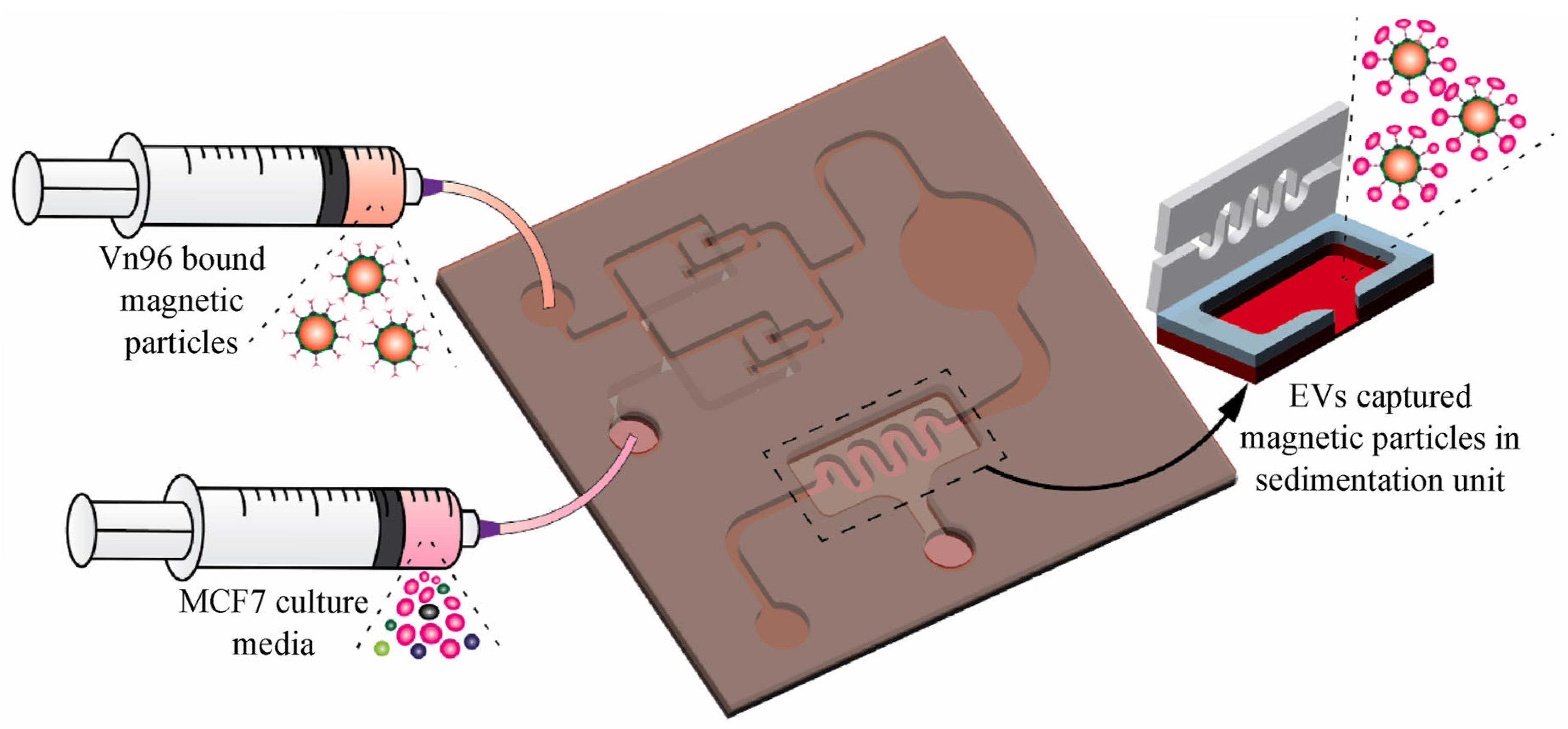
- Bathini, S.; Pakkiriswami, S.; Ouellette, R.J.; Ghosh, A.; Packirisamy, M. Magnetic particle based liquid biopsy chip for isolation of extracellular vesicles and characterization by gene amplification. Biosens. Bioelectron. 2021, 194, 113585. [Google Scholar] [CrossRef]
- Xu, C.; Thakur, A.; Li, Z.; Yang, T.; Zhao, C.; Li, Y.; Lee, Y.; Wu, C.M.L. Determination of glioma cells’ malignancy and their response to TMZ via detecting exosomal BIGH3 by a TiO2-CTFE-AuNIs plasmonic biosensor. Chem. Eng. J. 2021, 415, 128948. [Google Scholar] [CrossRef]
- Liyanage, T.; Alharbi, B.; Quan, L.; Esquela-Kerscher, A.; Slaughter, G. Plasmonic-Based Biosensor for the Early Diagnosis of Prostate Cancer. ACS Omega 2022, 7, 2411–2418. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Lehrich, B.M.; Zheng, S.; Lu, M. Emerging methods in biomarker identification for extracellular vesicle-based liquid biopsy. J. Extracell. Vesicles 2021, 10, e12090. [Google Scholar] [CrossRef] [PubMed]



| Tests (Company) ☐SC|☐D|☐PT | Cancers | Marker | Sample | Comments | Refs. |
|---|---|---|---|---|---|
| Guardant360 (Guardant Health, Palo Alto, CA, USA) ☑D | Lung (NSCLC), Breast, Colorectal, Prostate | 100 + ctDNA | Blood | Effective for advanced stage (stage III or IV) cancer. FDA approved in 2020. | [8,9,10] |
| FoundationOne Liquid (Foundation Medicine, Boston, MA, USA) ☑D | NSCLC | 70 + ctDNA | Blood | For patients with advanced cancer. FDA approved in 2020. | [11] |
| Epi pro Colon (Epigenomics AG Heidelberg, Germany) ☑SC, ☑D | colorectal cancer | methylated Septin 9 DNA | Blood | For individuals aged 50 and older who are at average risk for colorectal cancer, this is the first blood-based test available. FDA approved in 2016. | [12] |
| Cologuard (Exact Sciences, Madison, WI, USA) ☑D | colorectal cancer | DNA | Stool | The first non-invasive DNA screening test for colorectal cancer is intended to screen adults 45 years of age and older who are at average risk for colorectal cancer. FDA approved in 2014. | [13] |
| CELLSEARCH CTC kit (Menarini Silicon Biosystems, Inc., Huntingdon Valley, PA, USA) ☑D | Breast, Prostate and Colorectal | CTC | Blood | Test approved by Health Canada in 2010. First FDA-cleared test (2004) for the enumeration of circulating tumor cells in peripheral blood. | [14] |
| Laboratory-developed test (LDT) regulated by Clinical Laboratory Improvement Amendments (CLIA) | |||||
| Galleri (GRAIL, Menlo Park, CA, USA) ☑SC | Multiple cancer screening | ctDNA (methylation markers and machine learning for cancer detection) | Blood | [15] | |
| OncoBEAM (Sysmex Inostics, Baltimore, MD, USA) ☑SC, ☑PT | Lung, Colorectal, and other | ctDNA | Blood | [16,17] | |
| CancerSEEK (Johns Hopkins University, Baltimore, MD, USA) ☑SC | Ovarian, Liver, Stomach, and other. | ctDNA | Blood | ctDNA and protein markers for early cancer detection. | [18] |
| Pathfinder (Freenome, Brisbane, CA, USA) ☑SC | Colorectal cancer | ctDNA | Blood | a combination of DNA, RNA, and protein biomarkers. | [19] |
| Cancer Type | Exosomal Biomarker | Biomarker Type | Biofluid | Indication | Clinical Sample Size | Refs. |
|---|---|---|---|---|---|---|
| Lung cancer | miR-222-3p | miRNA | Serum | Prognosis | TP N = 50 | [56] |
| EGFR T790M | mRNA | Plasma | TP N = 84 [57] | [57,58] | ||
| miR-181-5p, miR-30a-3p, miR- Adenocarcinoma-specific: 30e-3p, and miR-361-5p were SCC specific: miR-10b-5p, miR-15b-5p, and miR-320b | miRNA | Plasma | TP N 46, HC N = 42, S = 60 | [59] | ||
| miR-193a-3p, miR-210-3p and miR-5100 | miRNA | Bone marrow and Plasma | Diagnosis and prognosis | TP N = 41, HC N = 30 | [60] | |
| circSATB2 | Circular RNA | Cell line H460, A549 and H1299 | [61] | |||
| NY-ESO-1 | Antigen | Plasma | [62] | |||
| TTF-1 and miR-21 | Protein | Serum | Diagnosis | NA | [63] | |
| Breast cancer | miR-1246 | miRNA | Plasma | Diagnosis | TP N = 46, HC N = 28 | [64] |
| miR-21, miR-105 and miR-222 | miRNA | Serum | Diagnosis | TP N = 53, HC N = 8 | [65] | |
| Glycoprotein (MUC1) | Protein | Cell line MCF7&MDA-MB-231 | [66] | |||
| PKG1, RALGAPA2, NFX1, TJP2 | Protein | Plasma | [67] | |||
| HER2 | Protein | Plasma | [68] | |||
| CD82 | Protein | Serum and plasma | Diagnosis | TP N = 80, BTP N = 80, HC N = 80 | [69] | |
| miR-375 | miRNA | Serum | Diagnosis | TP N = 17, HC N = 12 | [70] | |
| Gastric cancer | circSHKBP1 | Circular RNA | Serum | Diagnosis | TP N = 20, HC N = 20 | [71] |
| HOTTIP | long non-coding RNA | Serum | Diagnosis and prognosis | TP N = 126, HC N = 120 | [72] | |
| Early-stage GC | lncUEGC1 | long non-coding RNA | Plasma | Diagnosis | TP N = 10, HC N = 5 | [73] |
| Rectal cancer | miR-30d-5p, miR-181a-5p and miR-486-5p | miRNA | Plasma | Diagnosis and prognosis | TP N = 24, HC N = 5 | [74] |
| HCC | tRNA-ValTAC-3, tRNAGlyTCC-5, tRNA-ValAAC-5, and tRNA-GluCTC-5 | miRNA | Plasma | Diagnosis | TP N = 35, HC N = 11 | [75] |
| circUHRF1 | Circular RNA | Plasma | Diagnosis | TP N = 240, HC N = 20 | [76] | |
| Early-stage HCC | miR-21 and miR-10b | miRNA | Serum | Prognosis | TP N = 124 | [77] |
| Pancreatic cancer | KRAS | mRNA | Plasma | Diagnosis and prognosis | TP N = 127, HC N = 136 | [78,79] |
| CKAP4 | Protein | Serum | Diagnosis | TP N = 47, HC N = 18 | [80] | |
| Glypican 1(GPC1), Migration inhibition factor (MIF) | Protein | Serum | TP N = 71, HC N = 32 | [81] | ||
| Prostate cancer | AR-V7 | Androgen receptor | Plasma | Prognosis | TP N = 36 | [82] |
| miR-196a-5p and miR-501-3p | miRNA | Urine | Diagnosis | TP N = 48, HC N = 28 | [83] | |
| miR-1290 and miR-375 | miRNA | Plasma | TP N = 23, HC N = 50 | [84] | ||
| PSA | Antigen | Plasma | [85] | |||
| GGT1 | Gene | Serum | [86] | |||
| PTENP1 | Gene | Plasma | Diagnosis | TP N = 50, HC N = 60 | [87] | |
| Bladder cancer | lncLNMAT2 | long non-coding RNA | Serum and urine | Diagnosis and prognosis | TP N = 206, HC N = 120 | [88] |
| lncRNAs(SPRY4-IT1, MALAT1 and PCAT-1) | long non-coding RNA | Urine | Diagnosis and Prognosis | TP N = 184, HC N = 184 | [89] | |
| Colorectal | Glypican-1(miR-96-5p and miR-149) | miRNA | Plasma | [90] | ||
| CEA | Antigen | Serum | [91] | |||
| Cholangiocarcinoma | AMPN, VNN1, PIGR | Gene | Serum | [92] | ||
| Ovarian cancer | E-cadherin | Protein | Ascites | Diagnosis and prognosis | TP N = 35, HC N = 6 | [93] |
| miR-200b and miR-200c | miRNA | Serum | Diagnosis and prognosis | TP N = 163, BTP N = 20, HC N = 32 | [94] | |
| CD24, EpCAM, CA-125 | Protein | Plasma | [95] | |||
| Cervical cancer | let-7d-3p and miR-30d-5p | miRNA | Plasma | Diagnosis | NA | [96] |
| Multiple Melanoma | Ig-BCR | Cell receptor | Serum | Diagnosis | Serum of 5T33MM engrafted mice | [97] |
| let-7b and miR-18a | miRNA | Serum | Prognosis | TP N = 156, HC N = 5 | [98] | |
| PMSA3 and lncPMSA3-AS1 | Gene | Serum | Prognosis | Bortezomib resistance N = 12, bortezomib sensitivity N = 45 | [99] | |
| Melanoma | PD-L1 | Protein | Plasma | Diagnosis and prognosis | TP N = 44, HC N = 11 | [100] |
| BRAFV600E | Gene | Plasma | Prognosis | TP N = 12, HC N = 12 | [101] | |
| Glioblastoma | EGFR vIII | mRNA | Serum | Diagnostic | [102] |
| Optical Methods | Non-Optical Methods |
|---|---|
| Optical microscopy | Scanning electron microscope (SEM) |
| Dynamic Light Scattering (DLS) | Transmission Electron Microscopy (TEM) |
| Nano Tracking Analysis (NTA) | Atomic force microscopy (AFM) |
| Fluorescence microscopy | Impedance flow cytometry |
| Surface Plasmon Resonance (SPR) | Tunable Resistance Pulse Sensing (TRPS) |
| Localized Surface Plasmon Resonance (LSPR) | Mass spectroscopy |
| Biomarker | Cancer Type (Cell Line) | Sample | Detection Technique (Target Molecule) | Statistics | Refs. |
|---|---|---|---|---|---|
| CD63 | Glioma (U251) | Cell culture | SPR (Antibody) | 4.23 × 10−3 μg/mL | [156] |
| CD24 EpCAM | Ovarian (CaOV3) | Cell culture | SPR (Antibody) | 1000+ sites for inspection | [143] |
| EGFR PD-L1 | Non-small cell lung cancer (A549) | Cell culture, Serum | SPR (Antibody) | sensitivity of 9.258 × 103%/RIU and resolution of 8.311 × 10−6 RIU. | [146] |
| HER2 | Breast cancer (BT474) | Cell culture, Serum | (SPR) (Antibody) | LOD: 2070 Exosomes/μL | [145] |
| HSPG | Multiple myeloma | Serum | SPR (Protein) | LOD: 5 ng/mL | [142] |
| HSP90 | Breast cancer (MCF7) | Cell culture | LSPR (Vn96) | No data | [155] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neriya Hegade, K.P.; Bhat, R.B.; Packirisamy, M. Microfluidic Liquid Biopsy Minimally Invasive Cancer Diagnosis by Nano-Plasmonic Label-Free Detection of Extracellular Vesicles: Review. Int. J. Mol. Sci. 2025, 26, 6352. https://doi.org/10.3390/ijms26136352
Neriya Hegade KP, Bhat RB, Packirisamy M. Microfluidic Liquid Biopsy Minimally Invasive Cancer Diagnosis by Nano-Plasmonic Label-Free Detection of Extracellular Vesicles: Review. International Journal of Molecular Sciences. 2025; 26(13):6352. https://doi.org/10.3390/ijms26136352
Chicago/Turabian StyleNeriya Hegade, Keshava Praveena, Rama B. Bhat, and Muthukumaran Packirisamy. 2025. "Microfluidic Liquid Biopsy Minimally Invasive Cancer Diagnosis by Nano-Plasmonic Label-Free Detection of Extracellular Vesicles: Review" International Journal of Molecular Sciences 26, no. 13: 6352. https://doi.org/10.3390/ijms26136352
APA StyleNeriya Hegade, K. P., Bhat, R. B., & Packirisamy, M. (2025). Microfluidic Liquid Biopsy Minimally Invasive Cancer Diagnosis by Nano-Plasmonic Label-Free Detection of Extracellular Vesicles: Review. International Journal of Molecular Sciences, 26(13), 6352. https://doi.org/10.3390/ijms26136352