
Conformational Analysis and Structure-Altering Mutations of the HIV-1 Frameshifting Element



Abstract
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core FSE Structure | Experimental Method | FSE Length | Key Results | Reference |
|---|---|---|---|---|
| Simple hairpin loop with single upper stem | Mutagenesis and amino acid sequencing | 39 nt | Made mutations to slippery sequence, determined slippery sequence is necessary for frameshifting and the stem loop may also be influential | [26] |
| Stable upper stem with hairpin loop and possible triplex structure | Nuclease mapping and frameshift assays | 52 nt | Disruption of the proposed triplex-structure resulted in statistically significant decrease in frameshifting efficiency | [31] |
| Optical tweezers | 52 nt | Upper stem has heterogeneous refolding dynamics, pseudoknot-like triplex can form with truncated version of entire sequence but appears to be rare | [42] | |
| Two-stem helix with three-purine bulge | Mutagenesis and enzymatic probing | 52 nt | Mutations to the upper stem impacted frameshifting efficiency more than lower stem mutations | [32] |
| NMR | 45 nt | GC to AU mutations throughout FSE suggest stem stability crucial for frameshifting efficiency | [41] | |
| Two-stem helix with three-purine bulge | NMR | 41 nt | NMR structure correlates with chemical probing, reiterates upper stem is highly conserved | [43] |
| Cryo-EM, NMR, MD | 47 nt | Obtained model of HIV-1 RNA duplex, observed super helical twist and flipped out base | [44] | |
| Three-helix junction including core hairpin loop | SHAPE | Full genome | FSE upper stem is highly conserved, three-helix junction suggested in broader context | [34] |
| Frameshift assay | 52 nt | Mutations to the upper stem suggest stability of the first 3–4 base pairs of stem loop is primary determinant of frameshifting efficiency | [45] | |
| SHAPE | 140–160 nt | Identified dynamic switching between different conformations, suggested the global sequence context influences RNA structure | [20] | |
| NMR | 41 nt | RNA switches from three-helix junction to two-helix junction containing an upper and lower stem separated by a purine bulge, average inter-helical bend of 44° | [46] |
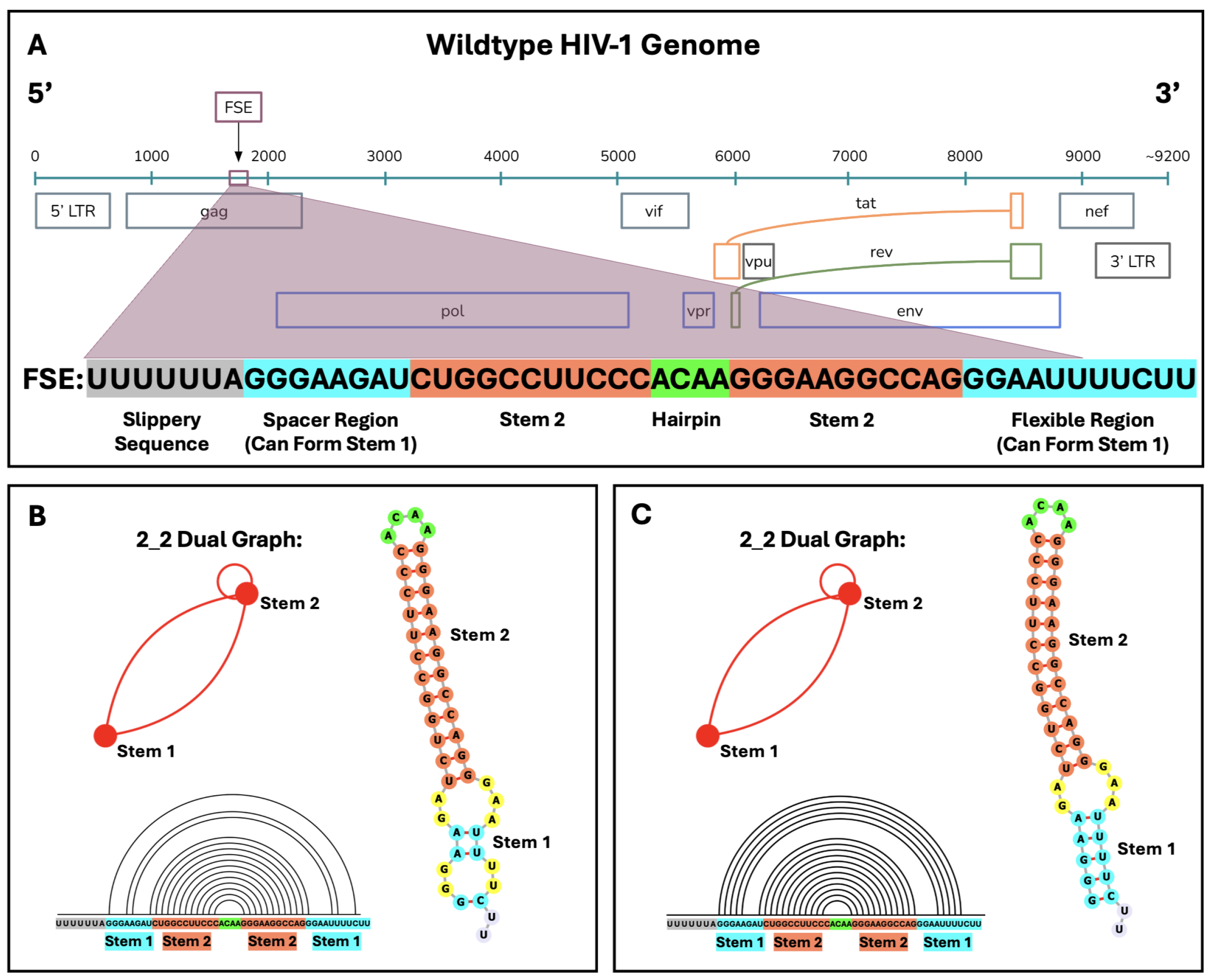
2. Results
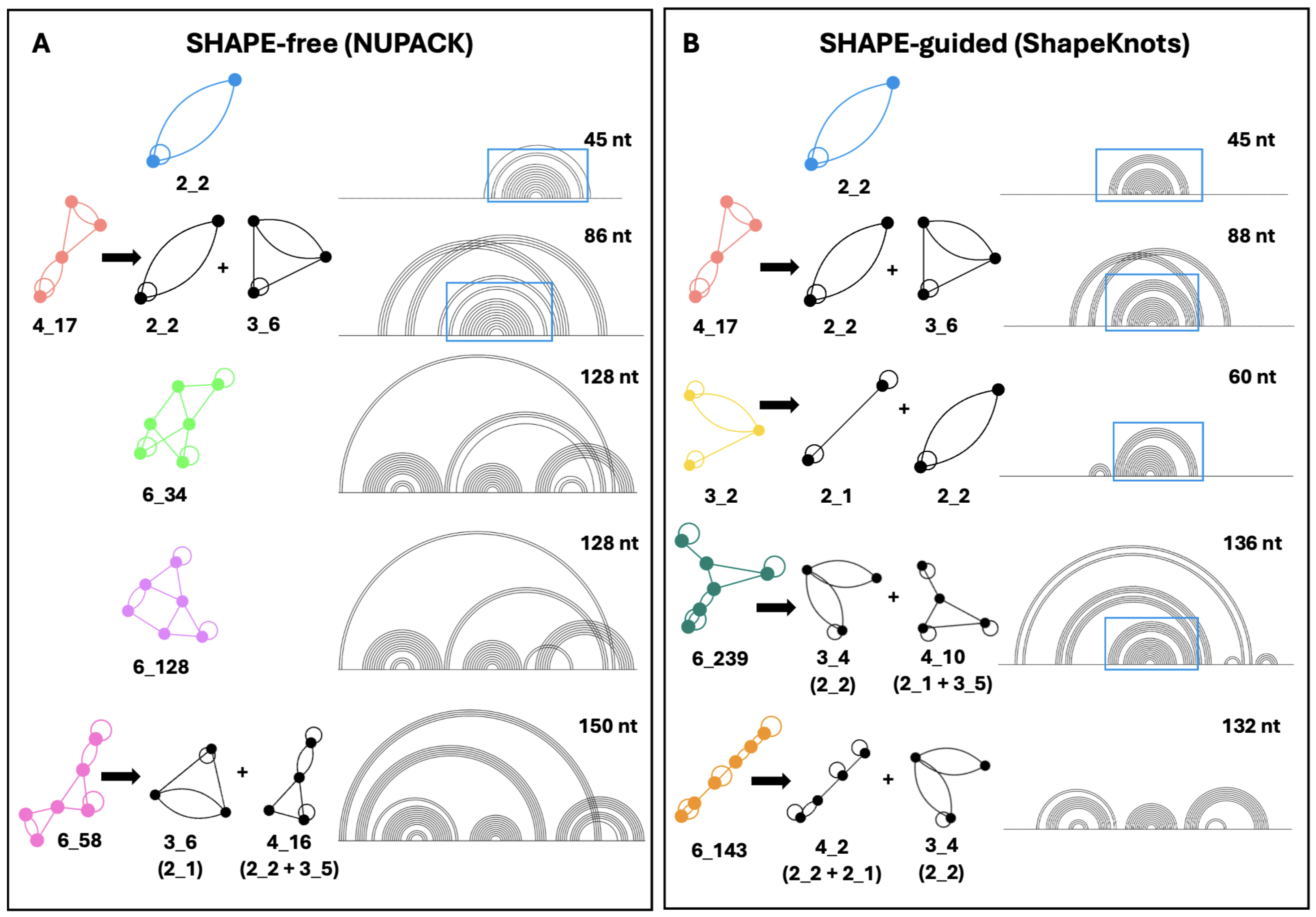
2.1. SHAPE-Free vs. SHAPE-Guided Characterization of Wildtype HIV-1 FSE Structure
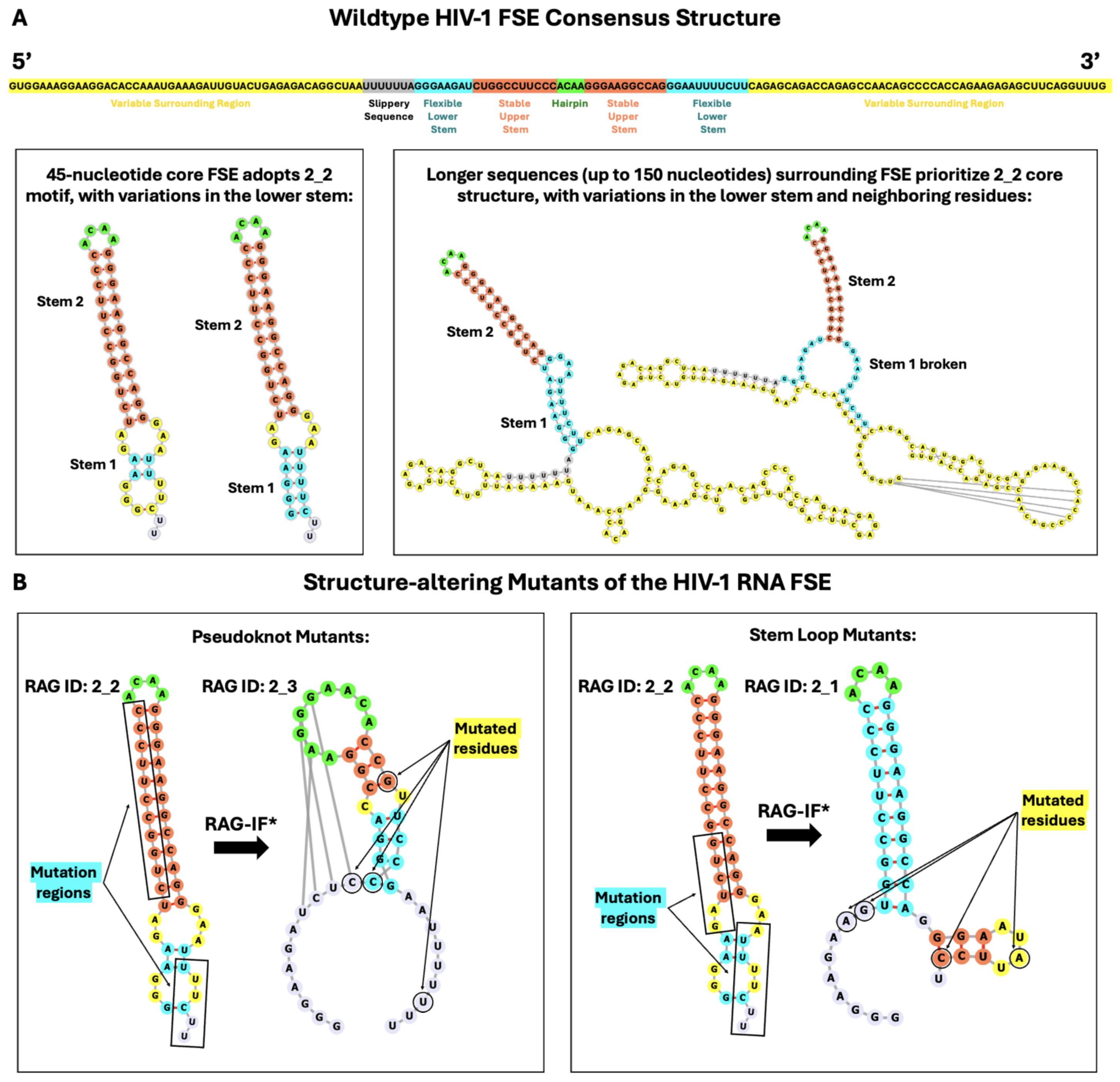
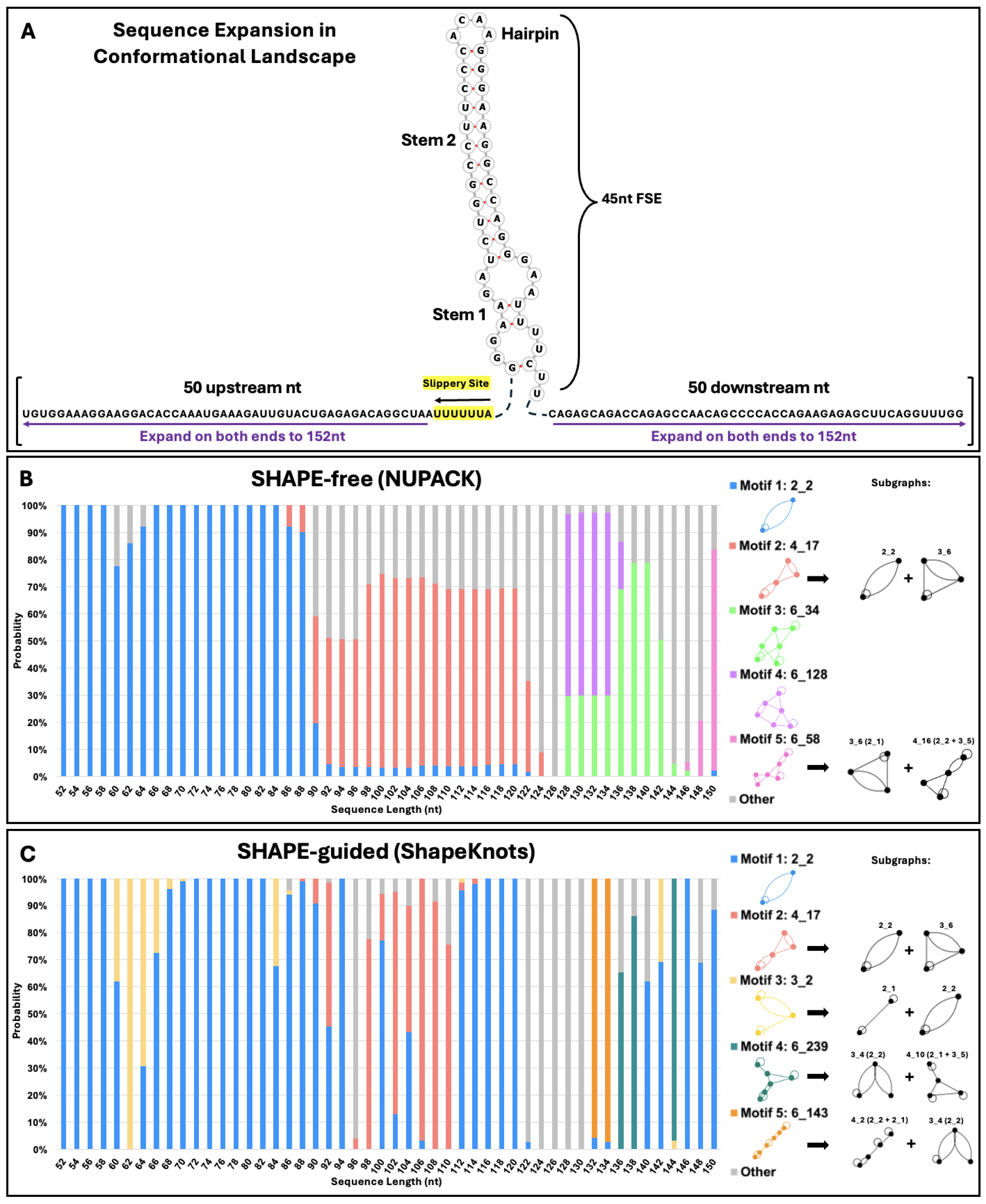
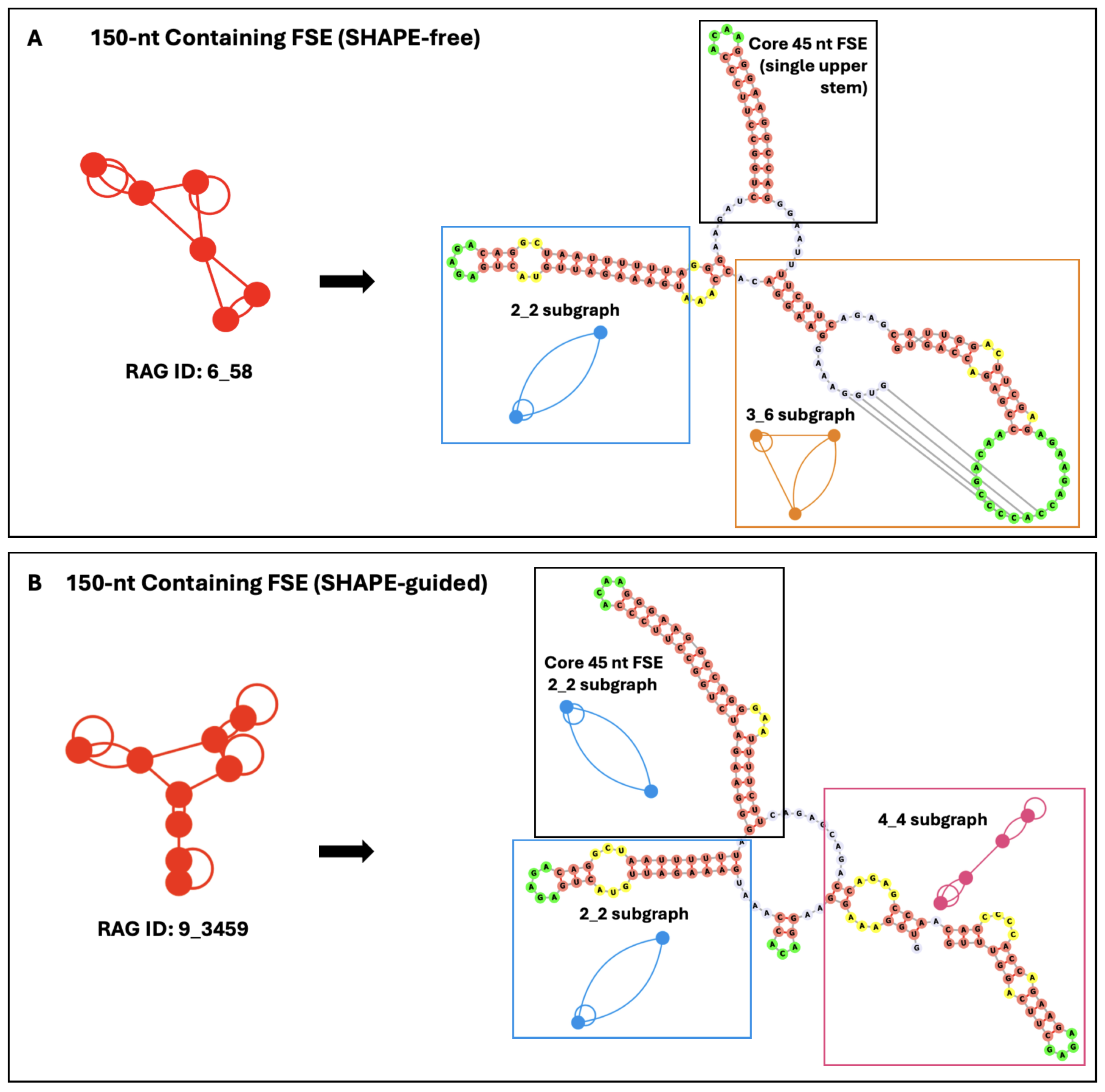
2.2. Conformational Analysis of HIV-1 FSE and Nearby Residues
2.3. Mutations for Target Graph 2_3 to Introduce Pseudoknot
2.4. Mutations for Target Graph 2_1 to Disrupt Lower Stem
3. Discussion
4. Materials and Methods
4.1. Identification of the HIV-1 RNA FSE
4.2. Secondary Structure Prediction Packages
4.3. RAG Dual Graphs
4.4. Conformational Landscapes and SHAPE Data
4.5. RAG-IF for Minimal Mutations
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van Schalkwyk, C.; Mahy, M.; Johnson, L.F.; Imai-Eaton, J.W. Updated data and methods for the 2023 UNAIDS HIV estimates. JAIDS 2024, 95, e1–e4. [Google Scholar] [CrossRef] [PubMed]
- Gumna, J.; Antczak, M.; Adamiak, R.W.; Bujnicki, J.M.; Chen, S.J.; Ding, F.; Ghosh, P.; Li, J.; Mukherjee, S.; Nithin, C.; et al. Computational Pipeline for Reference-Free Comparative Analysis of RNA 3D Structures Applied to SARS-CoV-2 UTR Models. Int. J. Mol. Sci. 2022, 23, 9630. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Q.; Wang, R.; Zhang, L.; Liu, Z. The frameshifting element in coronaviruses: Structure, function, and potential as a therapeutic target. Trends Sci. 2025, 46, 535–550. [Google Scholar] [CrossRef] [PubMed]
- Dinman, J.D. Programmed Ribosomal Frameshifting Goes Beyond Viruses: Organisms from all three kingdoms use frameshifting to regulate gene expression, perhaps signaling a paradigm shift. Microbe 2006, 1, 521–527. [Google Scholar]
- Caliskan, N.; Peske, F.; Rodnina, M.V. Changed in translation: mRNA recoding by -1 programmed ribosomal frameshifting. Trends Biochem. Sci. 2015, 40, 265–274. [Google Scholar] [CrossRef]
- Atkins, J.F.; Loughran, G.; Bhatt, P.R.; Firth, A.E.; Baranov, P.V. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016, 44, 7007–7078. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Zhuang, J.; Peltz, S.; Dougherty, J. Identification of a cellular factor that modulates HIV-1 programmed ribosomal frameshifting. J. Biol. Chem. 2010, 285, 19776–19784. [Google Scholar] [CrossRef]
- Anokhina, V.S.; Miller, B.L. Targeting Ribosomal Frameshifting as an Antiviral Strategy: From HIV-1 to SARS-CoV-2. Acc. Chem. Res. 2021, 54, 3349–3361. [Google Scholar] [CrossRef]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e14. [Google Scholar] [CrossRef]
- Schlick, T.; Zhu, Q.; Jain, S.; Yan, S. Structure-altering mutations of the SARS-CoV-2 frameshifting RNA element. Biophys. J. 2021, 120, 1040–1053. [Google Scholar] [CrossRef]
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Zheludev, I.N.; Hagey, R.J.; Haslecker, R.; Hou, Y.J.; Kretsch, R.; Pintilie, G.D.; Rangan, R.; Kladwang, W.; Li, S.; et al. Cryo-EM and antisense targeting of the 28-kDa frameshift stimulation element from the SARS-CoV-2 RNA genome. Nat. Struct. Mol. Biol. 2021, 28, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Kelly, J.A.; Olson, A.N.; Neupane, K.; Munshi, S.; San Emeterio, J.; Pollack, L.; Woodside, M.T.; Dinman, J.D. Structural and functional conservation of the programmed -1 ribosomal frameshift signal of SARS coronavirus 2 (SARS-CoV-2). J. Biol. Chem. 2020, 295, 10741–10748. [Google Scholar] [CrossRef] [PubMed]
- Lan, T.C.T.; Allan, M.F.; Malsick, L.E.; Woo, J.Z.; Zhu, C.; Zhang, F.; Khandwala, S.; Nyeo, S.S.Y.; Sun, Y.; Guo, J.U.; et al. Secondary structural ensembles of the SARS-CoV-2 RNA genome in infected cells. Nat. Commun. 2022, 13, 1128. [Google Scholar] [CrossRef]
- Huston, N.C.; Wan, H.; Strine, M.S.; de Cesaris Araujo Tavares, R.; Wilen, C.B.; Pyle, A.M. Comprehensive in vivo secondary structure of the SARS-CoV-2 genome reveals novel regulatory motifs and mechanisms. Mol. Cell 2021, 81, 584–598.e5. [Google Scholar] [CrossRef]
- Jones, C.P.; Ferré-D’Amaré, A.R. Crystal structure of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) frameshifting pseudoknot. RNA 2021, 28, 239–249. [Google Scholar] [CrossRef]
- Weichseldorfer, M.; Reitz, M.; Latinovic, O.S. Past HIV-1 medications and the current status of combined antiretroviral therapy options for HIV-1 patients. Pharmaceutics 2021, 13, 1798. [Google Scholar] [CrossRef]
- Deeks, S.G. Treatment of antiretroviral-drug-resistant HIV-1 infection. Lancet 2003, 362, 2002–2011. [Google Scholar] [CrossRef]
- Brierley, I.; Dos Ramos, F.J. Programmed ribosomal frameshifting in HIV-1 and the SARS–CoV. Virus Res. 2006, 119, 29–42. [Google Scholar] [CrossRef]
- Low, J.T.; Garcia-Miranda, P.; Mouzakis, K.D.; Gorelick, R.J.; Butcher, S.E.; Weeks, K.M. Structure and dynamics of the HIV-1 frameshift element RNA. Biochemistry 2014, 53, 4282–4291. [Google Scholar] [CrossRef]
- Shehu-Xhilaga, M.; Crowe, S.M.; Mak, J. Maintenance of the Gag/Gag-Pol ratio is important for human immunodeficiency virus type 1 RNA dimerization and viral infectivity. J. Virol. 2001, 75, 1834–1841. [Google Scholar] [CrossRef] [PubMed]
- Hilimire, T.A.; Chamberlain, J.M.; Anokhina, V.; Bennett, R.P.; Swart, O.; Myers, J.R.; Ashton, J.M.; Stewart, R.A.; Featherston, A.L.; Gates, K.; et al. HIV-1 frameshift RNA-targeted triazoles inhibit propagation of replication-competent and multi-drug-resistant HIV in human cells. ACS Chem. Biol. 2017, 12, 1674–1682. [Google Scholar] [CrossRef] [PubMed]
- German Advisory Committee Blood (Arbeitskreis Blut); Subgroup ‘Assessment of Pathogens Transmissible by Blood’. Human immunodeficiency virus (HIV). Trans. Med. Hemother. 2016, 43, 203. [Google Scholar] [CrossRef] [PubMed]
- Biswas, P.; Jiang, X.; Pacchia, A.L.; Dougherty, J.P.; Peltz, S.W. The human immunodeficiency virus type 1 ribosomal frameshifting site is an invariant sequence determinant and an important target for antiviral therapy. J. Virol. 2004, 78, 2082–2087. [Google Scholar] [CrossRef]
- Wilson, W.; Braddock, M.; Adams, S.E.; Rathjen, P.D.; Kingsman, S.M.; Kingsman, A.J. HIV expression strategies: Ribosomal frameshifting is directed by a short sequence in both mammalian and yeast systems. Cell 1988, 55, 1159–1169. [Google Scholar] [CrossRef]
- Jacks, T.; Madhani, H.D.; Masiarz, F.R.; Varmus, H.E. Signals for ribosomal frameshifting in the Rous sarcoma virus gag-pol region. Cell 1988, 55, 447–458. [Google Scholar] [CrossRef]
- Lee, S.; Yan, S.; Dey, A.; Laederach, A.; Schlick, T. A Cascade of Conformational Switches in SARS-CoV-2 Frameshifting: Coregulation by Upstream and Downstream Elements. Biochemistry 2025, 64, 953–966. [Google Scholar] [CrossRef]
- Dey, A.; Yan, S.; Schlick, T.; Laederach, A. Abolished frameshifting for predicted structure-stabilizing SARS-CoV-2 mutants: Implications to alternative conformations and their statistical structural analyses. RNA 2024, 30, 1437–1450. [Google Scholar] [CrossRef]
- Schlick, T.; Zhu, Q.; Dey, A.; Jain, S.; Yan, S.; Laederach, A. To knot or not to knot: Multiple conformations of the SARS-CoV-2 frameshifting RNA element. J. Amer. Chem. Soc. 2021, 143, 11404–11422. [Google Scholar] [CrossRef]
- Le, S.Y.; Shapiro, B.A.; Chen, J.H.; Nussinov, R.; Maizel, J.V. RNA pseudoknots downstream of the frameshift sites of retroviruses. Genet. Anal. Biomol. Eng. 1991, 8, 191–205. [Google Scholar]
- Dinman, J.D.; Richter, S.; Plant, E.P.; Taylor, R.C.; Hammell, A.B.; Rana, T.M. The frameshift signal of HIV-1 involves a potential intramolecular triplex RNA structure. Proc. Natl. Acad. Sci. USA 2002, 99, 5331–5336. [Google Scholar] [CrossRef] [PubMed]
- Dulude, D.; Baril, M.; Brakier-Gingras, L. Characterization of the frameshift stimulatory signal controlling a programmed −1 ribosomal frameshift in the human immunodeficiency virus type 1. Nucleic Acids Res. 2002, 30, 5094–5102. [Google Scholar] [CrossRef] [PubMed]
- Baril, M.; Dulude, D.; Gendron, K.; Lemay, G.; Brakier-Gingras, L. Efficiency of a programmed −1 ribosomal frameshift in the different subtypes of the human immunodeficiency virus type 1 group M. RNA 2003, 9, 1246–1253. [Google Scholar] [CrossRef] [PubMed]
- Watts, J.M.; Dang, K.K.; Gorelick, R.J.; Leonard, C.W.; Bess, J.W., Jr.; Swanstrom, R.; Burch, C.L.; Weeks, K.M. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 2009, 460, 711–716. [Google Scholar] [CrossRef]
- Garcia-Miranda, P.; Becker, J.T.; Benner, B.E.; Blume, A.; Sherer, N.M.; Butcher, S.E. Stability of HIV frameshift site RNA correlates with frameshift efficiency and decreased virus infectivity. J. Virol. 2016, 90, 6906–6917. [Google Scholar] [CrossRef]
- Gan, H.H.; Pasquali, S.; Schlick, T. Exploring the repertoire of RNA secondary motifs using graph theory; implications for RNA design. Nucleic Acids Res. 2003, 31, 2926–2943. [Google Scholar] [CrossRef]
- Meng, G.; Tariq, M.; Jain, S.; Elmetwaly, S.; Schlick, T. RAG-Web: RNA structure prediction/design using RNA-As-Graphs. Bioinformatics 2020, 36, 647–648. [Google Scholar] [CrossRef]
- Yan, S.; Zhu, Q.; Jain, S.; Schlick, T. Length-dependent motions of SARS-CoV-2 frameshifting RNA pseudoknot and alternative conformations suggest avenues for frameshifting suppression. Nat. Commun. 2022, 13, 4284. [Google Scholar] [CrossRef]
- Yan, S.; Schlick, T. Heterogeneous and multiple conformational transition pathways between pseudoknots of the SARS-CoV-2 frameshift element. Proc. Natl. Acad. Sci. USA 2025, 122, e2417479122. [Google Scholar] [CrossRef]
- Yan, S.; Zhu, Q.; Hohl, J.; Dong, A.; Schlick, T. Evolution of coronavirus frameshifting elements: Competing stem networks explain conservation and variability. Proc. Natl. Acad. Sci. USA 2023, 120, e2221324120. [Google Scholar] [CrossRef]
- Staple, D.W.; Butcher, S.E. Solution structure and thermodynamic investigation of the HIV-1 frameshift inducing element. J. Mol. Biol. 2005, 349, 1011–1023. [Google Scholar] [CrossRef] [PubMed]
- Ritchie, D.B.; Cappellano, T.R.; Tittle, C.; Rezajooei, N.; Rouleau, L.; Sikkema, W.K.; Woodside, M.T. Conformational dynamics of the frameshift stimulatory structure in HIV-1. RNA 2017, 23, 1376–1384. [Google Scholar] [CrossRef] [PubMed]
- Gaudin, C.; Mazauric, M.H.; Traïkia, M.; Guittet, E.; Yoshizawa, S.; Fourmy, D. Structure of the RNA signal essential for translational frameshifting in HIV-1. J. Mol. Biol. 2005, 349, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Keane, S.C.; Su, Z.; Irobalieva, R.N.; Chen, M.; Van, V.; Sciandra, C.A.; Marchant, J.; Heng, X.; Schmid, M.F.; et al. Structure of the 30 kDa HIV-1 RNA dimerization signal by a hybrid cryo-EM, NMR, and molecular dynamics approach. Structure 2018, 26, 490–498. [Google Scholar] [CrossRef]
- Mouzakis, K.D.; Lang, A.L.; Vander Meulen, K.A.; Easterday, P.D.; Butcher, S.E. HIV-1 frameshift efficiency is primarily determined by the stability of base pairs positioned at the mRNA entrance channel of the ribosome. Nucleic Acids Res. 2013, 41, 1901–1913. [Google Scholar] [CrossRef]
- Mouzakis, K.D.; Dethoff, E.A.; Tonelli, M.; Al-Hashimi, H.; Butcher, S.E. Dynamic motions of the HIV-1 frameshift site RNA. Biophys. J. 2015, 108, 644–654. [Google Scholar] [CrossRef]
- Lai, D.; Proctor, J.R.; Zhu, J.Y.A.; Meyer, I.M. R-CHIE: A web server and R package for visualizing RNA secondary structures. Nucleic Acids Res. 2012, 40, e95. [Google Scholar] [CrossRef]
- Jain, S.; Bayrak, C.S.; Petingi, L.; Schlick, T. Dual graph partitioning highlights a small group of pseudoknot-containing RNA submotifs. Genes 2018, 9, 371. [Google Scholar] [CrossRef]
- Telenti, A.; Martinez, R.; Munoz, M.; Bleiber, G.; Greub, G.; Sanglard, D.; Peters, S. Analysis of natural variants of the human immunodeficiency virus type 1 gag-pol frameshift stem-loop structure. J. Virol. 2002, 76, 7868–7873. [Google Scholar] [CrossRef]
- Mazauric, M.H.; Seol, Y.; Yoshizawa, S.; Visscher, K.; Fourmy, D. Interaction of the HIV-1 frameshift signal with the ribosome. Nucleic Acids Res. 2009, 37, 7654–7664. [Google Scholar] [CrossRef]
- Giedroc, D.P.; Cornish, P.V. Frameshifting RNA pseudoknots: Structure and mechanism. Virus Res. 2009, 139, 193–208. [Google Scholar] [CrossRef] [PubMed]
- Seah, Y.L. Molecular Dynamics Simulation of RNA Ribosomal Frameshift Stimulatory Elements. Ph.D. Thesis, Nanyang Technological University, Singapore, 2017. [Google Scholar]
- Dirks, R.M.; Pierce, N.A. A partition function algorithm for nucleic acid secondary structure including pseudoknots. J. Comput. Chem. 2003, 24, 1664–1677. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Kato, Y.; Hamada, M.; Akutsu, T.; Asai, K. IPknot: Fast and accurate prediction of RNA secondary structures with pseudoknots using integer programming. Bioinformatics 2011, 27, i85–i93. [Google Scholar] [CrossRef] [PubMed]
- Rivas, E.; Eddy, S.R. A dynamic programming algorithm for RNA structure prediction including pseudoknots. J. Mol. Biol. 1999, 285, 2053–2068. [Google Scholar] [CrossRef]
- Hajdin, C.E.; Bellaousov, S.; Huggins, W.; Leonard, C.W.; Mathews, D.H.; Weeks, K.M. Accurate SHAPE-directed RNA secondary structure modeling, including pseudoknots. Proc. Natl. Acad. Sci. USA 2013, 110, 5498–5503. [Google Scholar] [CrossRef]
- Jain, S.; Tao, Y.; Schlick, T. Inverse folding with RNA-As-Graphs produces a large pool of candidate sequences with target topologies. J. Struct. Biol. 2020, 209, 107438. [Google Scholar] [CrossRef]
- Jain, S.; Saju, S.; Petingi, L.; Schlick, T. An extended dual graph library and partitioning algorithm applicable to pseudoknotted RNA structures. Methods 2019, 162, 74–84. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Newton, K.; Yan, S.; Schlick, T. Conformational Analysis and Structure-Altering Mutations of the HIV-1 Frameshifting Element. Int. J. Mol. Sci. 2025, 26, 6297. https://doi.org/10.3390/ijms26136297
Newton K, Yan S, Schlick T. Conformational Analysis and Structure-Altering Mutations of the HIV-1 Frameshifting Element. International Journal of Molecular Sciences. 2025; 26(13):6297. https://doi.org/10.3390/ijms26136297
Chicago/Turabian StyleNewton, Katelyn, Shuting Yan, and Tamar Schlick. 2025. "Conformational Analysis and Structure-Altering Mutations of the HIV-1 Frameshifting Element" International Journal of Molecular Sciences 26, no. 13: 6297. https://doi.org/10.3390/ijms26136297
APA StyleNewton, K., Yan, S., & Schlick, T. (2025). Conformational Analysis and Structure-Altering Mutations of the HIV-1 Frameshifting Element. International Journal of Molecular Sciences, 26(13), 6297. https://doi.org/10.3390/ijms26136297