
The Emerging Role of the Molecular Chaperone Clusterin in Parkinson’s Disease

 , , and
, , and {kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Clusterin: Biogenesis, Distribution, and Chaperone Activity
1.2. Parkinson’s Disease: Clinical Presentation and Neuropathological Features
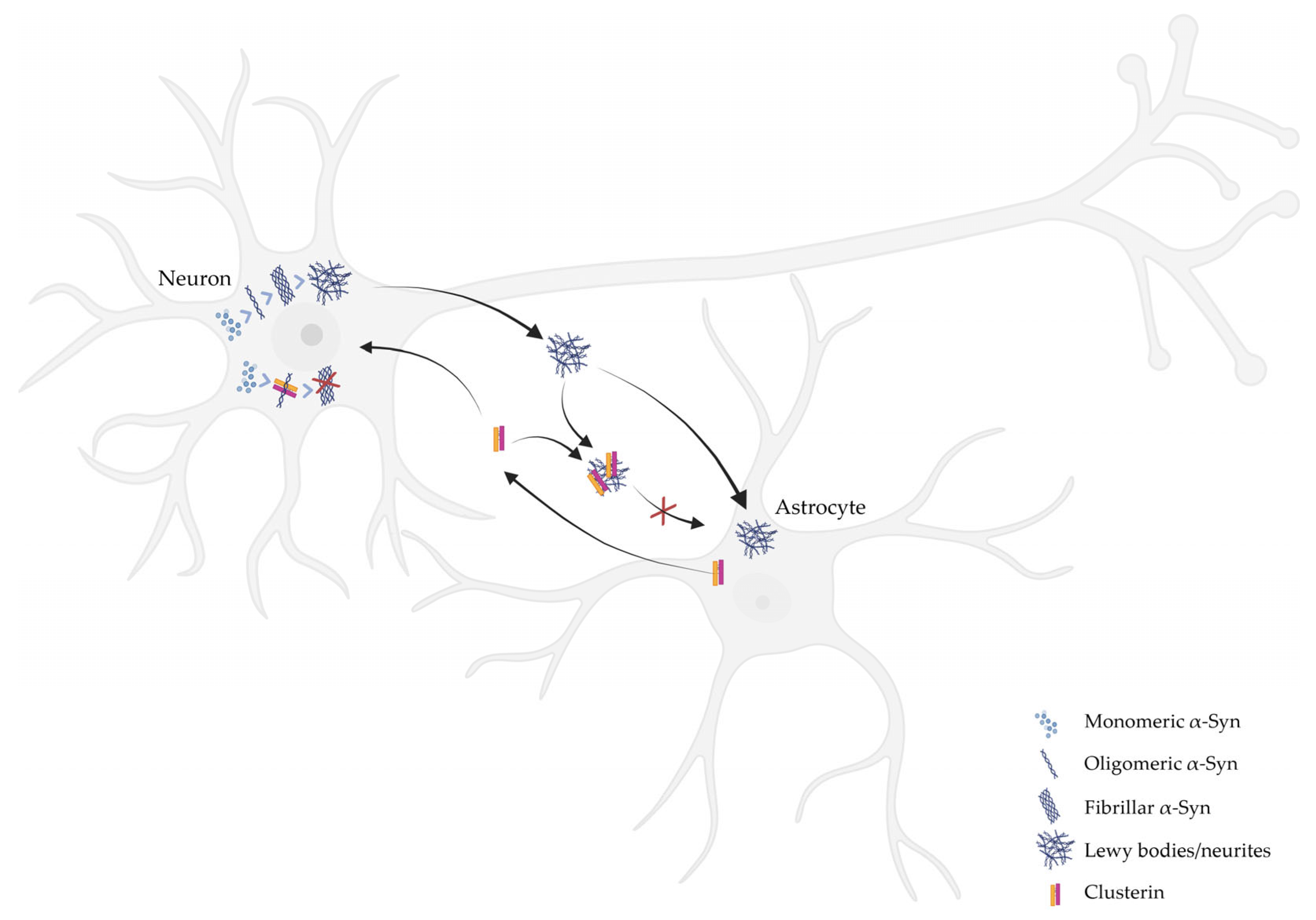
2. The Role of CLU on the Aggregation of α-Syn
3. The Role of CLU on the Clearance of α-Syn Aggregates by Glial Cells
4. CLU as a Biomarker for PD
4.1. CLU in CSF
4.2. CLU in Blood
5. Conclusions: Can CLU Be a Therapeutic Target for PD?
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ApoER2 | Apolipoprotein E Receptor 2 |
| α-Syn | Alpha-Synuclein |
| AD | Alzheimer’s Disease |
| Aβ | Amyloid-β |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| CLU | Clusterin |
| ER | Endoplasmic Reticulum |
| Hsp | Heat Shock Protein |
| iCLU | Intracellular CLU |
| LRRK2 | Leucine-Rich Repeat Kinase 2 |
| LBs | Lewy Bodies |
| LNs | Lewy Neurites |
| PD | Parkinson’s Disease |
| PTMs | Post-Translational Modifications |
| PFFs | Pre-Formed Fibrils |
| sCLU | Secreted CLU |
| TDP-43 | TAR DNA-Binding Protein 43 |
| VLDLR | Very Low-Density Lipoprotein Receptor |
References
- Gu, J.; He, Y.; He, C.; Zhang, Q.; Huang, Q.; Bai, S.; Wang, R.; You, Q.; Wang, L. Advances in the Structures, Mechanisms and Targeting of Molecular Chaperones. Signal Transduct. Target. Ther. 2025, 10, 84. [Google Scholar] [CrossRef]
- Geraghty, N.J.; Satapathy, S.; Kelly, M.; Cheng, F.; Lee, A.; Wilson, M.R. Expanding the Family of Extracellular Chaperones: Identification of Human Plasma Proteins with Chaperone Activity. Protein Sci. 2021, 30, 2272–2286. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Shah, O.P.; Sharma, L.; Gulati, M.; Behl, T.; Khalid, A.; Mohan, S.; Najmi, A.; Zoghebi, K. Molecular Chaperones as Therapeutic Target: Hallmark of Neurodegenerative Disorders. Mol. Neurobiol. 2024, 61, 4750–4767. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Doh-ura, K.; Wakisaka, Y.; Iwaki, T. Clusterin/Apolipoprotein J Is Associated with Cortical Lewy Bodies: Immunohistochemical Study in Cases with α-Synucleinopathies. Acta Neuropathol. 2002, 104, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Herring, S.K.; Moon, H.J.; Rawal, P.; Chhibber, A.; Zhao, L. Brain Clusterin Protein Isoforms and Mitochondrial Localization. eLife 2019, 8, e48255. [Google Scholar] [CrossRef]
- Choi-miura, N.H.; Takahashi, Y.; Nakano, Y.; Tobe, T.; Tomita, M. Identification of the Disulfide Bonds in Human Plasma Protein SP-40,40 (Apolipoprotein-J). J. Biochem. 1992, 112, 557–561. [Google Scholar] [CrossRef]
- Kapron, J.T.; Hilliard, G.M.; Lakins, J.N.; Tenniswood, M.P.R.; West, K.A.; Carr, S.A.; Crabb, J.W. Identification and Characterization of Glycosylation Sites in Human Serum Clusterin. Protein Sci. 1997, 6, 2120–2133. [Google Scholar] [CrossRef]
- Rohne, P.; Prochnow, H.; Koch-Brandt, C. The CLU-Files: Disentanglement of a Mystery. Biomol. Concepts 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Satapathy, S.; Dabbs, R.A.; Wilson, M.R. Rapid High-Yield Expression and Purification of Fully Post-Translationally Modified Recombinant Clusterin and Mutants. Sci. Rep. 2020, 10, 14243. [Google Scholar] [CrossRef] [PubMed]
- Rohne, P.; Prochnow, H.; Wolf, S.; Renner, B.; Koch-Brandt, C. The Chaperone Activity of Clusterin Is Dependent on Glycosylation and Redox Environment. Cell. Physiol. Biochem. 2014, 34, 1626–1639. [Google Scholar] [CrossRef]
- Prochnow, H.; Gollan, R.; Rohne, P.; Hassemer, M.; Koch-Brandt, C.; Baiersdörfer, M. Non-Secreted Clusterin Isoforms Are Translated in Rare Amounts from Distinct Human MRNA Variants and Do Not Affect Bax-Mediated Apoptosis or the NF-ΚB Signaling Pathway. PLoS ONE 2013, 8, e75303. [Google Scholar] [CrossRef]
- Leskov, K.S.; Klokov, D.Y.; Li, J.; Kinsella, T.J.; Boothman, D.A. Synthesis and Functional Analyses of Nuclear Clusterin, a Cell Death Protein. J. Biol. Chem. 2003, 278, 11590–11600. [Google Scholar] [CrossRef] [PubMed]
- Foster, E.M.; Dangla-Valls, A.; Lovestone, S.; Ribe, E.M.; Buckley, N.J. Clusterin in Alzheimer’s Disease: Mechanisms, Genetics, and Lessons from Other Pathologies. Front. Neurosci. 2019, 13, 164. [Google Scholar] [CrossRef]
- Nizard, P.; Tetley, S.; Le Dréan, Y.; Watrin, T.; Le Goff, P.; Wilson, M.R.; Michel, D. Stress-Induced Retrotranslocation of Clusterin/ApoJ into the Cytosol. Traffic 2007, 8, 554–565. [Google Scholar] [CrossRef]
- Gregory, J.M.; Whiten, D.R.; Brown, R.A.; Barros, T.P.; Kumita, J.R.; Yerbury, J.J.; Satapathy, S.; McDade, K.; Smith, C.; Luheshi, L.M.; et al. Clusterin Protects Neurons against Intracellular Proteotoxicity. Acta Neuropathol. Commun. 2017, 5, 81. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zoubeidi, A.; Beraldi, E.; Gleave, M.E. GRP78 Regulates Clusterin Stability, Retrotranslocation and Mitochondrial Localization under ER Stress in Prostate Cancer. Oncogene 2013, 32, 1933–1942. [Google Scholar] [CrossRef]
- Wong, P.; Taillefer, D.; Lakins, J.; Pineault, J.; Chader, G.; Tenniswood, M. Molecular Characterization of Human TRPM-2/Clusterin, a Gene Associated with Sperm Maturation, Apoptosis and Neurodegeneration. Eur. J. Biochem. 1994, 221, 917–925. [Google Scholar] [CrossRef]
- Han, Z.; Wang, Z.; Cheng, G.; Liu, B.; Li, P.; Li, J.; Wang, W.; Yin, C.; Zhang, W. Presence, Localization, and Origin of Clusterin in Normal Human Spermatozoa. J. Assist. Reprod. Genet. 2012, 29, 751–757. [Google Scholar] [CrossRef]
- Janiszewska, E.; Kmieciak, A.; Kacperczyk, M.; Witkowska, A.; Kratz, E.M. The Influence of Clusterin Glycosylation Variability on Selected Pathophysiological Processes in the Human Body. Oxid. Med. Cell Longev. 2022, 2022, 7657876. [Google Scholar] [CrossRef]
- Janiszewska, E.; Kokot, I.; Gilowska, I.; Faundez, R.; Kratz, E.M. The Possible Association of Clusterin Fucosylation Changes with Male Fertility Disorders. Sci. Rep. 2021, 11, 15674. [Google Scholar] [CrossRef]
- Filippini, A.; Carini, G.; Barbon, A.; Gennarelli, M.; Russo, I. Astrocytes Carrying LRRK2 G2019S Exhibit Increased Levels of Clusterin Chaperone via MiR-22-5p and Reduced Ability to Take up α-Synuclein Fibrils. Acta Neuropathol. Commun. 2025, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Filippini, A.; Mutti, V.; Faustini, G.; Longhena, F.; Ramazzina, I.; Rizzi, F.; Kaganovich, A.; Roosen, D.A.; Landeck, N.; Duffy, M.; et al. Extracellular Clusterin Limits the Uptake of α-Synuclein Fibrils by Murine and Human Astrocytes. Glia 2021, 69, 681–696. [Google Scholar] [CrossRef]
- Pasinetti, G.M.; Johnson, S.A.; Oda, T.; Rozovsky, I.; Finch, C.E. Clusterin (SGP-2): A Multifunctional Glycoprotein with Regional Expression in Astrocytes and Neurons of the Adult Rat Brain. J. Comp. Neurol. 1994, 339, 387–400. [Google Scholar] [CrossRef] [PubMed]
- Trougakos, I.P.; Gonos, E.S. Regulation of Clusterin/Apolipoprotein J, a Functional Homologue to the Small Heat Shock Proteins, by Oxidative Stress in Ageing and Age-Related Diseases. Free Radic. Res. 2006, 40, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Blaschuk, O.; Burdzy, K.; Fritz, I.B. Purification and Characterization of a Cell-Aggregating Factor (Clusterin), the Major Glycoprotein in Ram Rete Testis Fluid. J. Biol. Chem. 1983, 258, 7714–7720. [Google Scholar] [CrossRef]
- Rodríguez-Rivera, C.; Garcia, M.M.; Molina-Álvarez, M.; González-Martín, C.; Goicoechea, C. Clusterin: Always Protecting. Synthesis, Function and Potential Issues. Biomed. Pharmacother. 2021, 134, 111174. [Google Scholar] [CrossRef]
- Peix, L.; Evans, I.C.; Pearce, D.R.; Simpson, J.K.; Maher, T.M.; McAnulty, R.J. Diverse Functions of Clusterin Promote and Protect against the Development of Pulmonary Fibrosis. Sci. Rep. 2018, 8, 1906. [Google Scholar] [CrossRef]
- Flanagan, L.; Whyte, L.; Chatterjee, N.; Tenniswood, M. Effects of Clusterin Over-Expression on Metastatic Progression and Therapy in Breast Cancer. BMC Cancer 2010, 10, 107. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Bracher, A.; Hartl, F.U. The Chaperone Clusterin in Neurodegeneration−friend or Foe? BioEssays 2022, 44, e2100287. [Google Scholar] [CrossRef]
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin Is an ATP—Independent Chaperone with Very Broad Substrate Specificity that Stabilizes Stressed Proteins in a Folding-Competent State. Biochemistry 2000, 39, 15953–15960. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, A.R.; Yerbury, J.J.; Berghofer, P.; Greguric, I.; Katsifis, A.; Dobson, C.M.; Wilson, M.R. Clusterin Facilitates in Vivo Clearance of Extracellular Misfolded Proteins. Cell. Mol. Life Sci. 2011, 68, 3919–3931. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H. Extracellular ATP and Other Nucleotides—Ubiquitous Triggers of Intercellular Messenger Release. Purinergic Signal 2016, 12, 25–57. [Google Scholar] [CrossRef] [PubMed]
- Bloem, B.R.; Okun, M.S.; Klein, C. Parkinson’s Disease. Lancet 2021, 397, 2284–2303. [Google Scholar] [CrossRef] [PubMed]
- Periñán, M.T.; Brolin, K.; Bandres-Ciga, S.; Blauwendraat, C.; Klein, C.; Gan-Or, Z.; Singleton, A.; Gomez-Garre, P.; Swanberg, M.; Mir, P.; et al. Effect Modification between Genes and Environment and Parkinson’s Disease Risk. Ann. Neurol. 2022, 92, 715–724. [Google Scholar] [CrossRef]
- Wüllner, U.; Borghammer, P.; Choe, C.U.; Csoti, I.; Falkenburger, B.; Gasser, T.; Lingor, P.; Riederer, P. The Heterogeneity of Parkinson’s Disease. J. Neural Transm. 2023, 130, 827–838. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef]
- Cuenca-Bermejo, L.; Almela, P.; Navarro-Zaragoza, J.; Villalba, E.F.; González-Cuello, A.M.; Laorden, M.L.; Herrero, M.T. Cardiac Changes in Parkinson’s Disease: Lessons from Clinical and Experimental Evidence. Int. J. Mol. Sci. 2021, 22, 13488. [Google Scholar] [CrossRef]
- Bourdenx, M.; Koulakiotis, N.S.; Sanoudou, D.; Bezard, E.; Dehay, B.; Tsarbopoulos, A. Protein Aggregation and Neurodegeneration in Prototypical Neurodegenerative Diseases: Examples of Amyloidopathies, Tauopathies and Synucleinopathies. Prog. Neurobiol. 2017, 155, 171–193. [Google Scholar] [CrossRef]
- Srinivasan, E.; Chandrasekhar, G.; Chandrasekar, P.; Anbarasu, K.; Vickram, A.S.; Karunakaran, R.; Rajasekaran, R.; Srikumar, P.S. Alpha-Synuclein Aggregation in Parkinson’s Disease. Front. Med. 2021, 8, 736978. [Google Scholar] [CrossRef]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-Synuclein in Parkinson’s Disease and Other Synucleinopathies: From Overt Neurodegeneration Back to Early Synaptic Dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef]
- Vargas, K.J.; Schrod, N.; Davis, T.; Fernandez-Busnadiego, R.; Taguchi, Y.V.; Laugks, U.; Lucic, V.; Chandra, S.S. Synucleins Have Multiple Effects on Presynaptic Architecture. Cell Rep. 2017, 18, 1732–1745. [Google Scholar] [CrossRef]
- Gao, V.; Briano, J.A.; Komer, L.E.; Burré, J. Functional and Pathological Effects of α-Synuclein on Synaptic SNARE Complexes. J. Mol. Biol. 2023, 435, 167714. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Thorne, N.J.; Tumbarello, D.A. The Relationship of Alpha-Synuclein to Mitochondrial Dynamics and Quality Control. Front. Mol. Neurosci. 2022, 15, 947191. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Sang, J.C.; Rodrigues, M.; Carr, A.R.; Horrocks, M.H.; De, S.; Bongiovanni, M.N.; Flagmeier, P.; Dobson, C.M.; Wales, D.J.; et al. Mapping Surface Hydrophobicity of α-Synuclein Oligomers at the Nanoscale. Nano Lett. 2018, 18, 1497–1505. [Google Scholar] [CrossRef] [PubMed]
- Vilar, M.; Chou, H.T.; Lührs, T.; Maji, S.K.; Riek-Loher, D.; Verel, R.; Manning, G.; Stahlberg, H.; Riek, R. The Fold of α-Synuclein Fibrils. Proc. Natl. Acad. Sci. USA 2008, 105, 8637–8642. [Google Scholar] [CrossRef]
- Paleologou, K.E.; El-Agnaf, O.M.A. α-Synuclein Aggregation and Modulating Factors. Subcell. Biochem. 2012, 65, 109–164. [Google Scholar] [CrossRef]
- Won, S.J.; Fong, R.; Butler, N.; Sanchez, J.; Zhang, Y.; Wong, C.; Tambou Nzoutchoum, O.; Huynh, A.; Pan, J.; Swanson, R.A. Neuronal Oxidative Stress Promotes α-Synuclein Aggregation In Vivo. Antioxidants 2022, 11, 2466. [Google Scholar] [CrossRef]
- Filippini, A.; Gennarelli, M.; Russo, I. α-Synuclein and Glia in Parkinson’s Disease: A Beneficial or a Detrimental Duet for the Endo-Lysosomal System? Cell Mol. Neurobiol. 2019, 39, 161–168. [Google Scholar] [CrossRef]
- Russo, I.; Bubacco, L.; Greggio, E. LRRK2 and Neuroinflammation: Partners in Crime in Parkinson’s Disease? J. Neuroinflammation 2014, 11, 52. [Google Scholar] [CrossRef]
- Ramos-González, E.J.; Bitzer-Quintero, O.K.; Ortiz, G.; Hernández-Cruz, J.J.; Ramírez-Jirano, L.J. Relationship between Inflammation and Oxidative Stress and Its Effect on Multiple Sclerosis. Neurología 2024, 39, 292–301. [Google Scholar] [CrossRef]
- Lema Tomé, C.M.; Tyson, T.; Rey, N.L.; Grathwohl, S.; Britschgi, M.; Brundin, P. Inflammation and α-Synuclein’s Prion-like Behavior in Parkinson’s Disease—Is There a Link? Mol. Neurobiol. 2013, 47, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.J.; Choi, M.; Kim, J.T.; Kim, D.K.; Jung, M.K.; Kim, C.; Kim, T.K.; Lee, J.S.; Jung, B.C.; Shin, S.J.; et al. TNF-α Promotes α-Synuclein Propagation through Stimulation of Senescence-Associated Lysosomal Exocytosis. Exp. Mol. Med. 2022, 54, 788–800. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.K.; Bae, E.J.; Jung, B.C.; Choi, M.; Shin, S.J.; Park, S.J.; Kim, J.T.; Jung, M.K.; Ulusoy, A.; Song, M.Y.; et al. Inflammation Promotes Synucleinopathy Propagation. Exp. Mol. Med. 2022, 54, 2148–2161. [Google Scholar] [CrossRef] [PubMed]
- Laursen, A.L.S.; Olesen, M.V.; Folke, J.; Brudek, T.; Knecht, L.H.; Sotty, F.; Lambertsen, K.L.; Fog, K.; Dalgaard, L.T.; Aznar, S. Systemic Inflammation Activates Coagulation and Immune Cell Infiltration Pathways in Brains with Propagating α-Synuclein Fibril Aggregates. Mol. Cell. Neurosci. 2024, 129, 103931. [Google Scholar] [CrossRef]
- McLean, P.J.; Kawamata, H.; Shariff, S.; Hewett, J.; Sharma, N.; Ueda, K.; Breakefield, X.O.; Hyman, B.T. TorsinA and Heat Shock Proteins Act as Molecular Chaperones: Suppression of α-Synuclein Aggregation. J. Neurochem. 2002, 83, 846–854. [Google Scholar] [CrossRef]
- Mohammed, S.; Russo, I.; Ramazzina, I. Uncovering the Role of Natural and Synthetic Small Molecules in Counteracting the Burden of α-Synuclein Aggregates and Related Toxicity in Different Models of Parkinson’s Disease. Int. J. Mol. Sci. 2023, 24, 13370. [Google Scholar] [CrossRef]
- Cardinale, A.; Calabrese, V.; de Iure, A.; Picconi, B. Alpha-Synuclein as a Prominent Actor in the Inflammatory Synaptopathy of Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 6517. [Google Scholar] [CrossRef]
- Hu, S.; Tan, J.; Qin, L.; Lv, L.; Yan, W.; Zhang, H.; Tang, B.S.; Wang, C. Molecular Chaperones and Parkinson’s Disease. Neurobiol. Dis. 2021, 160, 105527. [Google Scholar] [CrossRef]
- Zhou, Y.; Gu, G.; Goodlett, D.R.; Zhang, T.; Pan, C.; Montine, T.J.; Montine, K.S.; Aebersold, R.H.; Zhang, J. Analysis of α-Synuclein-Associated Proteins by Quantitative Proteomics. J. Biol. Chem. 2004, 279, 39155–39164. [Google Scholar] [CrossRef]
- Lenzi, C.; Ramazzina, I.; Russo, I.; Filippini, A.; Bettuzzi, S.; Rizzi, F. The Down-Regulation of Clusterin Expression Enhances the Asynuclein Aggregation Process. Int. J. Mol. Sci. 2020, 21, 7181. [Google Scholar] [CrossRef]
- Yuste-Checa, P.; Trinkaus, V.A.; Riera-Tur, I.; Imamoglu, R.; Schaller, T.F.; Wang, H.; Dudanova, I.; Hipp, M.S.; Bracher, A.; Hartl, F.U. The Extracellular Chaperone Clusterin Enhances Tau Aggregate Seeding in a Cellular Model. Nat. Commun. 2021, 12, 4863. [Google Scholar] [CrossRef] [PubMed]
- Leeb, C.; Eresheim, C.; Nimpf, J. Clusterin Is a Ligand for Apolipoprotein e Receptor 2 (ApoER2) and Very Low Density Lipoprotein Receptor (VLDLR) and Signals via the Reelin-Signaling Pathway. J. Biol. Chem. 2014, 289, 35732–35742. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Park, J.; Kim, K.; Kim, M.; Cho, S.R. Reelin Alleviates Mesenchymal Stem Cell Senescence and Reduces Pathological α-Synuclein Expression in an in Vitro Model of Parkinson’s Disease. Genes 2021, 12, 1066. [Google Scholar] [CrossRef]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carrá, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, X.M. Clusterin Binds to Aβ1-42 Oligomers with High Affinity and Interferes with Peptide Aggregation by Inhibiting Primary and Secondary Nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Sens, J.P.; Kang, S.S.; Baker, K.E.; Berry, T.J.; Kurti, A.; Daughrity, L.; Jansen-West, K.R.; Dickson, D.W.; Petrucelli, L.; et al. Astrocyte-Derived Clusterin Suppresses Amyloid Formation in Vivo. Mol. Neurodegener. 2020, 15, 71. [Google Scholar] [CrossRef]
- Wojtas, A.M.; Carlomagno, Y.; Sens, J.P.; Kang, S.S.; Jensen, T.D.; Kurti, A.; Baker, K.E.; Berry, T.J.; Phillips, V.R.; Castanedes, M.C.; et al. Clusterin Ameliorates Tau Pathology in Vivo by Inhibiting Fibril Formation. Acta Neuropathol. Commun. 2020, 8, 210. [Google Scholar] [CrossRef]
- Bieri, G.; Gitler, A.D.; Brahic, M. Internalization, Axonal Transport and Release of Fibrillar Forms of Alpha-Synuclein. Neurobiol. Dis. 2018, 109, 219–225. [Google Scholar] [CrossRef] [PubMed]
- da Fonseca, T.L.; Villar-Piqué, A.; Outeiro, T.F. The Interplay between Alpha-Synuclein Clearance and Spreading. Biomolecules 2015, 5, 435–471. [Google Scholar] [CrossRef]
- Deyell, J.S.; Sriparna, M.; Ying, M.; Mao, X. The Interplay between α-Synuclein and Microglia in α-Synucleinopathies. Int. J. Mol. Sci. 2023, 24, 2477. [Google Scholar] [CrossRef]
- Lindström, V.; Gustafsson, G.; Sanders, L.H.; Howlett, E.H.; Sigvardson, J.; Kasrayan, A.; Ingelsson, M.; Bergström, J.; Erlandsson, A. Extensive Uptake of α-Synuclein Oligomers in Astrocytes Results in Sustained Intracellular Deposits and Mitochondrial Damage. Mol. Cell. Neurosci. 2017, 82, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Loria, F.; Vargas, J.Y.; Bousset, L.; Syan, S.; Salles, A.; Melki, R.; Zurzolo, C. α-Synuclein Transfer between Neurons and Astrocytes Indicates That Astrocytes Play a Role in Degradation Rather than in Spreading. Acta Neuropathol. 2017, 134, 789–808. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.N.; Brooks, M.M.; Chakrabarty, P.; Saha, K.; Khoshbouei, H.; Golde, T.E.; Giasson, B.I. Proteolysis of α-Synuclein Fibrils in the Lysosomal Pathway Limits Induction of Inclusion Pathology. J. Neurochem. 2017, 140, 951–964. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, P.W.; Nadarajah, C.J.; Kanan, M.F.; Patterson, J.N.; Novotny, B.; Lawrence, J.H.; King, M.W.; Brase, L.; Inman, C.E.; Yuede, C.M.; et al. An Astrocyte BMAL1-BAG3 Axis Protects against Alpha-Synuclein and Tau Pathology. Neuron 2023, 111, 1286–1303. [Google Scholar] [CrossRef]
- Mulder, S.D.; Nielsen, H.M.; Blankenstein, M.A.; Eikelenboom, P.; Veerhuis, R. Apolipoproteins E and J Interfere with Amyloid-Beta Uptake by Primary Human Astrocytes and Microglia in Vitro. Glia 2014, 62, 493–503. [Google Scholar] [CrossRef]
- Nielsen, H.M.; Mulder, S.D.; Beliën, J.A.M.; Musters, R.J.P.; Eikelenboom, P.; Veerhuis, R. Astrocytic Aβ1-42 Uptake Is Determined by Aβ-Aggregation State and the Presence of Amyloid-Associated Proteins. Glia 2010, 58, 1235–1246. [Google Scholar] [CrossRef]
- Zhou, J.; Singh, N.; Galske, J.; Hudobenko, J.; Hu, X.; Yan, R. BACE1 Regulates Expression of Clusterin in Astrocytes for Enhancing Clearance of β-Amyloid Peptides. Mol. Neurodegener. 2023, 18, 31. [Google Scholar] [CrossRef]
- Renkawek, K.; Stege, G.J.J.; Bosman, G.J.C.G.M. Dementia, Gliosis and Expression of the Small Heat Shock Proteins Hsp27 and AB-Crystallin in Parkinson’s Disease. Neuroreport 1999, 10, 1747–1751. [Google Scholar] [CrossRef]
- Wilhelmus, M.M.M.; Otte-Höller, I.; Wesseling, P.; De Waal, R.M.W.; Boelens, W.C.; Verbeek, M.M. Specific Association of Small Heat Shock Proteins with the Pathological Hallmarks of Alzheimer’s Disease Brains. Neuropathol. Appl. Neurobiol. 2006, 32, 119–130. [Google Scholar] [CrossRef]
- Du, X.; Chen, Z.; Shui, W. Clusterin: Structure, Function and Roles in Disease. Int. J. Med. Sci. 2025, 22, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Vranová, H.P.; Mareš, J.; Nevrlý, M.; Stejskal, D.; Zapletalová, J.; Hluštík, P.; Kaňovský, P. CSF Markers of Neurodegeneration in Parkinson’s Disease. J. Neural Transm. 2010, 117, 1358–1393. [Google Scholar] [CrossRef]
- Vranová, H.P.; Hényková, E.; Kaiserová, M.; Menšíková, K.; Vaštík, M.; Mareš, J.; Hluštík, P.; Zapletalová, J.; Strnad, M.; Stejskal, D.; et al. Tau Protein, Beta-Amyloid1-42 and Clusterin CSF Levels in the Differential Diagnosis of Parkinsonian Syndrome with Dementia. J. Neurol. Sci. 2014, 343, 178–183. [Google Scholar] [CrossRef] [PubMed]
- Vranova, H.P.; Hényková, E.; Mareš, J.; Kaiserová, M.; Menšíková, K.; Vaštík, M.; Hluštík, P.; Zapletalová, J.; Strnad, M.; Stejskal, D.; et al. Clusterin CSF Levels in Differential Diagnosis of Neurodegenerative Disorders. J. Neurol. Sci. 2016, 361, 294–299. [Google Scholar] [CrossRef]
- Maarouf, C.L.; Beach, T.G.; Adler, C.H.; Shill, H.A.; Sabbagh, M.N.; Wu, T.; Walker, D.G.; Kokjohn, T.A.; Roher, A.E. Cerebrospinal Fluid Biomarkers of Neuropathologically Diagnosed Parkinson’s Disease Subjects. Neurol. Res. 2012, 34, 646–652. [Google Scholar] [CrossRef]
- Paslawski, W.; Zareba-Paslawska, J.; Zhang, X.; Hölzl, K.; Wadensten, H.; Shariatgorji, M.; Janelidze, S.; Hansson, O.; Forsgren, L.; Andrén, P.E.; et al. α-Synuclein−lipoprotein Interactions and Elevated ApoE Level in Cerebrospinal Fluid from Parkinson’s Disease Patients. Proc. Natl. Acad. Sci. USA 2019, 116, 15226–15235. [Google Scholar] [CrossRef]
- Paslawski, W.; Svenningsson, P. Elevated ApoE, ApoJ and Lipoprotein-Bound α-Synuclein Levels in Cerebrospinal Fluid from Parkinson’s Disease Patients—Validation in the BioFIND Cohort. Park. Relat. Disord. 2023, 116, 105765. [Google Scholar] [CrossRef]
- Lidström, A.M.; Hesse, C.; Rosengren, L.; Fredman, P.; Davidsson, P. Normal Levels of Clusterin in Cerebrospinal Fluid in Alzheimer’s Disease, and No Change after Acute Ischemic Stroke. J. Alzheimer’s Dis. 2001, 3, 377–385. [Google Scholar] [CrossRef]
- Van Dijk, K.D.; Jongbloed, W.; Heijst, J.A.; Teunissen, C.E.; Groenewegen, H.J.; Berendse, H.W.; van de Berg, W.D.J.; Veerhuis, R. Cerebrospinal Fluid and Plasma Clusterin Levels in Parkinson’s Disease. Park. Relat. Disord. 2013, 19, 839–843. [Google Scholar] [CrossRef]
- Koníčková, D.; Menšíková, K.; Klíčová, K.; Chudáčková, M.; Kaiserová, M.; Přikrylová, H.; Otruba, P.; Nevrlý, M.; Hluštík, P.; Hényková, E.; et al. Cerebrospinal Fluid and Blood Serum Biomarkers in Neurodegenerative Proteinopathies: A Prospective, Open, Cross-Correlation Study. J. Neurochem. 2023, 167, 690–705. [Google Scholar] [CrossRef]
- Zhang, X.; Yin, X.; Yu, H.; Liu, X.; Yang, F.; Yao, J.; Jin, H.; Yang, P. Quantitative Proteomic Analysis of Serum Proteins in Patients with Parkinson’s Disease Using an Isobaric Tag for Relative and Absolute Quantification Labeling, Two-Dimensional Liquid Chromatography, and Tandem Mass Spectrometry. Analyst 2012, 137, 4628–4635. [Google Scholar] [CrossRef] [PubMed]
- Mnich, K.; Moghaddam, S.; Browne, P.; Counihan, T.; Fitzgerald, S.P.; Martin, K.; Richardson, C.; Samali, A.; Gorman, A.M. Endoplasmic Reticulum Stress-Regulated Chaperones as a Serum Biomarker Panel for Parkinson’s Disease. Mol. Neurobiol. 2023, 60, 2743–2757. [Google Scholar] [CrossRef]
- Jiang, R.; Rong, C.; Ke, R.; Meng, S.; Yan, X.; Ke, H.; Wu, S.; Azim, A. Differential Proteomic Analysis of Serum Exosomes Reveals Alterations in Progression of Parkinson Disease. Medicine 2019, 98, e17478. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Berg, D.; Hu, M.T.; Pilotto, A.; Borroni, B.; Davis, J.J.; Tofaris, G.K. Validation of α-Synuclein in L1CAM-Immunocaptured Exosomes as a Biomarker for the Stratification of Parkinsonian Syndromes. Mov. Disord. 2021, 36, 1326–1335. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum Neuronal Exosomes Predict and Differentiate Parkinson’s Disease from Atypical Parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef]
- Polimeno, L.; Niccoli Asabella, A.; Mazzocca, A.; De Fazio, G.; Polimeno, R.; Buquicchio, R.; Lavelli, V.; Rubini, G. Plasma Levels of Clusterin Are Representative of the Early Phase of the Neurodegenerative Process in Parkinson’s Disease. J. Clin. Mol. Med. 2018, 1, 1–5. [Google Scholar] [CrossRef]
- Lin, Y.; Lu, L.; Zhou, M.; Liu, H.Q.; Ye, P.; Zhang, W.; Qiu, J.; Zhang, Z.; Yang, X.; Ding, L.; et al. Association of CLU Gene Polymorphism with Parkinson’s Disease in the Chinese Han Population. J. Gene Med. 2021, 23, e3302. [Google Scholar] [CrossRef]
- Kitamura, Y.; Kojima, M.; Kurosawa, T.; Sasaki, R.; Ichihara, S.; Hiraku, Y.; Tomimoto, H.; Murata, M.; Oikawa, S. Proteomic Profiling of Exosomal Proteins for Blood-Based Biomarkers in Parkinson’s Disease. Neuroscience 2018, 392, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Whiten, D.R.; Cox, D.; Horrocks, M.H.; Taylor, C.G.; De, S.; Flagmeier, P.; Tosatto, L.; Kumita, J.R.; Ecroyd, H.; Dobson, C.M.; et al. Single-Molecule Characterization of the Interactions between Extracellular Chaperones and Toxic α-Synuclein Oligomers. Cell Rep. 2018, 23, 3492–3500. [Google Scholar] [CrossRef]
- Pan, T.; Kondo, S.; Le, W.; Jankovic, J. The Role of Autophagy-Lysosome Pathway in Neurodegeneration Associated with Parkinson’s Disease. Brain 2008, 131, 1969–1978. [Google Scholar] [CrossRef]
- Joshi, N.; Raveendran, A.; Nagotu, S. Chaperones and Proteostasis: Role in Parkinson’s Disease. Diseases 2020, 8, 24. [Google Scholar] [CrossRef] [PubMed]
- Silvestro, S.; Raffaele, I.; Mazzon, E. Modulating Stress Proteins in Response to Therapeutic Interventions for Parkinson’sDisease. Int. J. Mol. Sci. 2023, 24, 16233. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Zhang, S.; Wang, X.; Xiao, C.; Cui, G.; Yang, X. Secretory Clusterin Inhibits Dopamine Neuron Apoptosis in MPTP Mice by Preserving Autophagy Activity. Neuroscience 2024, 540, 237–250. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carini, G.; Mohammed, S.; Filippini, A.; Ramazzina, I.; Russo, I. The Emerging Role of the Molecular Chaperone Clusterin in Parkinson’s Disease. Int. J. Mol. Sci. 2025, 26, 6351. https://doi.org/10.3390/ijms26136351
Carini G, Mohammed S, Filippini A, Ramazzina I, Russo I. The Emerging Role of the Molecular Chaperone Clusterin in Parkinson’s Disease. International Journal of Molecular Sciences. 2025; 26(13):6351. https://doi.org/10.3390/ijms26136351
Chicago/Turabian StyleCarini, Giulia, Salihu Mohammed, Alice Filippini, Ileana Ramazzina, and Isabella Russo. 2025. "The Emerging Role of the Molecular Chaperone Clusterin in Parkinson’s Disease" International Journal of Molecular Sciences 26, no. 13: 6351. https://doi.org/10.3390/ijms26136351
APA StyleCarini, G., Mohammed, S., Filippini, A., Ramazzina, I., & Russo, I. (2025). The Emerging Role of the Molecular Chaperone Clusterin in Parkinson’s Disease. International Journal of Molecular Sciences, 26(13), 6351. https://doi.org/10.3390/ijms26136351