
GPX1 and RCN1 as New Endoplasmic Reticulum Stress-Related Biomarkers in Multiple Sclerosis Brain Tissue and Their Involvement in the APP-CD74 Pathway: An Integrated Study Combining Machine Learning and Multi-Omics

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Result
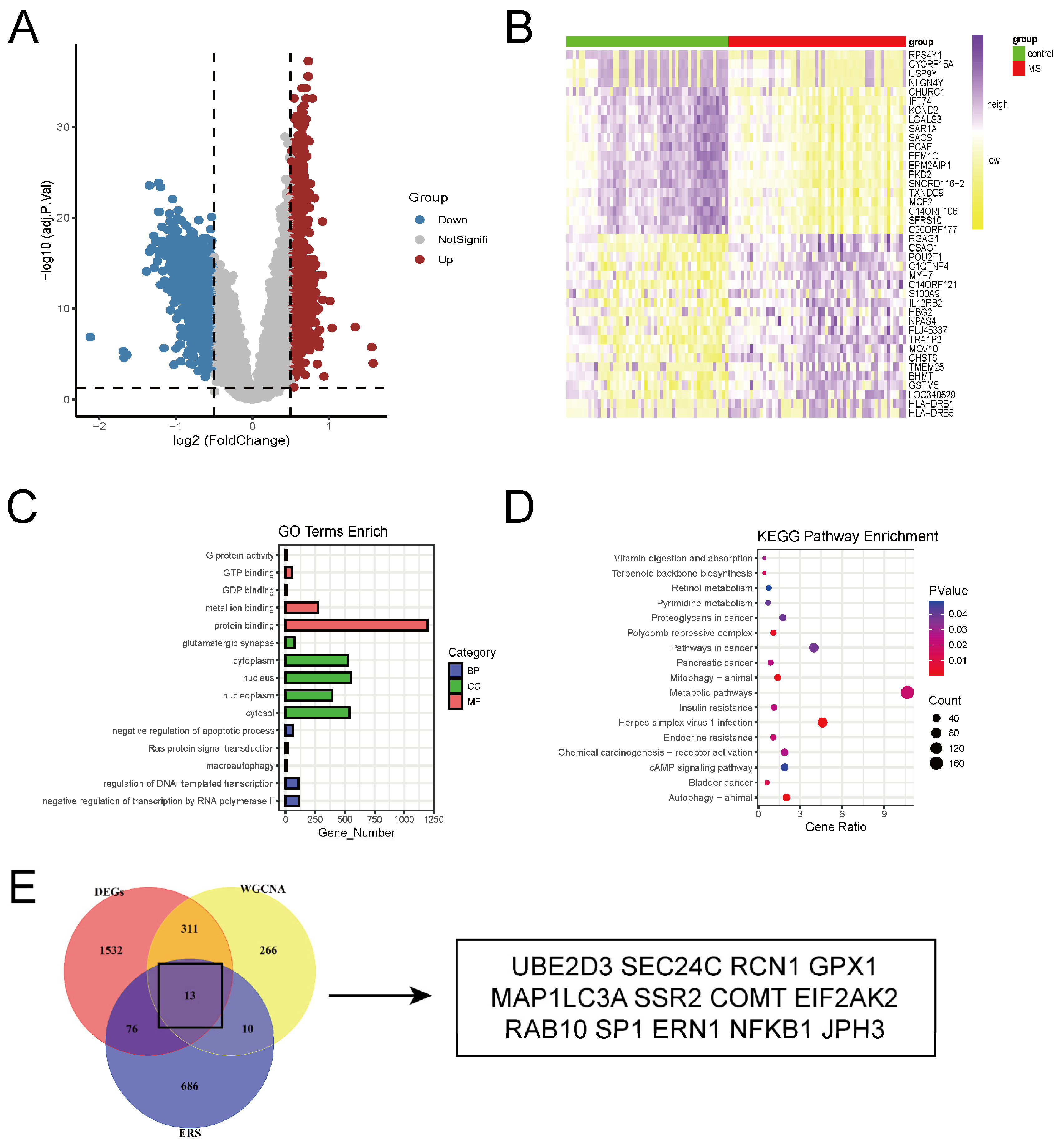
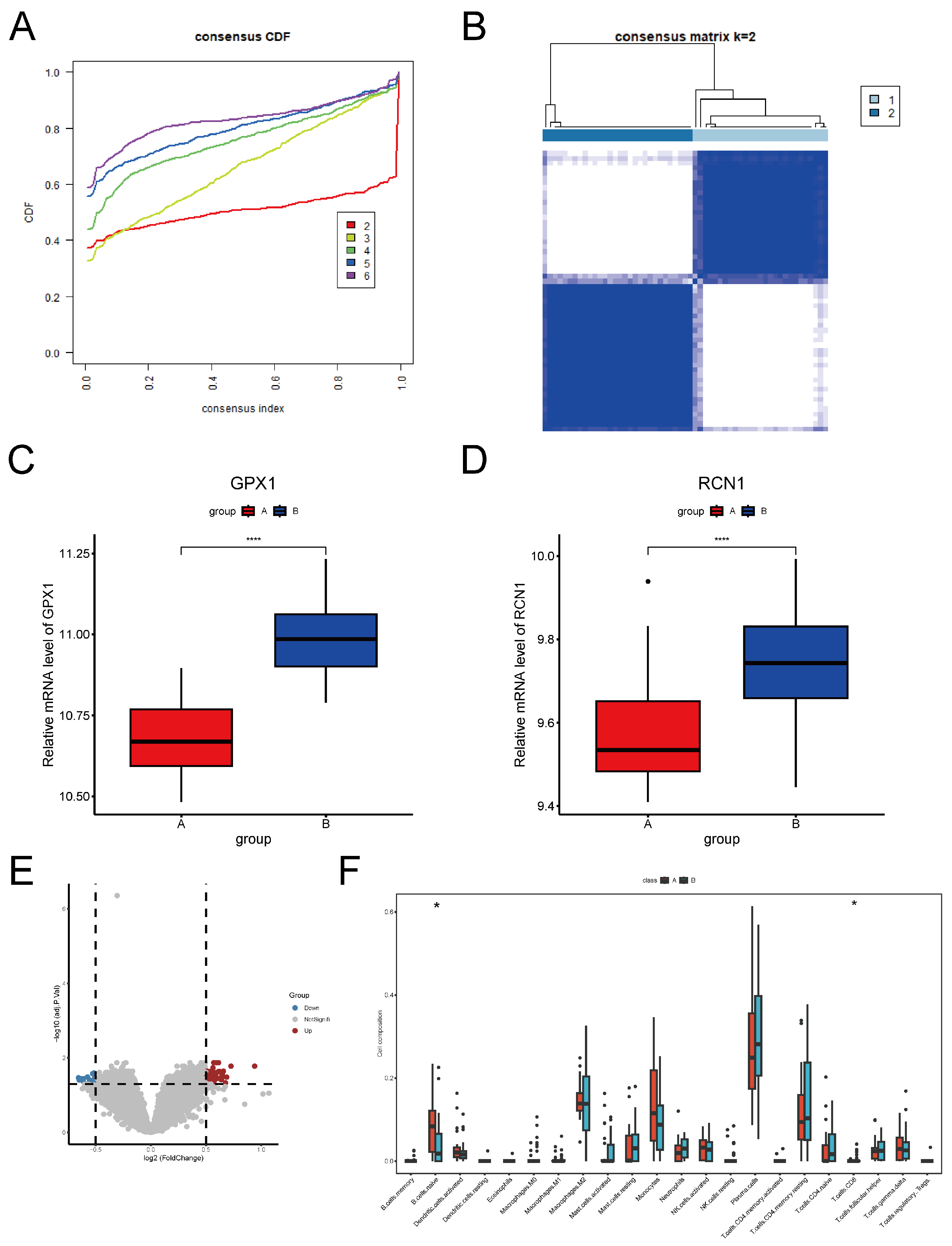
2.1. Identification of DEGs Between Brain Tissues of Multiple Sclerosis Patients and Controls
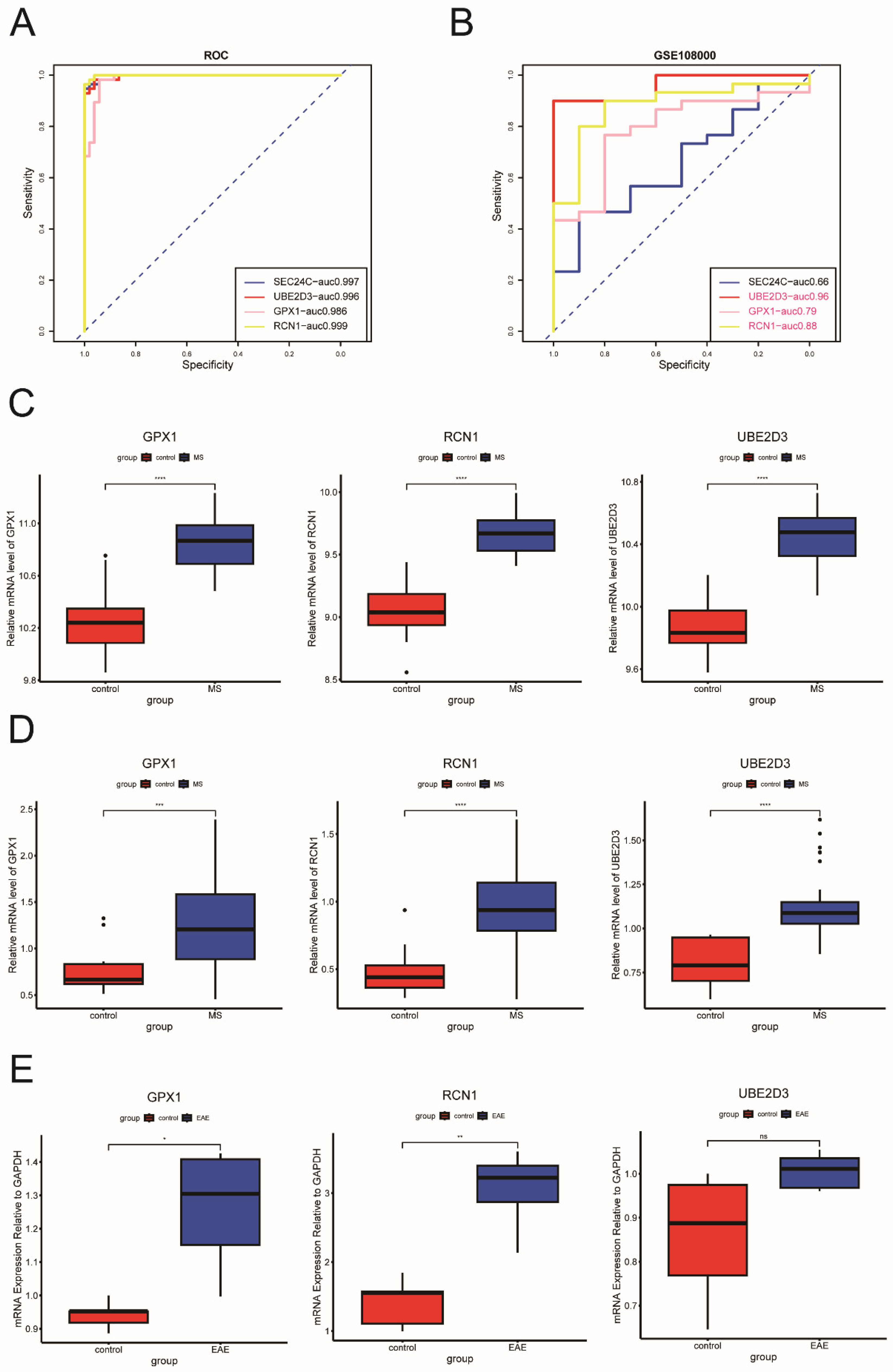
2.2. Identification and Validation of Key ERS-DEGs
2.3. Immune Infiltration Analysis in MS
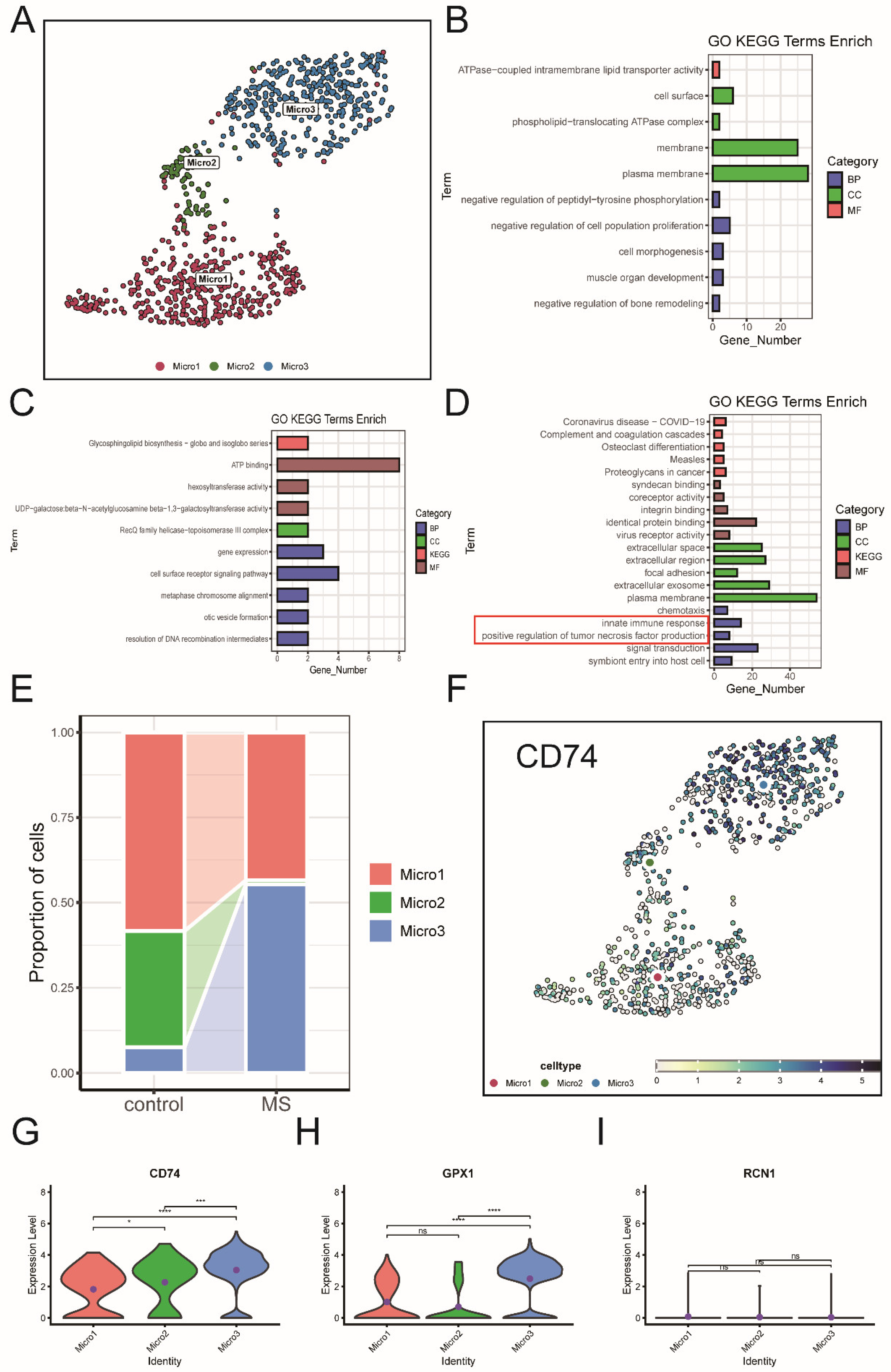
2.4. Unsupervised Clustering and Analysis of MS
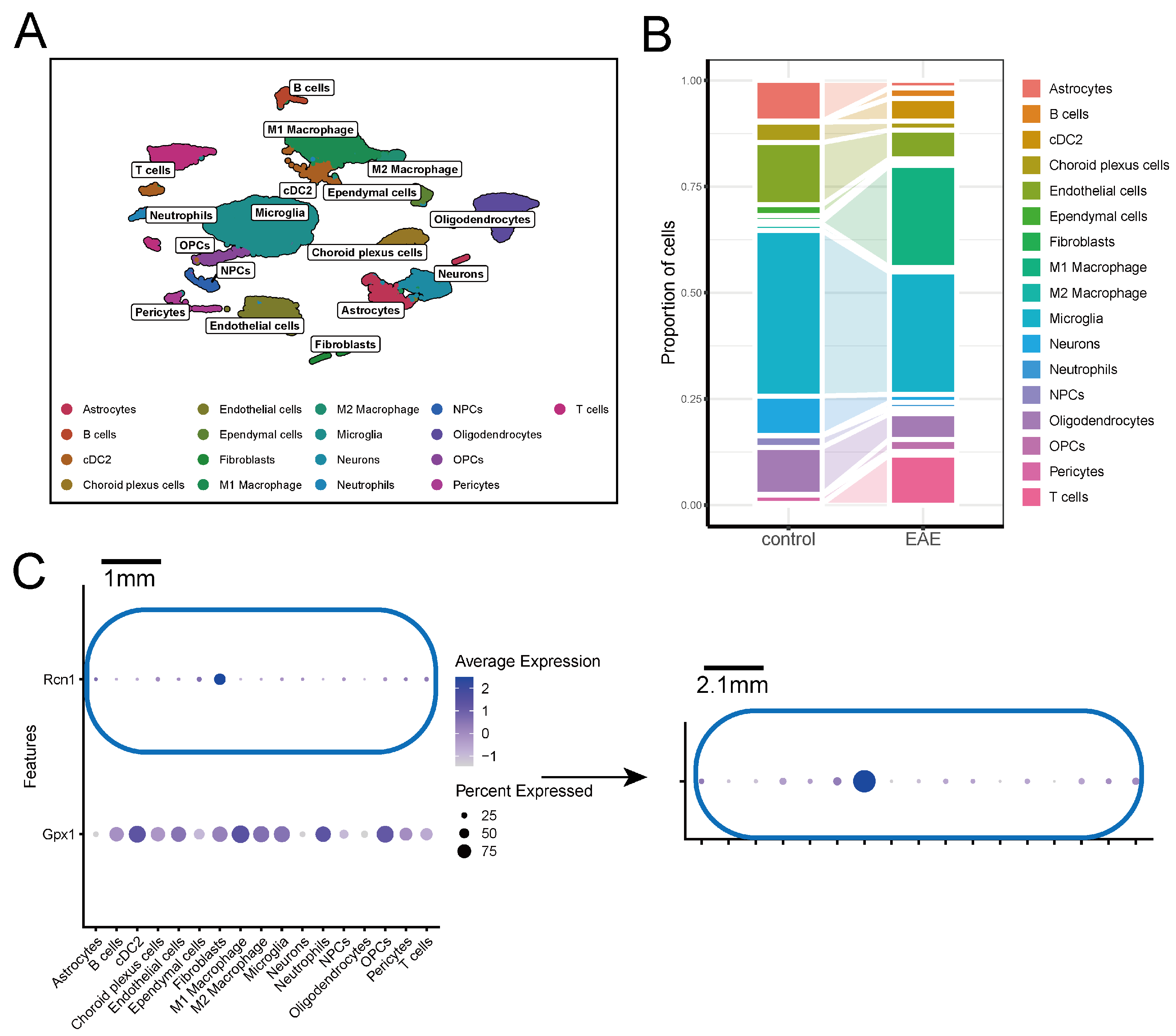
2.5. Expression of Key ERS-DEGs in Single-Cell Data
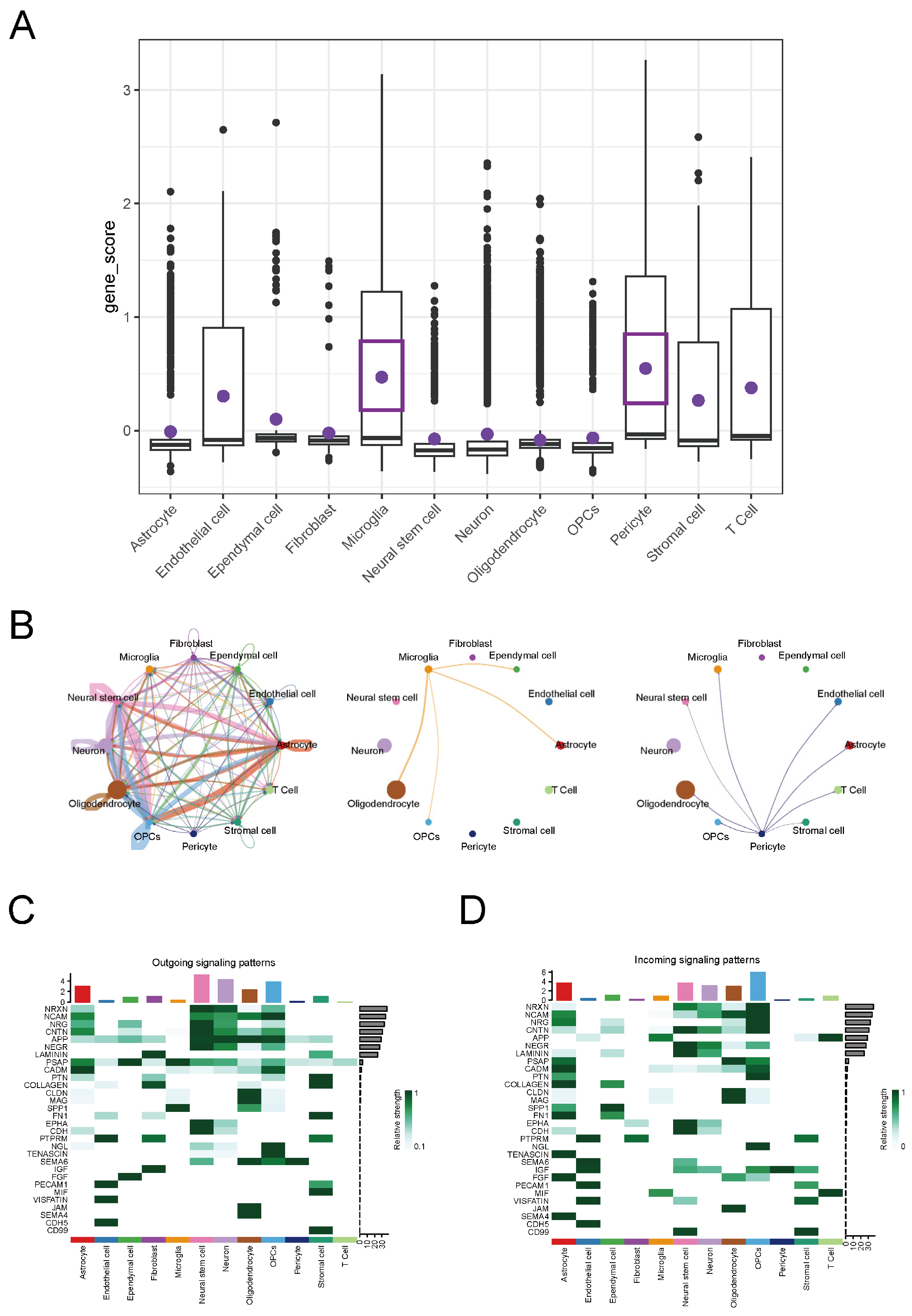
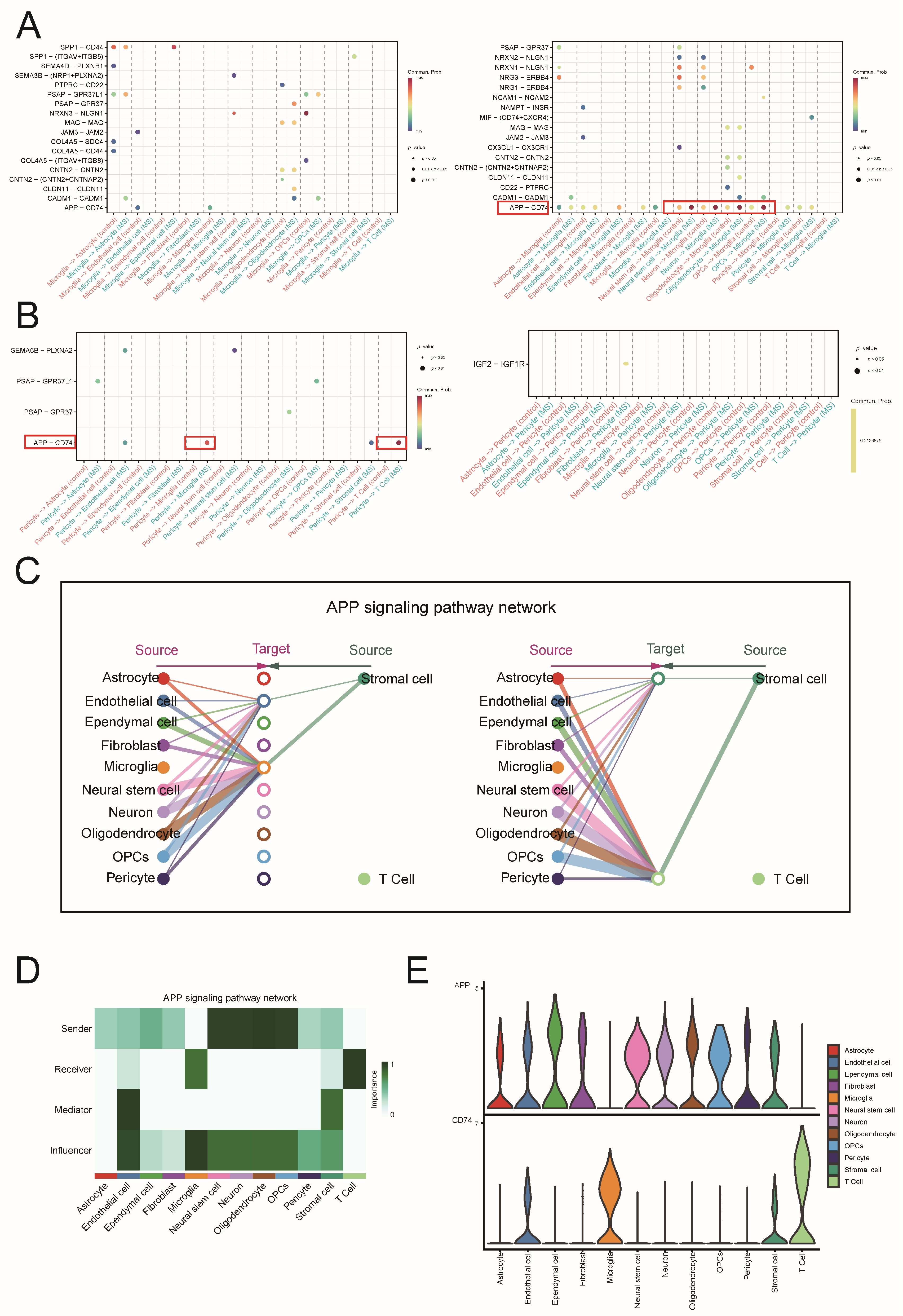
2.6. Analysis of Cell Communication in MS
2.7. Conducting Separate Analyses of Cell Communication via the APP-CD74 Pathway in MS and the EAE Mouse Model
2.8. Functional Roles of Microglia and Pericytes via the APP-CD74 Pathway in MS
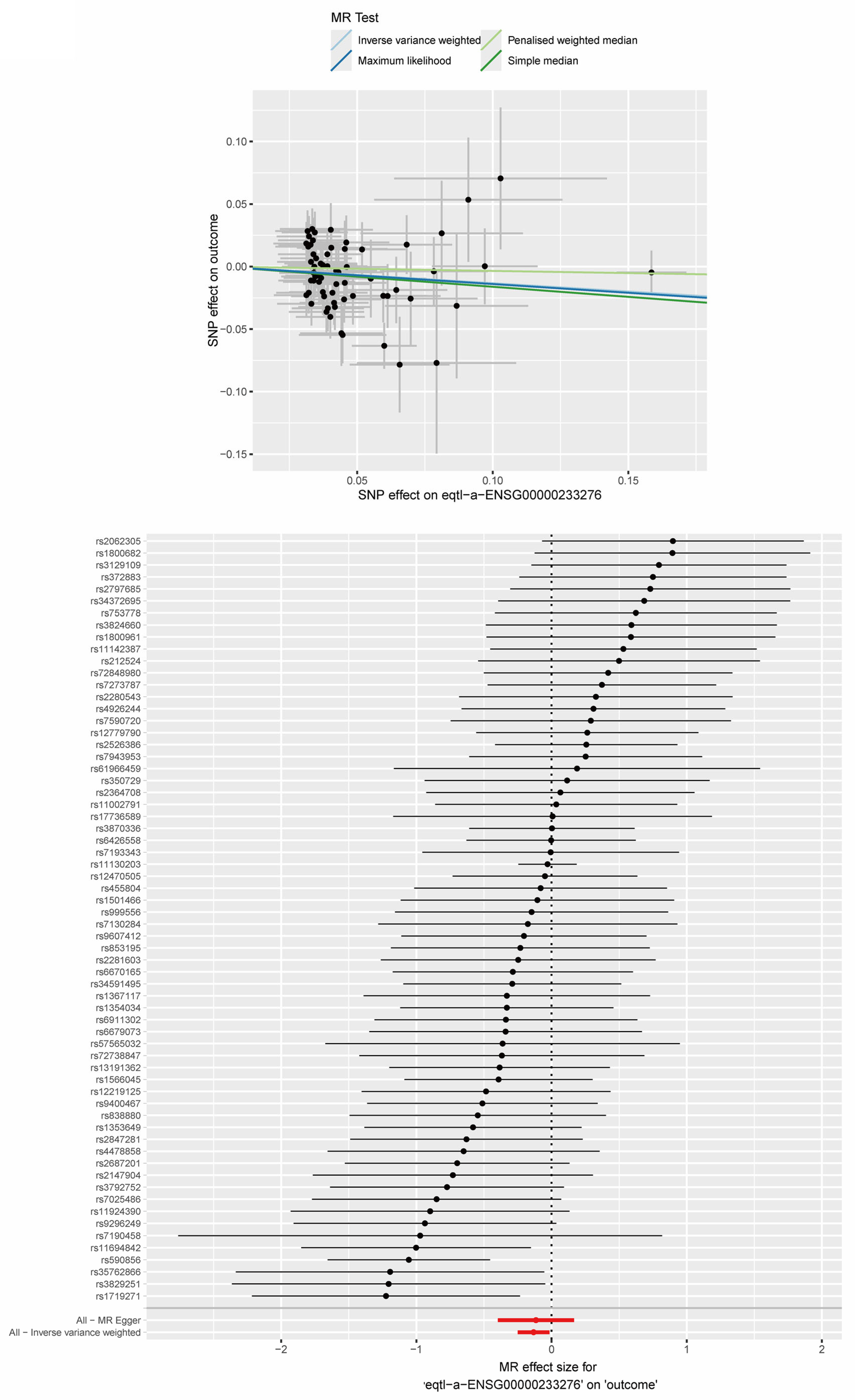
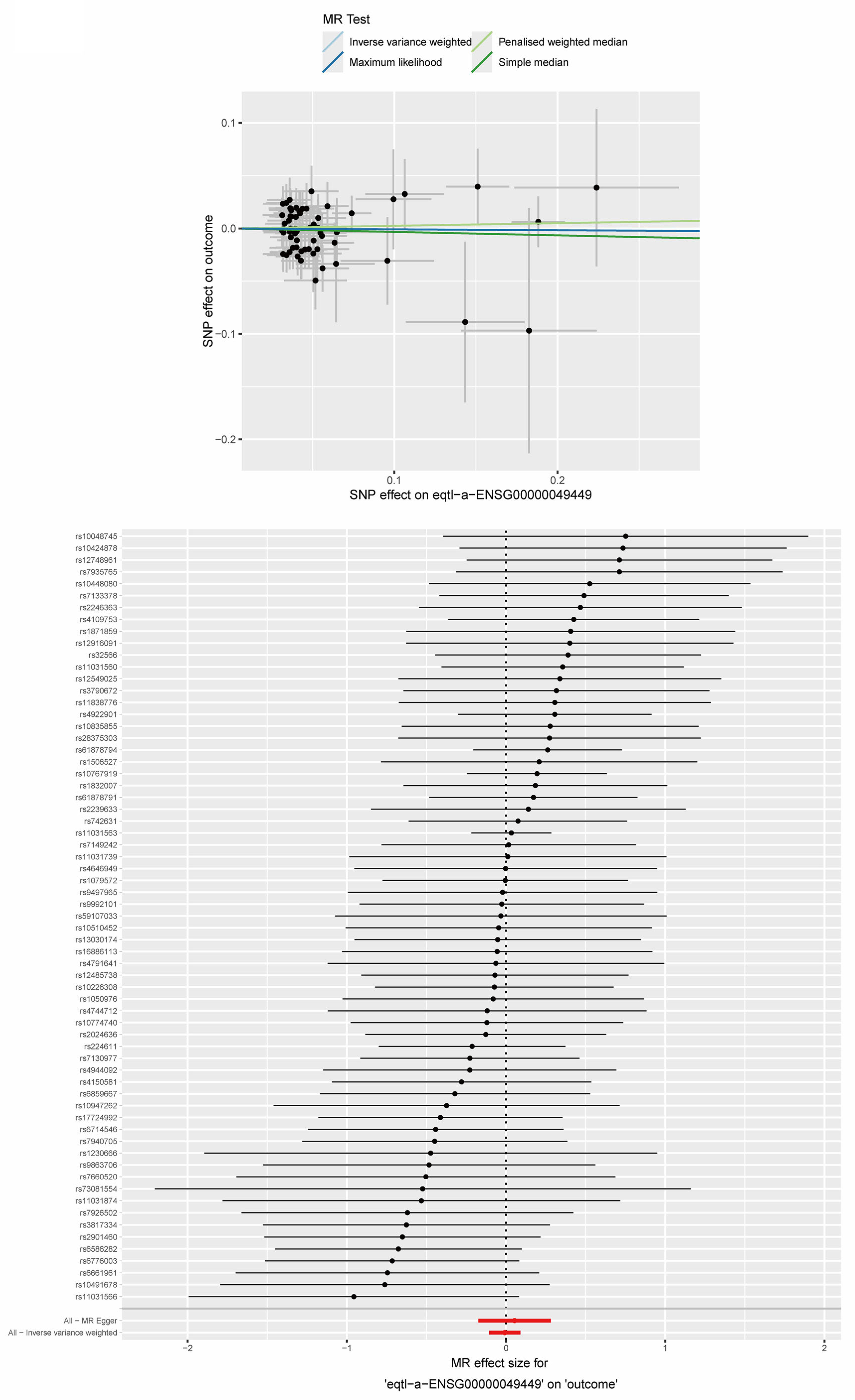
2.9. MR Analysis Between Key ERS-DEGs and MS
3. Discussion
4. Materials and Methods
4.1. Data Acquisition
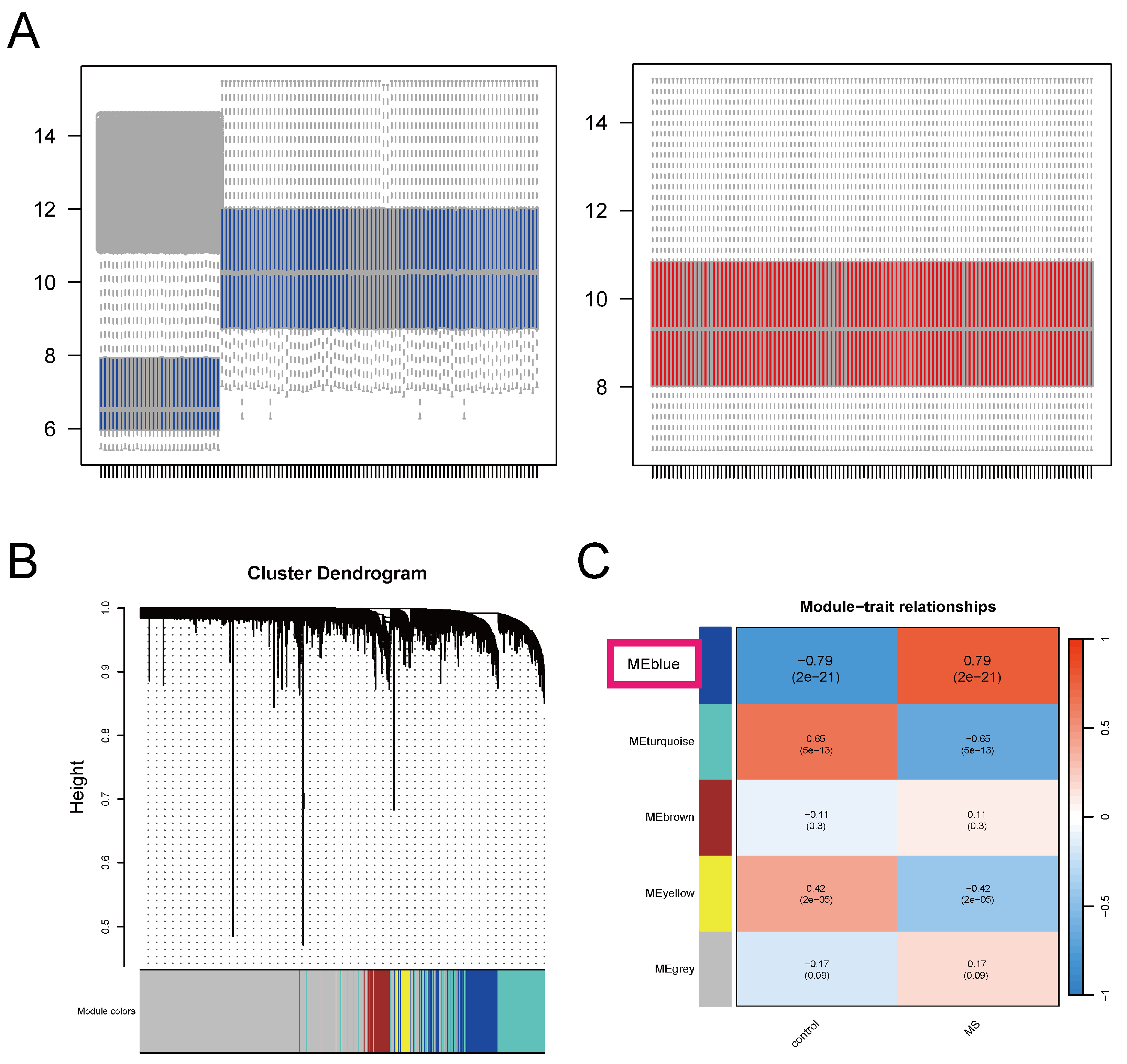
4.2. Weighted Gene Co-Expression Network Analysis
4.3. Identification of Differentially Expressed Genes (DEGs)
4.4. Identification and Validation of ERS-DEGs Associated with Clinical Traits in MS
4.5. Immune Infiltration Analysis in Multiple Sclerosis Using CIBERSORT
4.6. Subclustering Analysis Using Key ERS-DEGs
4.7. Processing and Clustering of Single-Nucleus RNA and Single-Cell RNA Data
4.8. Scoring of Key ERS-DEGs in snRNA-seq
4.9. Cell Communication Analysis
4.10. Collection of GWAS Data and Two-Sample Mendelian Randomization Analysis
4.11. Immunohistochemistry and Immunofluorescence
4.12. Establishment of EAE Model
4.13. RNA Extraction and qRT-PCR
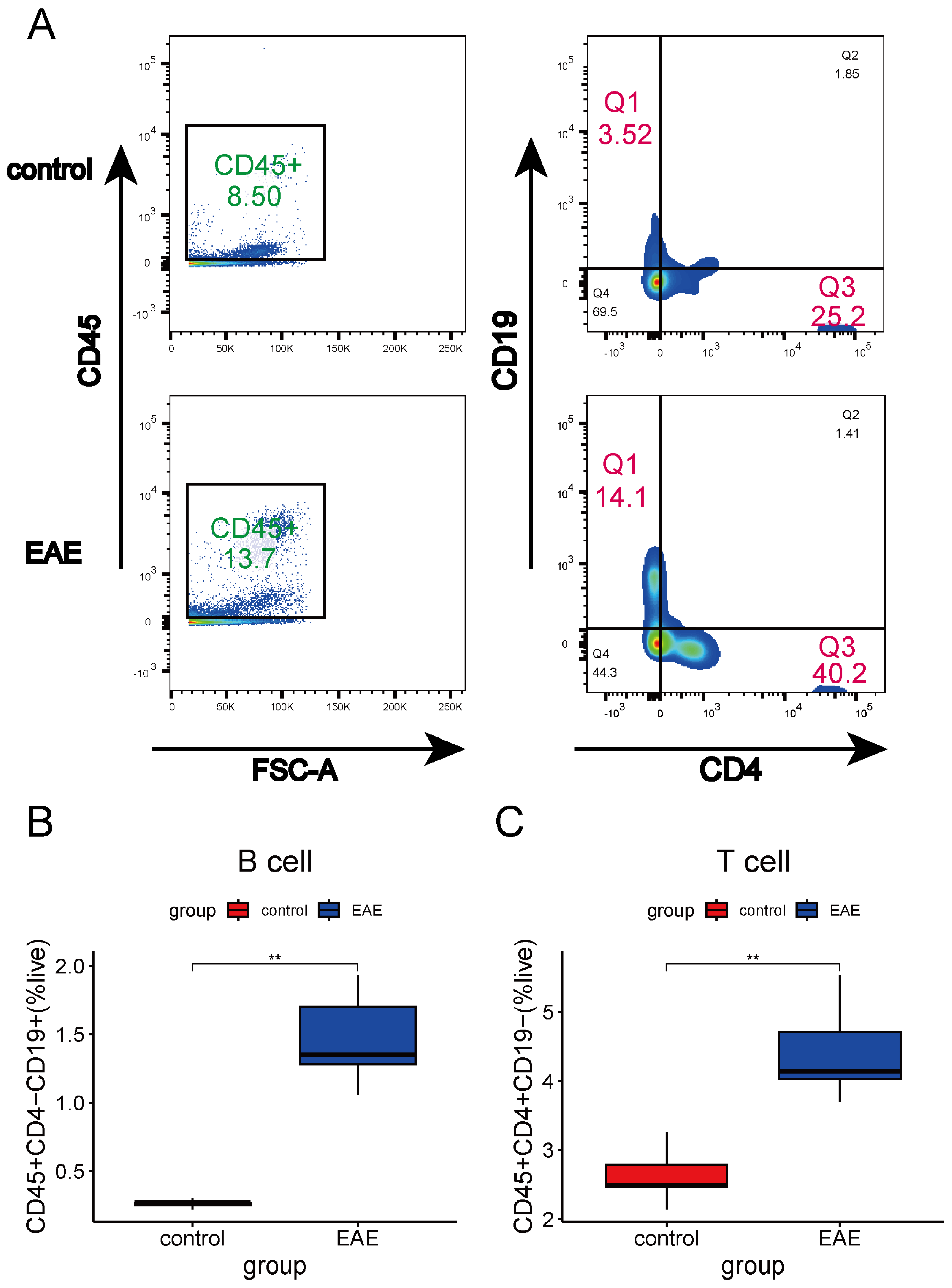
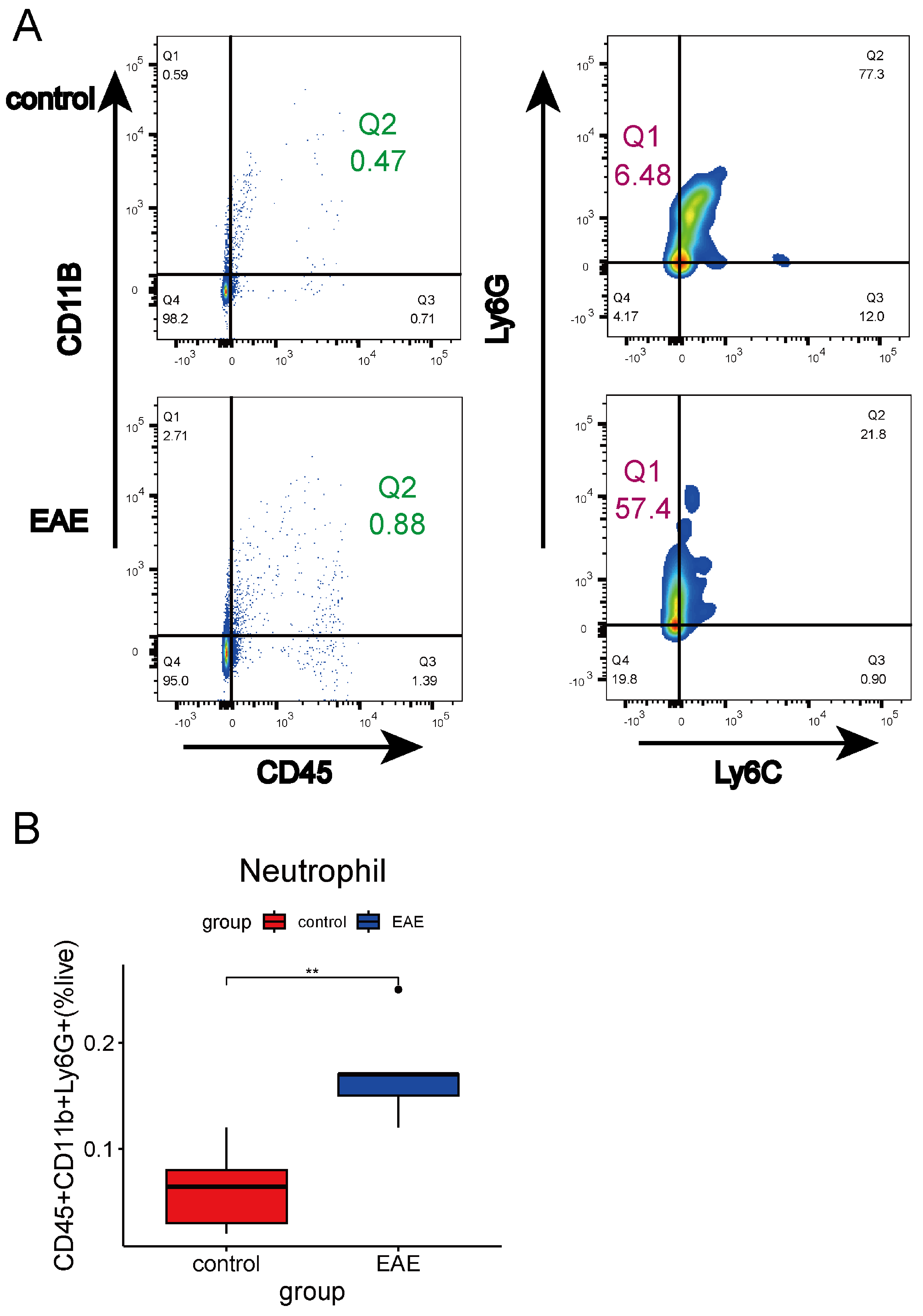
4.14. Flow Cytometry Analysis
4.15. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dobson, R.; Giovannoni, G. Multiple sclerosis—A review. Eur. J. Neurol. 2018, 26, 27–40. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Cognitive impairment in multiple sclerosis: From phenomenology to neurobiological mechanisms. J. Neural Transm. 2024, 131, 871–899. [Google Scholar] [CrossRef]
- Hunter, S.F. Overview and diagnosis of multiple sclerosis. Am. J. Manag Care 2016, 22 (Suppl. 6), s141–s150. [Google Scholar] [PubMed]
- Garg, N.; Smith, T.W. An update on immunopathogenesis, diagnosis, and treatment of multiple sclerosis. Brain Behav. 2015, 5, e00362. [Google Scholar] [CrossRef]
- Konen, F.F.; Möhn, N.; Witte, T.; Schefzyk, M.; Wiestler, M.; Lovric, S.; Hufendiek, K.; Schwenkenbecher, P.; Sühs, K.W.; Friese, M.A.; et al. Treatment of autoimmunity: The impact of disease-modifying therapies in multiple sclerosis and comorbid autoimmune disorders. Autoimmun. Rev. 2023, 22, 103312. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Shi, C.; He, M.; Xiong, S.; Xia, X. Endoplasmic reticulum stress: Molecular mechanism and therapeutic targets. Signal Transduct. Target. Ther. 2023, 8, 352. [Google Scholar] [CrossRef]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Fujii, C.; Zorumski, C.F.; Izumi, Y. Endoplasmic reticulum stress, autophagy, neuroinflammation, and sigma 1 receptors as contributors to depression and its treatment. Neural Regen. Res. 2024, 19, 2202–2211. [Google Scholar] [CrossRef]
- Kou, J.J.; Shi, J.Z.; He, Y.Y.; Hao, J.J.; Zhang, H.Y.; Luo, D.M.; Song, J.K.; Yan, Y.; Xie, X.M.; Du, G.H.; et al. Luteolin alleviates cognitive impairment in Alzheimer’s disease mouse model via inhibiting endoplasmic reticulum stress-dependent neuroinflammation. Acta Pharmacol. Sin. 2022, 43, 840–849. [Google Scholar] [CrossRef]
- Nussbaum, Y.I.; Manjunath, Y.; Suvilesh, K.N.; Warren, W.C.; Shyu, C.R.; Kaifi, J.T.; Ciorba, M.A.; Mitchem, J.B. Current and Prospective Methods for Assessing Anti-Tumor Immunity in Colorectal Cancer. Int. J. Mol. Sci. 2021, 22, 4802. [Google Scholar] [CrossRef]
- Pan, M.; Zhou, J.; Jiao, C.; Ge, J. Bioinformatics analysis of the endoplasmic reticulum stress-related prognostic model and immune cell infiltration in acute myeloid leukemia. Hematology 2023, 28, 2221101. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Baizán, N.; Ribas, L.; Piferrer, F. Improved biomarker discovery through a plot twist in transcriptomic data analysis. BMC Biol. 2022, 20, 208. [Google Scholar] [CrossRef]
- Katz Sand, I. Classification, diagnosis, and differential diagnosis of multiple sclerosis. Curr. Opin. Neurol. 2015, 28, 193–205. [Google Scholar] [CrossRef]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Pan, X.; Wei, G.; Hua, Y. Research progress of glutathione peroxidase family (GPX) in redoxidation. Front. Pharmacol. 2023, 14, 1147414. [Google Scholar] [CrossRef]
- Zou, M.; Liang, Q.; Zhang, W.; Zhu, Y.; Xu, Y. Endoplasmic reticulum stress related genome-wide Mendelian randomization identifies therapeutic genes for ulcerative colitis and Crohn’s disease. Front. Genet. 2023, 14, 1270085. [Google Scholar] [CrossRef]
- Sharma, G.; Shin, E.J.; Sharma, N.; Nah, S.Y.; Mai, H.N.; Nguyen, B.T.; Jeong, J.H.; Lei, X.G.; Kim, H.C. Glutathione peroxidase-1 and neuromodulation: Novel potentials of an old enzyme. Food Chem. Toxicol. 2021, 148, 111945. [Google Scholar] [CrossRef]
- Gomez, J.; Areeb, Z.; Stuart, S.F.; Nguyen, H.P.T.; Paradiso, L.; Zulkifli, A.; Madan, S.; Rajagopal, V.; Montgomery, M.K.; Gan, H.K.; et al. EGFRvIII Promotes Cell Survival during Endoplasmic Reticulum Stress through a Reticulocalbin 1-Dependent Mechanism. Cancers 2021, 13, 1198. [Google Scholar] [CrossRef]
- Deng, S.; Pan, Y.; An, N.; Chen, F.; Chen, H.; Wang, H.; Xu, X.; Liu, R.; Yang, L.; Wang, X.; et al. Downregulation of RCN1 promotes pyroptosis in acute myeloid leukemia cells. Mol. Oncol. 2023, 17, 2584–2602. [Google Scholar] [CrossRef]
- Newman, A.M.; Liu, C.L.; Green, M.R.; Gentles, A.J.; Feng, W.; Xu, Y.; Hoang, C.D.; Diehn, M.; Alizadeh, A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 2015, 12, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Jelcic, I.; Al Nimer, F.; Wang, J.; Lentsch, V.; Planas, R.; Jelcic, I.; Madjovski, A.; Ruhrmann, S.; Faigle, W.; Frauenknecht, K.; et al. Memory B Cells Activate Brain-Homing, Autoreactive CD4(+) T Cells in Multiple Sclerosis. Cell 2018, 175, 85–100.e123. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Qi, C.; Bai, Y. The immunometabolic landscape of bone marrow cells in multiple sclerosis. FASEB J. 2023, 37, e23267. [Google Scholar] [CrossRef]
- Peng, Y.; Zhou, M.; Yang, H.; Qu, R.; Qiu, Y.; Hao, J.; Bi, H.; Guo, D. Regulatory Mechanism of M1/M2 Macrophage Polarization in the Development of Autoimmune Diseases. Mediat. Inflamm. 2023, 2023, 8821610. [Google Scholar] [CrossRef] [PubMed]
- Jensen, I.J.; Jensen, S.N.; Sjaastad, F.V.; Gibson-Corley, K.N.; Dileepan, T.; Griffith, T.S.; Mangalam, A.K.; Badovinac, V.P. Sepsis impedes EAE disease development and diminishes autoantigen-specific naive CD4 T cells. Elife 2020, 9, e55800. [Google Scholar] [CrossRef]
- De Bondt, M.; Hellings, N.; Opdenakker, G.; Struyf, S. Neutrophils: Underestimated Players in the Pathogenesis of Multiple Sclerosis (MS). Int. J. Mol. Sci. 2020, 21, 4558. [Google Scholar] [CrossRef]
- Kinzel, S.; Weber, M.S. B Cell-Directed Therapeutics in Multiple Sclerosis: Rationale and Clinical Evidence. CNS Drugs 2016, 30, 1137–1148. [Google Scholar] [CrossRef]
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The Role of Natural Killer Cells in Autoimmune Diseases. Front. Immunol. 2021, 12, 622306. [Google Scholar] [CrossRef]
- Piacente, F.; Bottero, M.; Benzi, A.; Vigo, T.; Uccelli, A.; Bruzzone, S.; Ferrara, G. Neuroprotective Potential of Dendritic Cells and Sirtuins in Multiple Sclerosis. Int. J. Mol. Sci. 2022, 23, 4352. [Google Scholar] [CrossRef]
- Bo, N.; Yilin, H.; Chaoyue, Y.; Lu, L.; Yuan, Y. Acrylamide induces NLRP3 inflammasome activation via oxidative stress- and endoplasmic reticulum stress-mediated MAPK pathway in HepG2 cells. Food Chem. Toxicol. 2020, 145, 111679. [Google Scholar] [CrossRef]
- Ming, S.; Tian, J.; Ma, K.; Pei, C.; Li, L.; Wang, Z.; Fang, Z.; Liu, M.; Dong, H.; Li, W.; et al. Oxalate-induced apoptosis through ERS-ROS-NF-κB signalling pathway in renal tubular epithelial cell. Mol. Med. 2022, 28, 88. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.R.; Shen, S.Y.; Zhai, M.Y.; Shen, Z.Q.; Li, W.; Liang, L.F.; Yin, S.Y.; Han, Q.Q.; Li, B.; Zhang, Y.Q.; et al. Augmented microglial endoplasmic reticulum-mitochondria contacts mediate depression-like behavior in mice induced by chronic social defeat stress. Nat. Commun. 2024, 15, 5199. [Google Scholar] [CrossRef]
- Thangaraj, A.; Sil, S.; Tripathi, A.; Chivero, E.T.; Periyasamy, P.; Buch, S. Targeting endoplasmic reticulum stress and autophagy as therapeutic approaches for neurological diseases. Int. Rev. Cell Mol. Biol. 2020, 350, 285–325. [Google Scholar]
- Chung, Y.R.; Choi, J.A.; Koh, J.Y.; Yoon, Y.H. Ursodeoxycholic Acid Attenuates Endoplasmic Reticulum Stress-Related Retinal Pericyte Loss in Streptozotocin-Induced Diabetic Mice. J. Diabetes Res. 2017, 2017, 1763292. [Google Scholar] [CrossRef]
- Zhu, X.; Jia, Y.; Zhao, Z.; Zhang, X.; Zhao, Y.; Gui, S.; Yang, X.A. Cell signaling communication within papillary craniopharyngioma revealed by an integrated analysis of single-cell RNA sequencing and bulk RNA sequencing. J. Transl. Med. 2025, 23, 124. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, C.; Tang, G.; Wang, N.; Feng, Y. Single-cell transcriptome profiling highlights the role of APP in blood vessels in assessing the risk of patients with proliferative diabetic retinopathy developing Alzheimer’s disease. Front. Cell Dev. Biol. 2023, 11, 1328979. [Google Scholar] [CrossRef]
- De Schepper, S.; Ge, J.Z.; Crowley, G.; Ferreira, L.S.S.; Garceau, D.; Toomey, C.E.; Sokolova, D.; Rueda-Carrasco, J.; Shin, S.H.; Kim, J.S.; et al. Perivascular cells induce microglial phagocytic states and synaptic engulfment via SPP1 in mouse models of Alzheimer’s disease. Nat. Neurosci. 2023, 26, 406–415. [Google Scholar] [CrossRef]
- Li, Y.Y.; Guo, D.D.; Duan, R.N.; Li, Y. Interactions between Beta-Amyloid and Pericytes in Alzheimer’s Disease. Front. Biosci. (Landmark Ed.) 2024, 29, 136. [Google Scholar] [CrossRef] [PubMed]
- Pietroboni, A.M.; Caprioli, M.; Carandini, T.; Scarioni, M.; Ghezzi, L.; Arighi, A.; Cioffi, S.; Cinnante, C.; Fenoglio, C.; Oldoni, E.; et al. CSF β-amyloid predicts prognosis in patients with multiple sclerosis. Mult. Scler. 2019, 25, 1223–1231. [Google Scholar] [CrossRef]
- Yong, V.W. Microglia in multiple sclerosis: Protectors turn destroyers. Neuron 2022, 110, 3534–3548. [Google Scholar] [CrossRef]
- Jang, B.I.; Jung, J.Y.; Koh, S.A.; Lee, K.H. The Important Role of GPX1 and NF-κB Signaling Pathway in Human Gastric Cancer: Implications for Cell Proliferation and Invasion. Cancer Genom. Proteom. 2024, 21, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Lalle, G.; Lautraite, R.; Bouherrou, K.; Plaschka, M.; Pignata, A.; Voisin, A.; Twardowski, J.; Perrin-Niquet, M.; Stéphan, P.; Durget, S.; et al. NF-κB subunits RelA and c-Rel selectively control CD4+ T cell function in multiple sclerosis and cancer. J. Exp. Med. 2024, 221. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, Q.; Wen, B.; Wu, N.; He, B.; Chen, J. Berberine Reduces Aβ(42) Deposition and Tau Hyperphosphorylation via Ameliorating Endoplasmic Reticulum Stress. Front. Pharmacol. 2021, 12, 640758. [Google Scholar]
- Matsuda, S.; Matsuda, Y.; D’Adamio, L. CD74 interacts with APP and suppresses the production of Abeta. Mol. Neurodegener. 2009, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, M.; Ge, Y.; Chen, J.; Ma, J.; Wang, C.; Sun, M.; Wang, L.; Yao, S.; Yao, C. β-amyloid protein induces mitophagy-dependent ferroptosis through the CD36/PINK/PARKIN pathway leading to blood-brain barrier destruction in Alzheimer’s disease. Cell Biosci. 2022, 12, 69. [Google Scholar] [CrossRef]
- Shi, H.; Koronyo, Y.; Rentsendorj, A.; Regis, G.C.; Sheyn, J.; Fuchs, D.T.; Kramerov, A.A.; Ljubimov, A.V.; Dumitrascu, O.M.; Rodriguez, A.R.; et al. Identification of early pericyte loss and vascular amyloidosis in Alzheimer’s disease retina. Acta Neuropathol. 2020, 139, 813–836. [Google Scholar] [CrossRef]
- Abdi, M.; Pasbakhsh, P.; Shabani, M.; Nekoonam, S.; Sadeghi, A.; Fathi, F.; Abouzaripour, M.; Mohamed, W.; Zibara, K.; Kashani, I.R.; et al. Metformin Therapy Attenuates Pro-inflammatory Microglia by Inhibiting NF-κB in Cuprizone Demyelinating Mouse Model of Multiple Sclerosis. Neurotox. Res. 2021, 39, 1732–1746. [Google Scholar] [CrossRef]
- Xie, Y.; Li, J.; Tao, Q.; Wu, Y.; Liu, Z.; Zeng, C.; Chen, Y. Identification of glutamine metabolism-related gene signature to predict colorectal cancer prognosis. J. Cancer 2024, 15, 3199–3214. [Google Scholar] [CrossRef]
- Rogers, T.J. Kappa Opioid Receptor Expression and Function in Cells of the Immune System. Handb. Exp. Pharmacol. 2022, 271, 419–433. [Google Scholar]
- Shariq, M.; Khan, M.F.; Raj, R.; Ahsan, N.; Kumar, P. PRKAA2, MTOR, and TFEB in the regulation of lysosomal damage response and autophagy. J. Mol. Med. 2024, 102, 287–311. [Google Scholar] [CrossRef]
- Giacometti, J.; Grubić-Kezele, T. Olive Leaf Polyphenols Attenuate the Clinical Course of Experimental Autoimmune Encephalomyelitis and Provide Neuroprotection by Reducing Oxidative Stress, Regulating Microglia and SIRT1, and Preserving Myelin Integrity. Oxid. Med. Cell Longev. 2020, 2020, 6125638. [Google Scholar] [CrossRef] [PubMed]
- Enz, L.S.; Zeis, T.; Schmid, D.; Geier, F.; van der Meer, F.; Steiner, G.; Certa, U.; Binder, T.M.C.; Stadelmann, C.; Martin, R.; et al. Increased HLA-DR expression and cortical demyelination in MS links with HLA-DR15. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e656. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.W.; Durrenberger, P.; Aricò, E.; James, R.; Cruciani, C.; Reeves, C.; Roncaroli, F.; Nicholas, R.; Reynolds, R. Meningeal inflammation changes the balance of TNF signalling in cortical grey matter in multiple sclerosis. J. Neuroinflamm. 2019, 16, 259. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, D.A.E.; van Scheppingen, J.; van der Poel, M.; Bossers, K.; Schuurman, K.G.; van Eden, C.G.; Hol, E.M.; Hamann, J.; Huitinga, I. Gene Expression Profiling of Multiple Sclerosis Pathology Identifies Early Patterns of Demyelination Surrounding Chronic Active Lesions. Front. Immunol. 2017, 8, 1810. [Google Scholar] [CrossRef] [PubMed]
- Jäkel, S.; Agirre, E.; Falcão, A.M.; van Bruggen, D.; Lee, K.W.; Knuesel, I.; Malhotra, D.; Ffrench-Constant, C.; Williams, A.; Castelo-Branco, G. Altered human oligodendrocyte heterogeneity in multiple sclerosis. Nature 2019, 566, 543–547. [Google Scholar] [CrossRef]
- Drake, S.S.; Zaman, A.; Gianfelice, C.; Hua, E.M.; Heale, K.; Afanasiev, E.; Klement, W.; Stratton, J.A.; Prat, A.; Zandee, S.; et al. Senolytic treatment diminishes microglia and decreases severity of experimental autoimmune encephalomyelitis. J. Neuroinflamm. 2024, 21, 283. [Google Scholar] [CrossRef]
- Zhang, Q.; Guan, G.; Cheng, P.; Cheng, W.; Yang, L.; Wu, A. Characterization of an endoplasmic reticulum stress-related signature to evaluate immune features and predict prognosis in glioma. J. Cell Mol. Med. 2021, 25, 3870–3884. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Xie, R.; Liu, L.; Lu, X.; He, C.; Li, G. Identification of the diagnostic genes and immune cell infiltration characteristics of gastric cancer using bioinformatics analysis and machine learning. Front. Genet. 2022, 13, 1067524. [Google Scholar] [CrossRef]
- Hao, Y.; Stuart, T.; Kowalski, M.H.; Choudhary, S.; Hoffman, P.; Hartman, A.; Srivastava, A.; Molla, G.; Madad, S.; Fernandez-Granda, C.; et al. Dictionary learning for integrative, multimodal and scalable single-cell analysis. Nat. Biotechnol. 2024, 42, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An updated database of manually curated cell markers in human/mouse and web tools based on scRNA-seq data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Carmona, E. Generating publication ready visualizations for Single Cell transcriptomics using SCpubr. bioRxiv 2022. [Google Scholar] [CrossRef]
- Jin, S.; Plikus, M.V.; Nie, Q. CellChat for systematic analysis of cell-cell communication from single-cell transcriptomics. Nat. Protoc. 2025, 20, 180–219. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiao, Z.; Wang, Y.; Ma, X.; Zhang, X.; Wu, J.; Li, A.; Wang, C.; Xiu, X.; Zhang, S.; Lang, X.; et al. GPX1 and RCN1 as New Endoplasmic Reticulum Stress-Related Biomarkers in Multiple Sclerosis Brain Tissue and Their Involvement in the APP-CD74 Pathway: An Integrated Study Combining Machine Learning and Multi-Omics. Int. J. Mol. Sci. 2025, 26, 6286. https://doi.org/10.3390/ijms26136286
Qiao Z, Wang Y, Ma X, Zhang X, Wu J, Li A, Wang C, Xiu X, Zhang S, Lang X, et al. GPX1 and RCN1 as New Endoplasmic Reticulum Stress-Related Biomarkers in Multiple Sclerosis Brain Tissue and Their Involvement in the APP-CD74 Pathway: An Integrated Study Combining Machine Learning and Multi-Omics. International Journal of Molecular Sciences. 2025; 26(13):6286. https://doi.org/10.3390/ijms26136286
Chicago/Turabian StyleQiao, Zhixin, Yanping Wang, Xiaoru Ma, Xiyu Zhang, Junfeng Wu, Anqi Li, Chao Wang, Xin Xiu, Sifan Zhang, Xiujuan Lang, and et al. 2025. "GPX1 and RCN1 as New Endoplasmic Reticulum Stress-Related Biomarkers in Multiple Sclerosis Brain Tissue and Their Involvement in the APP-CD74 Pathway: An Integrated Study Combining Machine Learning and Multi-Omics" International Journal of Molecular Sciences 26, no. 13: 6286. https://doi.org/10.3390/ijms26136286
APA StyleQiao, Z., Wang, Y., Ma, X., Zhang, X., Wu, J., Li, A., Wang, C., Xiu, X., Zhang, S., Lang, X., Liu, X., Sun, B., Li, H., & Liu, Y. (2025). GPX1 and RCN1 as New Endoplasmic Reticulum Stress-Related Biomarkers in Multiple Sclerosis Brain Tissue and Their Involvement in the APP-CD74 Pathway: An Integrated Study Combining Machine Learning and Multi-Omics. International Journal of Molecular Sciences, 26(13), 6286. https://doi.org/10.3390/ijms26136286