
Genetic Dissection of Drought Tolerance in Maize Through GWAS of Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity
 , , and
, , and 
Abstract
1. Introduction
2. Results
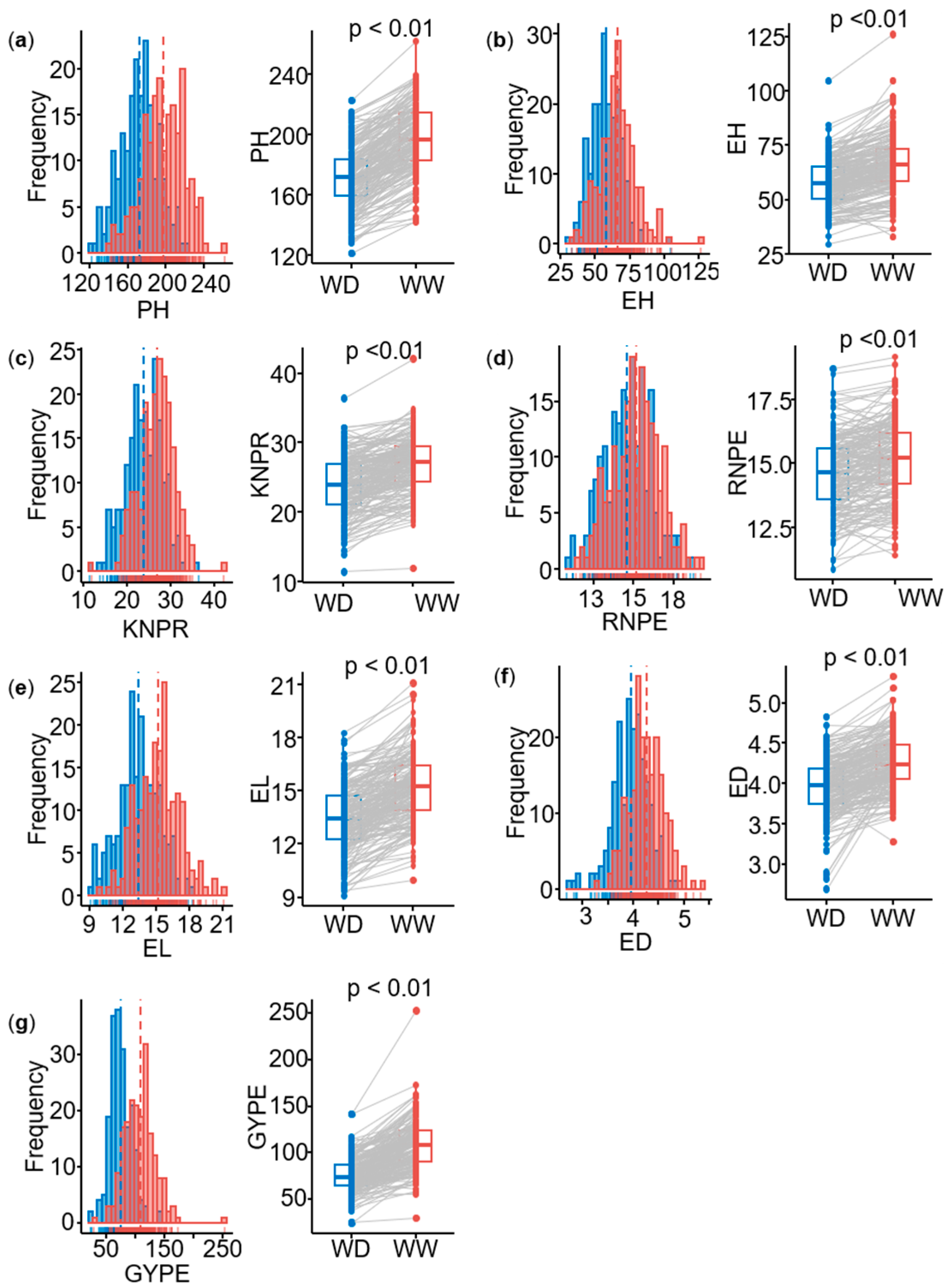
2.1. Phenotypic Variation and Heritability of Agronomic Traits
2.2. Correlation Analysis Among Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity
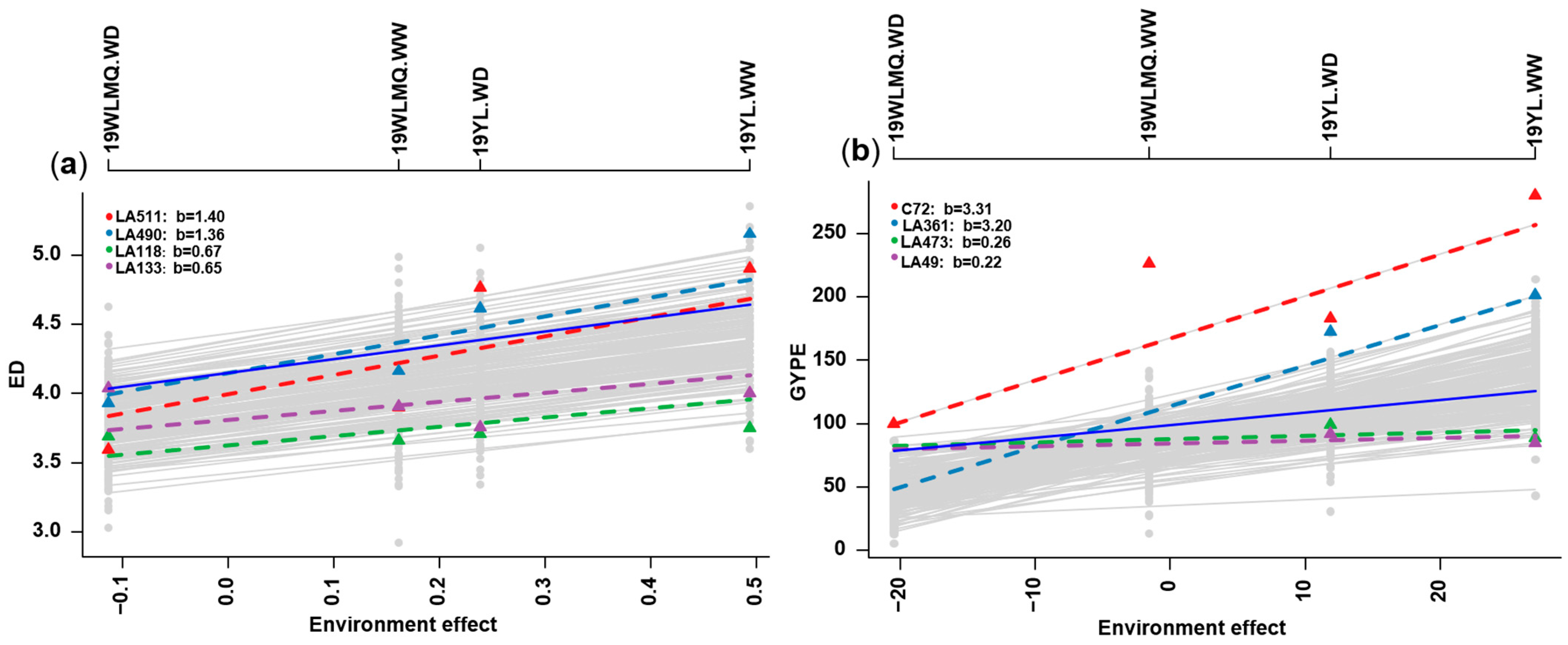
2.3. Phenotypic Plasticity Estimation Through Finlay–Wilkinson Regression
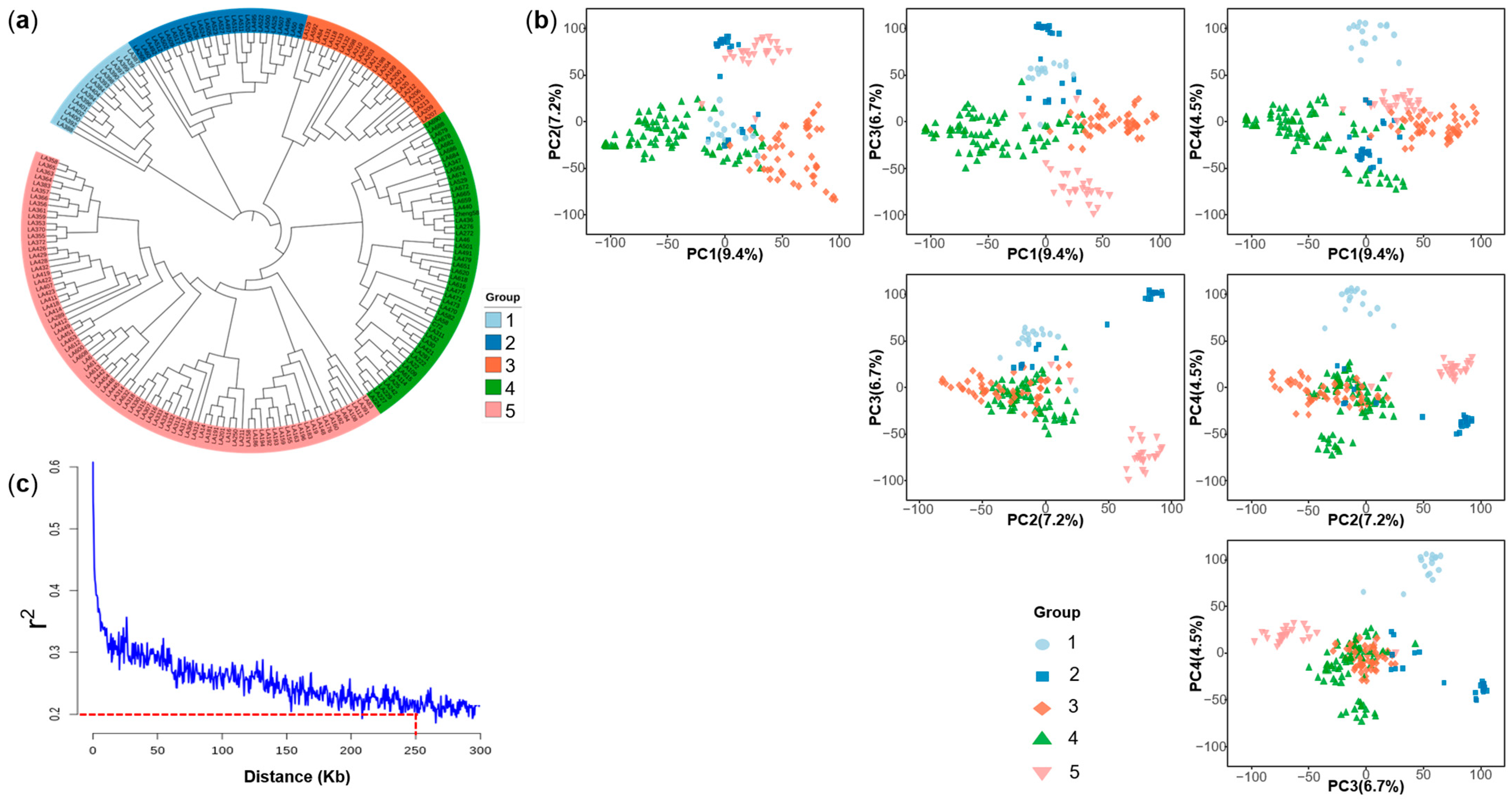
2.4. Population Structure and Linkage Disequilibrium of the Multi-Parental DH Population
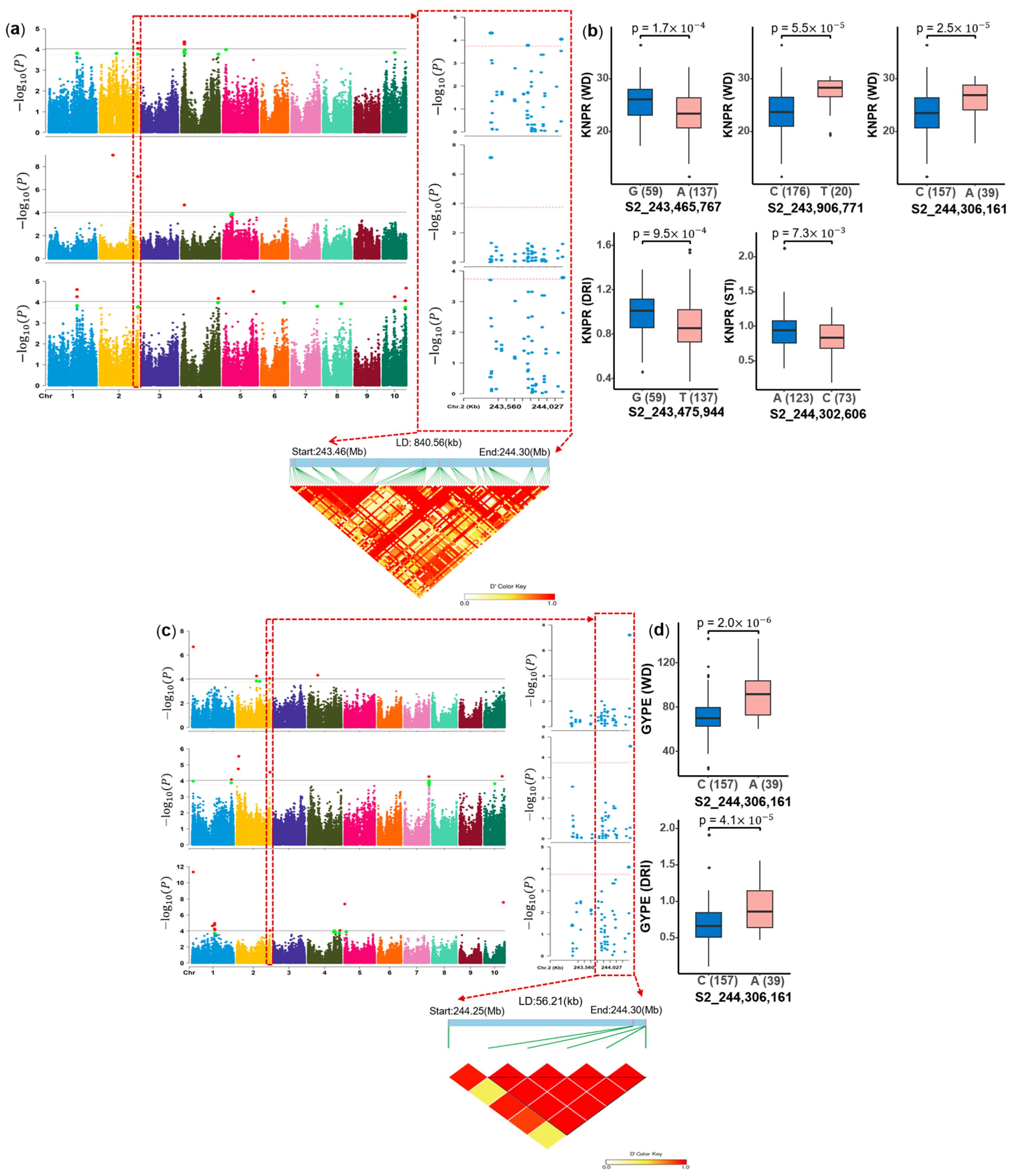
2.5. Genome-Wide Association Study (GWAS) for Three Trait Groups
2.5.1. GWAS for Seven Agronomic Traits Under Water-Deficit (WD) and Well-Watered (WW) Conditions
2.5.2. GWAS for Stress Tolerance Indices
2.5.3. GWAS for Phenotypic Plasticity
2.5.4. QTL Identified for Agronomic Traits, Stress Tolerance Indices and Phenotypic Plasticity
2.6. Candidate Genes Functional Annotation
2.7. Mining of Candidate Genes and Identification of Polymorphisms
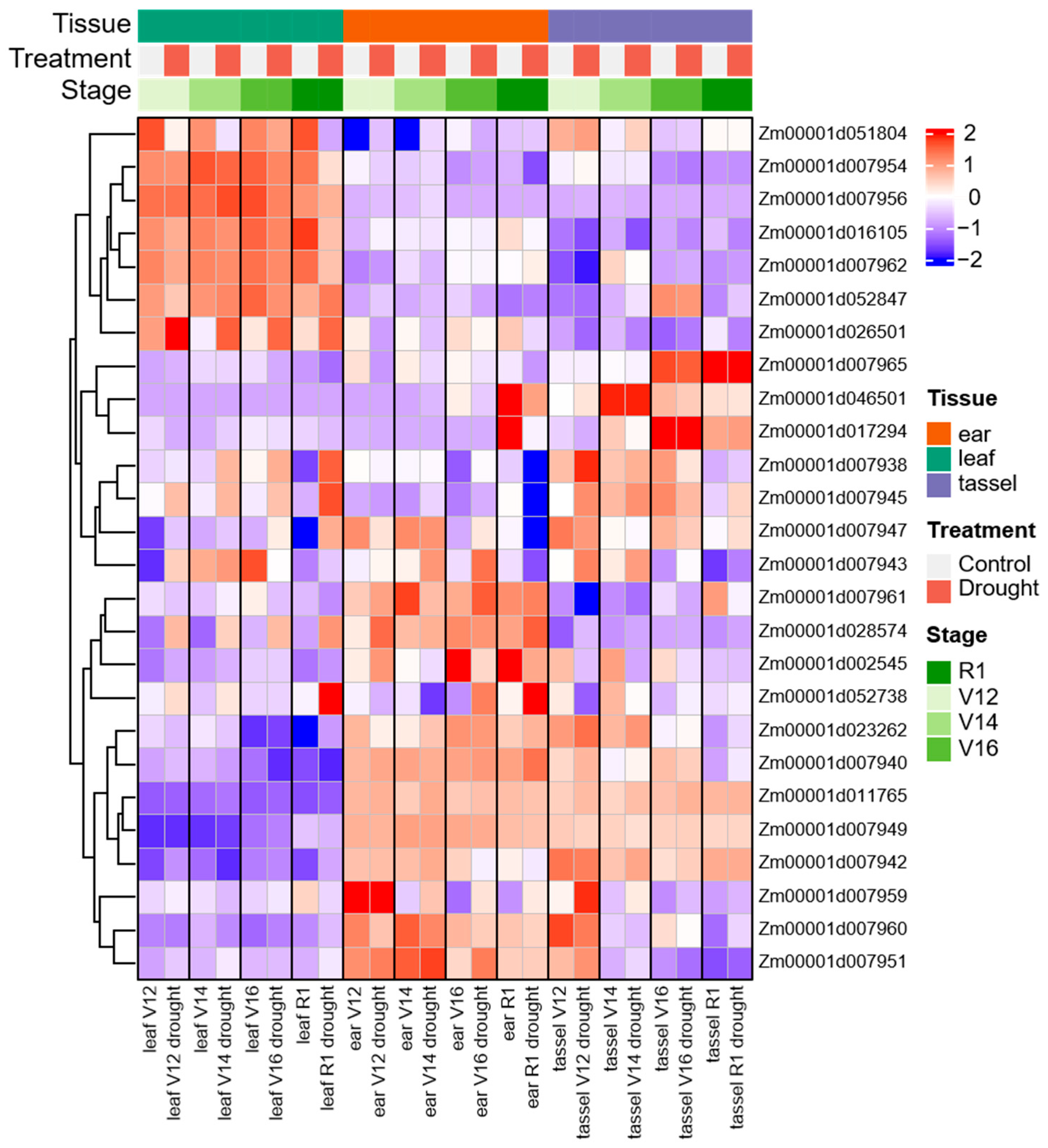
2.8. Analysis of Transcript Expression in Different Tissues and Under Drought Stress
3. Discussion
3.1. Combining Agronomic Traits, Stress Tolerance Indices and Phenotypic Plasticity Provides a New Vision for Drought Tolerance-Related Phenotypic Diversity
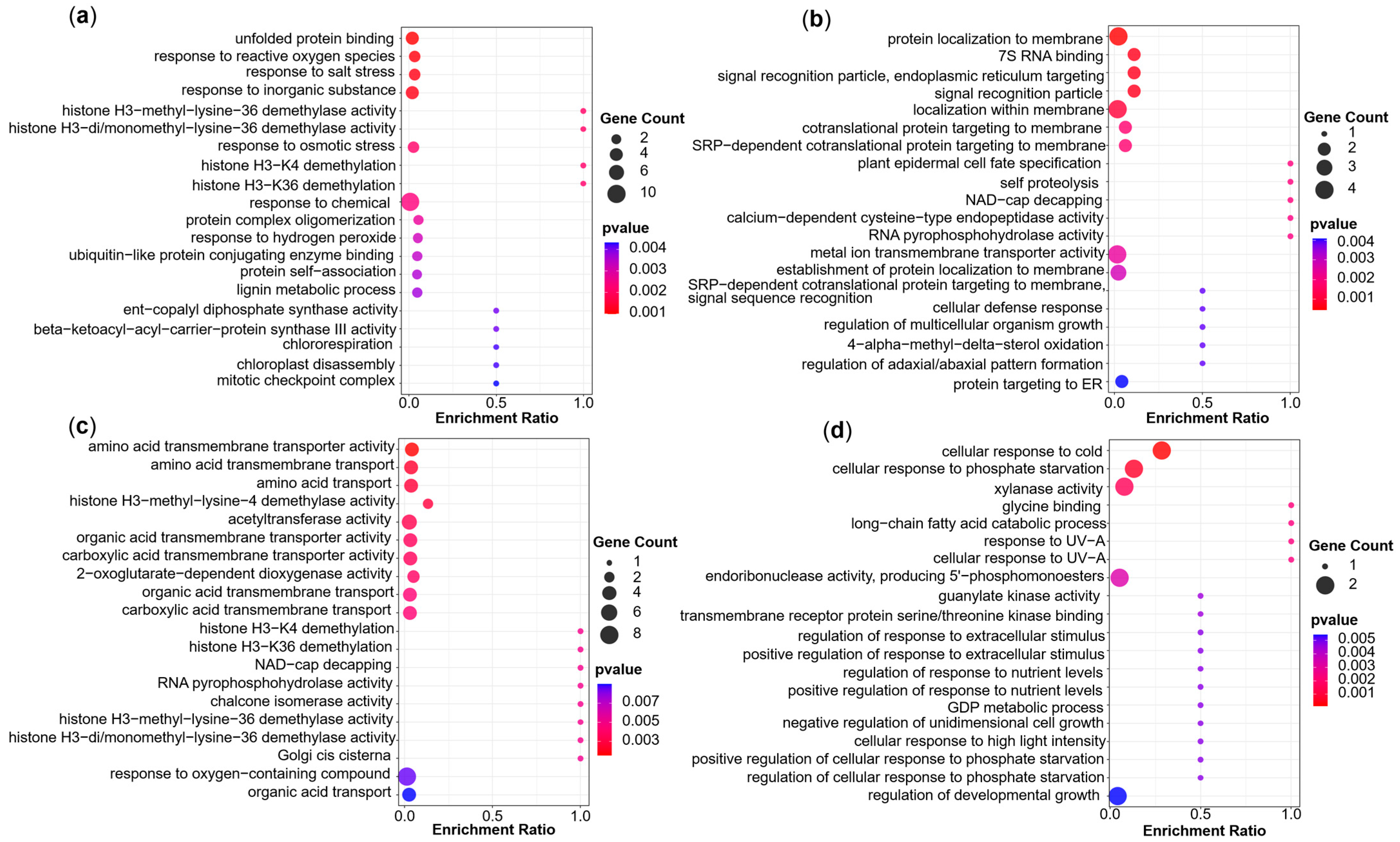
3.2. GO Enrichment Analysis of Candidate Genes Showed Significant Differences Between Agronomic Traits, Stress Tolerance Indices and Phenotypic Plasticity
3.3. Drought Tolerance Candidate Genes Were Identified by GWAS at the Harvest Stage
4. Materials and Methods
4.1. Plant Materials and Field Experiments
4.2. Phenotypic Evaluation
4.3. Statistical Analysis
4.4. Finlay–Wilkinson Regression Analysis
4.5. Genotyping
4.6. Population Analysis and Linkage Disequilibrium (LD) Analysis
4.7. Genome-Wide Association Study
4.8. Candidate Gene Identification and Functional Analysis
4.9. Expression Analysis of Potential Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, N.; Feng, Z.; Gao, H.; He, Q.; Zeng, W. Elucidating the influence and mechanism of different phenols on the properties, food quality and function of maize starch. Food Chem. 2024, 449, 139191. [Google Scholar] [CrossRef] [PubMed]
- Hartinger, T.; Gruber, T.; Fliegerova, K.; Terler, G.; Zebeli, Q. Mixed ensiling with by-products and silage additives significantly valorizes drought-impaired whole-crop corn. Anim. Feed Sci. Technol. 2024, 309, 115899. [Google Scholar] [CrossRef]
- Choudhary, M.; Singh, A.; Gupta, M.; Rakshit, S. Enabling technologies for utilization of maize as a bioenergy feedstock. Biofuels Bioprod. Biorefining-Biofpr 2020, 14, 402–416. [Google Scholar] [CrossRef]
- Tenorio Berrío, R.; Nelissen, H.; Inzé, D.; Dubois, M. Increasing yield on dry fields: Molecular pathways with growing potential. Plant J. 2022, 109, 323–341. [Google Scholar] [CrossRef]
- Saez-Cigarruista, A.; Morales-Guevara, D.; Gordon-Mendoza, R.; Jaen-Villarreal, J.; Franco-Barrera, J.; Ramos-Manzane, F. Sensitivity of the corn (Zea mays L.) crop to different periods of controlled water deficit. Agron. Mesoam. 2024, 35, 55660. [Google Scholar] [CrossRef]
- Ibrahim, A.I.Z. Drought Tolerance Traits in Maize (Zea mays) at the Early Vegetative Growth Stage: Phenotyping and Gene Discovery via Genome-Wide Association Mapping. Ph.D. thesis, The University of Wisconsin – Madison, Madison, WI, USA, 2017. [Google Scholar]
- Qin, L.X.; Trouverie, J.; Chateau-Joubert, S.; Simond-Côte, E.; Thévenot, C.; Prioul, J.L. Involvement of the Ivr2-invertase in the perianth during maize kernel development under water stress. Plant Sci. 2004, 166, 371–379. [Google Scholar] [CrossRef]
- Dwyer, L.M.; Stewart, D.W.; Tollenaar, M. Analysis of maize leaf photosynthesis under drought stress. Can. J. Plant Sci. 1992, 72, 477–481. [Google Scholar] [CrossRef]
- Zhang, L.; Tang, H.; Xie, X.; Sun, B.; Liu, C. A Quantitative Index for Evaluating Maize Leaf Wilting and Its Sustainable Application in Drought Resistance Screening. Sustainability 2024, 16, 6129. [Google Scholar] [CrossRef]
- Ma, X.; Feng, F.; Wei, H.; Mei, H.; Xu, K.; Chen, S.; Li, T.; Liang, X.; Liu, H.; Luo, L. Genome-Wide Association Study for Plant Height and Grain Yield in Rice under Contrasting Moisture Regimes. Front. Plant Sci. 2016, 7, 1801. [Google Scholar] [CrossRef]
- Tang, H.; Zhang, R.; Wang, M.; Xie, X.; Zhang, L.; Zhang, X.; Liu, C.; Sun, B.; Qin, F.; Yang, X. QTL mapping for flowering time in a maize-teosinte population under well-watered and water-stressed conditions. Mol. Breed. 2023, 43, 67. [Google Scholar] [CrossRef]
- Li, H.; Jiang, Y.; Jia, S.; Zhao, G.; Wang, Y.; Yang, Q.; Liu, T.; Li, C.; Shao, R. Research Progress on Drought Stress Affecting Ear and Tassel Development of Maize. J. Maize Sci. 2020, 28, 90–95. [Google Scholar] [CrossRef]
- Mehmood, M.; Tanveer, N.A.; Joyia, F.A.; Ullah, I.; Mohamed, H.I. Effect of high temperature on pollen grains and yield in economically important crops: A review. Planta 2025, 261, 141. [Google Scholar] [CrossRef]
- Liu, S.; Qin, F. Genetic dissection of maize drought tolerance for trait improvement. Mol. Breed. 2021, 41, 8. [Google Scholar] [CrossRef]
- Kumar, A.; Adhikari, S.; Joshi, A.; Singh, N.K.; Bharati, A.; Rohit, R.; Prasad, B.; Dinkar, V.; Singh, A. Study of co-localization of QTL for drought tolerance of morphological traits in teosinte introgressed maize population. Cereal Res. Commun. 2024, 53, 821–832. [Google Scholar] [CrossRef]
- Ziyomo, C.; Bernardo, R. Drought Tolerance in Maize: Indirect Selection through Secondary Traits versus Genomewide Selection. Crop Sci. 2013, 53, 1269–1275. [Google Scholar] [CrossRef]
- Amadu, M.K.; Beyene, Y.; Chaikam, V.; Tongoona, P.B.; Danquah, E.Y.; Ifie, B.E.; Burgueno, J.; Prasanna, B.M.; Gowda, M. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and agronomic traits under drought and optimum conditions in maize. BMC Plant Biol. 2025, 25, 135. [Google Scholar] [CrossRef]
- Hu, X.; Wang, G.; Du, X.; Zhang, H.; Xu, Z.; Wang, J.; Chen, G.; Wang, B.; Li, X.; Chen, X.; et al. QTL analysis across multiple environments reveals promising chromosome regions associated with yield-related traits in maize under drought conditions. Crop J. 2021, 9, 759–766. [Google Scholar] [CrossRef]
- Chen, Z.; Cao, Z.; Tang, B.; Li, L. Mapping QTL for Several Drought Related Traits in Maize (Zea mays L.) under Field Condition. Acta Agric. Boreali-Sin. 2012, 27, 79–86. [Google Scholar] [CrossRef]
- Guo, J.; Su, G.; Zhang, J.; Wang, G. Genetic analysis and QTL mapping of maize yield and associate agronomic traits under semi-arid land condition. Afr. J. Biotechnol. 2008, 7, 1829–1838. [Google Scholar] [CrossRef]
- Pour-Aboughadareh, A.; Yousefian, M.; Moradkhani, H.; Vahed, M.M.; Poczai, P.; Siddique, K.H.M. iPASTIC: An online toolkit to estimate plant abiotic stress indices. Appl. Plant Sci. 2019, 7, e11278. [Google Scholar] [CrossRef]
- Kumar, A.; Bharti, B.; Kumar, J.; Santosh; Singh, G.P.; Jaiswal, J.P.; Prasad, R. Evaluation of drought tolerance indices for identification of drought tolerant and susceptible genotypes in wheat (Triticum aestivum L.). Electron. J. Plant Breed. 2020, 11, 727–734. [Google Scholar] [CrossRef]
- Papathanasiou, F.; Dordas, C.; Gekas, F.; Pankou, C.; Ninou, E.; Mylonas, I.; Tsantarmas, K.; Sistanis, I.; Sinapidou, E.; Lithourgidis, A.; et al. The use of stress tolerance indices for the selection of tolerant inbred lines and their correspondent hybrids under normal and water-stress conditions. In Proceedings of the 4th International Conference on Agriculture and Horticulture (AGRI) 2015, Amsterdam, The Netherlands, 15–17 February 2015; pp. 274–275. [Google Scholar]
- Nouraei, S.; Mia, M.S.; Liu, H.; Turner, N.C.; Yan, G. Genome-wide association study of drought tolerance in wheat (Triticum aestivum L.) identifies SNP markers and candidate genes. Mol. Genet. Genom. 2024, 299, 22. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, R.; Paudel, H.; Alharbi, S.A.; Ansari, M.J.; Poudel, M.R.; Neupane, M.P.; Solanki, P.; Kushwaha, U.K.S. Evaluating stress tolerance indices for their comparative validity to access terminal heat stress and heat drought tolerance of winter wheat (Triticum aestivum L.) genotypes. J. Agric. Food Res. 2024, 18, 101506. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, B.; Ye, F.; Chang, J.; Zhou, Y.; Wang, Y.; Zhang, W.; Zhang, X.; Xu, S.; Xue, J. Molecular mechanisms of drought resistance using genome-wide association mapping in maize (Zea mays L.). BMC Plant Biol. 2023, 23, 468. [Google Scholar] [CrossRef]
- Djabali, Y.; Rincent, R.; Martin, M.-L.; Blein-Nicolas, M. Plasticity QTLs specifically contribute to the genotype x water availability interaction in maize. Theor. Appl. Genet. 2023, 136, 228. [Google Scholar] [CrossRef]
- Zhao, H.; Savin, K.W.; Li, Y.; Breen, E.J.; Maharjan, P.; Tibbits, J.F.; Kant, S.; Hayden, M.J.; Daetwyler, H.D. Genome-wide association studies dissect the G x E interaction for agronomic traits in a worldwide collection of safflowers (Carthamus tinctorius L.). Mol. Breed. 2022, 42, 24. [Google Scholar] [CrossRef]
- Bradshaw, A.D. Evolutionary Significance of Phenotypic Plasticity in Plants. Adv. Genet. 1965, 13, 115–155. [Google Scholar] [CrossRef]
- Finlay, K.; Wilkinson, G. The analysis of adaptation in a plant-breeding programme. Aust. J. Agric. Res. 1963, 14, 742–764. [Google Scholar] [CrossRef]
- Kusmec, A.; Srinivasan, S.; Nettleton, D.; Schnable, P.S. Distinct genetic architectures for phenotype means and plasticities in Zea mays. Nat. Plants 2017, 3, 715–723. [Google Scholar] [CrossRef]
- Li, D.; Li, G.; Wang, H.; Guo, Y.; Wang, M.; Lu, X.; Luo, Z.; Zhu, X.; Weiss, T.M.; Roller, S.; et al. Genetic Dissection of Phosphorus Use Efficiency and Genotype-by-Environment Interaction in Maize. Int. J. Mol. Sci. 2022, 23, 13943. [Google Scholar] [CrossRef]
- Wei, J.; Guo, T.; Mu, Q.; Alladassi, B.M.E.; Mural, R.V.; Boyles, R.E.; Hoffmann, L.; Hayes, C.M.; Sigmon, B.; Thompson, A.M.; et al. Genetic and Environmental Patterns Underlying Phenotypic Plasticity in Flowering Time and Plant Height in Sorghum. Plant Cell Environ. 2024, 1, 15213. [Google Scholar] [CrossRef] [PubMed]
- Shenton, M. Plant GWAS. In Plant Omics: Advances in Big Data Biology; CABI: Tsukuba, Japan, 2022; pp. 181–189. [Google Scholar] [CrossRef]
- Dong, A.; Wang, N.; Zenda, T.; Zhai, X.; Zhong, Y.; Yang, Q.; Xing, Y.; Duan, H.; Yan, X. ZmDnaJ-ZmNCED6 module positively regulates drought tolerance via modulating stomatal closure in maize. Plant Physiol. Biochem. 2025, 218, 0981–9428. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Cheng, M.; Chen, Y.; Liu, B.; Wang, X.; Li, G.; Zhou, Y.; Luo, P.; Xi, Z.; Yong, H.; et al. Natural variations in the non-coding region of ZmNAC080308 contributes maintaining grain yield under drought stress in maize. BMC Plant Biol. 2021, 21, 305. [Google Scholar] [CrossRef]
- Wu, X.; Feng, H.; Wu, D.; Yan, S.; Zhang, P.; Wang, W.; Zhang, J.; Ye, J.; Dai, G.; Fan, Y.; et al. Using high-throughput multiple optical phenotyping to decipher the genetic architecture of maize drought tolerance. Genome Biol. 2021, 22, 185. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, B.; Yang, Z.; Liu, Y.; Yang, S.; Shi, Y.; Jiang, C.; Qin, F. Manipulating ZmEXPA4 expression ameliorates the drought-induced prolonged anthesis and silking interval in maize. Plant Cell 2021, 33, 2058–2071. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Liu, S.; Ferjani, A.; Li, J.; Yan, J.; Yang, X.; Qin, F. Genetic variation in ZmVPP1 contributes to drought tolerance in maize seedlings. Nat. Genet. 2016, 48, 1233–1241. [Google Scholar] [CrossRef]
- Li, G.; Zhou, Y.-H.; Li, H.-F.; Zhang, Y.-M. A multi-locus linear mixed model methodology for detecting small-effect QTLs for quantitative traits in MAGIC, NAM, and ROAM populations. Comput. Struct. Biotechnol. J. 2023, 21, 2241–2252. [Google Scholar] [CrossRef] [PubMed]
- Edward, S.B.; James, B.H.; Peter, J.B.; Charlotte, B.A.; Patrick, J.B.; Chris, B.; Elhan, E.; Sherry, F.-G.; Arturo, G.; Jeffrey, C.G.; et al. The Genetic Architecture of Maize Flowering Time. Science 2009, 325, 714–718. [Google Scholar] [CrossRef]
- Cook, J.P.; McMullen, M.D.; Holland, J.B.; Tian, F.; Bradbury, P.; Ross-Ibarra, J.; Buckler, E.S.; Flint-Garcia, S.A. Genetic Architecture of Maize Kernel Composition in the Nested Association Mapping and Inbred Association Panels. Plant Physiol. 2012, 158, 824–834. [Google Scholar] [CrossRef]
- Tibbs-Cortes, L.E.; Guo, T.; Andorf, C.M.; Li, X.; Yu, J. Comprehensive identification of genomic and environmental determinants of phenotypic plasticity in maize. Genome Res. 2024, 34, 1253–1263. [Google Scholar] [CrossRef]
- Kover, P.X.; Valdar, W.; Trakalo, J.; Scarcelli, N.; Ehrenreich, I.M.; Purugganan, M.D.; Durrant, C.; Mott, R. A Multiparent Advanced Generation Inter-Cross to Fine-Map Quantitative Traits in Arabidopsis thaliana. PLoS Genet. 2009, 5, e1000551. [Google Scholar] [CrossRef]
- Rida, S.; Maafi, O.; Lopez-Malvar, A.; Revilla, P.; Riache, M.; Djemel, A. Genetics of Germination and Seedling Traits under Drought Stress in a MAGIC Population of Maize. Plants 2021, 10, 1786. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Tong, H.; Yang, X.; Xu, S.; Pan, Q.; Qiao, F.; Raihan, M.S.; Luo, Y.; Liu, H.; Zhang, X.; et al. Genome-wide dissection of the maize ear genetic architecture using multiple populations. New Phytol. 2016, 210, 1095–1106. [Google Scholar] [CrossRef]
- Chen, G.; Xiao, Y.; Dai, S.; Dai, Z.; Wang, X.; Li, B.; Jaqueth, J.S.; Li, W.; Lai, Z.; Ding, J.; et al. Genetic basis of resistance to southern corn leaf blight in the maize multi-parent population and diversity panel. Plant Biotechnol. J. 2023, 21, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Swamy, N.; Kuchanur, P.H.; Patil, A.; Zaidi, P.H.; Vinayan, M.T.; Arunkumar, B.; Sowmya, H.C.; Dhanoji, M.M. Genetic gains for heat stress tolerance in maize through integration of rapid cycle genomic selection and doubled haploid technology. Plant Arch. 2024, 24, 2700–2714. [Google Scholar] [CrossRef]
- Fan, Z.; Lin, S.; Jiang, J.; Zeng, Y.; Meng, Y.; Ren, J.; Wu, P. Dual-Model GWAS Analysis and Genomic Selection of Maize Flowering Time-Related Traits. Genes 2024, 15, 740. [Google Scholar] [CrossRef]
- Huang, L.; Chen, L.; Wang, L.; Yang, Y.; Rao, Y.; Ren, D.; Dai, L.; Gao, Y.; Zou, W.; Lu, X.; et al. A Nck-associated protein 1-like protein affects drought sensitivity by its involvement in leaf epidermal development and stomatal closure in rice. Plant J. 2019, 98, 884–897. [Google Scholar] [CrossRef]
- Miao, J.; Li, X.; Li, X.; Tan, W.; You, A.; Wu, S.; Tao, Y.; Chen, C.; Wang, J.; Zhang, D.; et al. OsPP2C09, a negative regulatory factor in abscisic acid signalling, plays an essential role in balancing plant growth and drought tolerance in rice. New Phytol. 2020, 227, 1417–1433. [Google Scholar] [CrossRef]
- Wang, Y.; Wan, L.; Zhang, L.; Zhang, Z.; Zhang, H.; Quan, R.; Zhou, S.; Huang, R. An ethylene response factor OsWR1 responsive to drought stress transcriptionally activates wax synthesis related genes and increases wax production in rice. Plant Mol. Biol. 2012, 78, 275–288. [Google Scholar] [CrossRef]
- Shen, H.; Liu, C.; Zhang, Y.; Meng, X.; Zhou, X.; Chu, C.; Wang, X. OsWRKY30 is activated by MAP kinases to confer drought tolerance in rice. Plant Mol. Biol. 2012, 80, 241–253. [Google Scholar] [CrossRef]
- Verma, R.K.; Kumar, V.V.S.; Yadav, S.K.; Pushkar, S.; Rao, M.V.; Chinnusamy, V. Overexpression of ABA Receptor PYL10 Gene Confers Drought and Cold Tolerance to Indica Rice. Front. Plant Sci. 2019, 10, 01488. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Cui, Y.; Wang, M.; Xia, X. Overexpression of a novel MYB-related transcription factor, OsMYBR1, confers improved drought tolerance and decreased ABA sensitivity in rice. Biochem. Biophys. Res. Commun. 2017, 490, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- James, D.; Borphukan, B.; Fartyal, D.; Ram, B.; Singho, J.; Manna, M.; Sheri, V.; Panditi, V.; Yadav, R.; Achary, V.M.M.; et al. Concurrent Overexpression o f OsGS1;1 and OsGS2 Genes in Transgenic Rice (Oryza sativa L.): Impact on Tolerance to Abiotic Stresses. Front. Plant Sci. 2018, 9, 786. [Google Scholar] [CrossRef]
- Xiang, J.; Ran, J.; Zou, J.; Zhou, X.; Liu, A.; Zhang, X.; Peng, Y.; Tang, N.; Luo, G.; Chen, X. Heat shock factor OsHsfB2b negatively regulates drought and salt tolerance in rice. Plant Cell Rep. 2013, 32, 1795–1806. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, L.; Chen, Y.; Xiao, N.; Zhang, D.; Zhang, M.; Wang, W.; Zhang, C.; Zhang, A.; Li, H.; et al. Phytochrome interacting factor regulates stomatal aperture by coordinating red light and abscisic acid. Plant Cell 2022, 34, 4293–4312. [Google Scholar] [CrossRef]
- Singh, K.K.; Ghosh, S. Regulation of glutamine synthetase isoforms in two differentially drought-tolerant rice (Oryza sativa L.) cultivars under water deficit conditions. Plant Cell Rep. 2013, 32, 183–193. [Google Scholar] [CrossRef]
- Lo, S.-F.; Ho, T.-H.D.; Liu, Y.-L.; Jiang, M.-J.; Hsieh, K.-T.; Chen, K.-T.; Yu, L.-C.; Lee, M.-H.; Chen, C.-y.; Huang, T.-P.; et al. Ectopic expression of specific GA2 oxidase mutants promotes yield and stress tolerance in rice. Plant Biotechnol. J. 2017, 15, 850–864. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.A.; Du, H.; Ning, J.; Ye, H.; Xiong, L. Characterization of Glossy1-homologous genes in rice involved in leaf wax accumulation and drought resistance. Plant Mol. Biol. 2009, 70, 443–456. [Google Scholar] [CrossRef]
- Nepolean, T.; Firoz, H.; Kanika, A.; Rinku, S.; Kaliyugam, S.; Swati, M.; Sweta, M.; Namratha, P.M.; Sreelatha, D.; Rani, T.S.; et al. Functional mechanisms of drought tolerance in subtropical maize (Zea mays L.) identified using genome-wide association mapping. BMC Genom. 2014, 15, 1182. [Google Scholar] [CrossRef]
- Khan, S.U.; Zheng, Y.; Chachar, Z.; Zhang, X.; Zhou, G.; Zong, N.; Leng, P.; Zhao, J. Dissection of Maize Drought Tolerance at the Flowering Stage Using Genome-Wide Association Studies. Genes 2022, 13, 564. [Google Scholar] [CrossRef]
- Jiang, L.; Fan, M.; Guo, J. QTL Analysis of Plant Height and Yield of Maize under Drought and Irrigation Condition. Acta Agric. Boreali-Sin. 2007, 22, 86–90. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, X.; Sun, C.; Zhu, X.; Li, M.; Zhang, G.; Tian, Y.; Wang, Z. Mapping of QTL Associated with Drought Tolerance in a Semi-Automobile Rain Shelter in Maize (Zea mays L.). Agric. Sci. China 2011, 10, 987–996. [Google Scholar] [CrossRef]
- Kong, J.; Jiang, F.; Shaw, R.K.; Bi, Y.; Yin, X.; Pan, Y.; Gong, X.; Zong, H.; Ijaz, B.; Fan, X. Combined Genome-Wide Association Study and Linkage Analysis for Mining Candidate Genes for the Kernel Row Number in Maize (Zea mays L.). Plants 2024, 13, 3308. [Google Scholar] [CrossRef]
- Scharwies, J.D.; Clarke, T.; Zheng, Z.; Dinneny, A.; Birkeland, S.; Veltman, M.A.; Sturrock, C.J.; Banda, J.; Torres-Martinez, H.H.; Viana, W.G.; et al. Moisture-responsive root-branching pathways identified in diverse maize breeding germplasm. Science 2025, 387, 666–673. [Google Scholar] [CrossRef]
- Nikolic, A.; Andjelkovic, V.; Dodig, D.; Mladenovic Drinic, S.; Kravic, N.; Ignjatovic-Micic, D. Identification of QTLs for drought tolerance in maize, II: Yield and yield components. Genetika 2013, 45, 341–350. [Google Scholar] [CrossRef]
- Li, R.; Wang, Y.; Li, D.; Guo, Y.; Zhou, Z.; Zhang, M.; Zhang, Y.; Wuerschum, T.; Liu, W. Meta-Quantitative Trait Loci Analysis and Candidate Gene Mining for Drought Tolerance-Associated Traits in Maize (Zea mays L.). Int. J. Mol. Sci. 2024, 25, 4295. [Google Scholar] [CrossRef] [PubMed]
- Machado, I.P.; de Pontes, F.C.F.; Silveira, M.V.d.S.; Lobo, A.L.A.; Siqueira, M.J.S.; Fritsche-Neto, R.; Dovale, J.C. Unveiling early-stage responses of sensitive traits to water stress in tropical maize: A characterization study of a public panel. Euphytica 2025, 221, 2. [Google Scholar] [CrossRef]
- Lisboa, I.P.; McMechan, A.J.; Elmore, R.W.; Proctor, C.A.; Geyer, A.; Thomison, P.; Lindsey, A.; Zarnstorff, M.; Houx, J.; Ortez, O.A. Popcorn stand reduction in the United States Midwest: Insights on plant traits and grain yield. Field Crops Res. 2025, 329, 109955. [Google Scholar] [CrossRef]
- Salarpour, M.; Abdolshahi, R.; Pakniyat, H.; Heidari, B.; Aminizadeh, S. Mapping quantitative trait loci for drought tolerance/susceptibility indices and estimation of breeding values of doubled haploid lines in wheat (Triticum aestivum). Crop Pasture Sci. 2021, 72, 500–513. [Google Scholar] [CrossRef]
- Jaishreepriyanka, R.; Ravikesavan, R.; Iyanar, K.; Uma, D.; Senthil, N. Exploring the usefulness of drought tolerance indices in screening of maize inter-racial derivatives. Electron. J. Plant Breed. 2024, 15, 592–603. [Google Scholar] [CrossRef]
- Diouf, I.; Derivot, L.; Koussevitzky, S.; Carretero, Y.; Bitton, F.; Moreau, L.; Causse, M. Genetic basis of phenotypic plasticity and genotype x environment interactions in a multi-parental tomato population. J. Exp. Bot. 2020, 71, 5365–5376. [Google Scholar] [CrossRef] [PubMed]
- Fatiukha, A.; Deblieck, M.; Klymiuk, V.; Merchuk-Ovnat, L.; Peleg, Z.; Ordon, F.; Fahima, T.; Korol, A.; Saranga, Y.; Krugman, T. Genomic Architecture of Phenotypic Plasticity in Response to Water Stress in Tetraploid Wheat. Int. J. Mol. Sci. 2021, 22, 1723. [Google Scholar] [CrossRef]
- Lee, G.; Kim, J. Sufficient Light Intensity Is Required for the Drought Responses in Sweet Basil (Ocimum basilicum L.). Agronomy 2024, 14, 2101. [Google Scholar] [CrossRef]
- Barbhuiya, I.H.; Moulick, D.; Hossian, A.; Choudhury, S. Role of phosphate in drought stress regulation in developing rice (Oryza sativa L.) seedlings. Cereal Res. Commun. 2024, 52, 531–544. [Google Scholar] [CrossRef]
- Nagatoshi, Y.; Ikazaki, K.; Kobayashi, Y.; Mizuno, N.; Sugita, R.; Takebayashi, Y.; Kojima, M.; Sakakibara, H.; Kobayashi, N.I.; Tanoi, K.; et al. Phosphate starvation response precedes abscisic acid response under progressive mild drought in plants. Nat. Commun. 2023, 14, 5047. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Zhou, B.; Pei, T.; Meng, H.; Zhang, J.; Ma, F.; Wei, Q. Phosphate Transporter MdPHT1;7 Enhances Phosphorus Accumulation and Improves Low Phosphorus and Drought Tolerance. J. Plant Biol. 2021, 64, 403–416. [Google Scholar] [CrossRef]
- Aanniz, T.; El Baaboua, A.; Aboulaghras, S.; Bouyahya, A.; Benali, T.; Balahbib, A.; El Omari, N.; Butnariu, M.; Muzammil, K.; Yadav, K.K.; et al. Impact of water stress to plant epigenetic mechanisms in stress and adaptation. Physiol. Plant. 2025, 177, e70058. [Google Scholar] [CrossRef]
- Yadav, S.; Kalwan, G.; Gill, S.S.; Jain, P.K. The ABC transporters and their epigenetic regulation under drought stress in chickpea. Plant Physiol. Biochem. PPB 2025, 223, 109903. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, P.; Jing, H.; Zhou, X.F.; Zhao, B.; Li, Y.; Jin, J.B. JMJ27-mediated histone H3K9 demethylation positively regulates drought-stress responses in Arabidopsis. New Phytol. 2021, 232, 221–236. [Google Scholar] [CrossRef]
- Rai, V.K. Role of amino acids in plant responses to stresses. Biol. Plant. 2002, 45, 481–487. [Google Scholar] [CrossRef]
- Diniz, A.L.; Rodrigues da Silva, D.I.; Lembke, C.G.; Lamas Costa, M.D.-B.; ten-Caten, F.; Li, F.; Vilela, R.D.; Menossi, M.; Ware, D.; Endres, L.; et al. Amino Acid and Carbohydrate Metabolism Are Coordinated to Maintain Energetic Balance during Drought in Sugarcane. Int. J. Mol. Sci. 2020, 21, 9124. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Liu, X.; Zhou, Y.; Summers, R.M.; Zhang, Z. BLINK: A package for the next level of genome-wide association studies with both individuals and markers in the millions. Gigascience 2019, 8, giy154. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Zhang, J.; Zhao, Y.; Schmidt, R.H.; Mascher, M.; Reif, J.C.; Jiang, Y. A comprehensive overview and benchmarking analysis of fast algorithms for genome-wide association studies. bioRxiv 2023, 1–46. [Google Scholar] [CrossRef]
- Pe’er, I.; Yelensk, R.; Altshuler, D.; Daly, M.J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genet. Epidemiol. 2008, 32, 381–385. [Google Scholar] [CrossRef]
- Schnable, P.S.; Ware, D.; Fulton, R.S.; Stein, J.C.; Wei, F.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2009, 326, 1112–1115. [Google Scholar] [CrossRef]
- Frank, M.J.; Cartwright, H.N.; Smith, L.G. Three Brick genes have distinct functions in a common pathway promoting polarized cell division and cell morphogenesis in the maize leaf epidermis. Development 2003, 130, 753–762. [Google Scholar] [CrossRef]
- Ren, W.; Shi, Z.; Zhao, Y.; Zhang, Q.; Zhou, M.; Cheng, C.; Liu, M.; Zhao, B.; Guo, Y.; Du, H.; et al. Transcriptional analysis of maize elite inbred line Jing24 and the function of ZmMAPKKK21 in the response to drought stress. Agric. Commun. 2024, 2, 100063. [Google Scholar] [CrossRef]
- He, Z.; Zhong, J.; Sun, X.; Wang, B.; Terzaghi, W.; Dai, M. The Maize ABA Receptors ZmPYL8, 9, and 12 Facilitate Plant Drought Resistance. Front. Plant Sci. 2018, 9, 422. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Zhao, Y.; Zhang, J.; Chen, L.; Si, W.; Jiang, H. A maize heat shock factor ZmHsf11 negatively regulates heat stress tolerance in transgenic plants. BMC Plant Biol. 2022, 22, 406. [Google Scholar] [CrossRef]
- Wang BaoMei, W.B.; Liu Can, L.C.; Zhang DengFeng, Z.D.; He ChunMei, H.C.; Zhang JuRen, Z.J.; Li ZhaoXia, L.Z. Effects of maize organ-specific drought stress response on yields from transcriptome analysis. BMC Plant Biol. 2019, 19, 335. [Google Scholar] [CrossRef]
- He, K.; Zhang, Y.; Ren, W.; Chen, P.; Liu, J.; Mi, G.; Chen, F.; Pan, Q. QTL mapping and transcriptome analysis identify candidate genes influencing water-nitrogen interaction in maize. Crop J. 2023, 11, 1872–1883. [Google Scholar] [CrossRef]
- Xie, P.; Wu, Y.; Xie, Q. Evolution of cereal floral architecture and threshability. Trends Plant Sci. 2023, 28, 1438–1450. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Liu, H.; Xiu, Z.-h.; Yang, H.-h.; Wang, H.; Wang, Y.; Tan, B.-C. Defective kernel 58 encodes an Rrp15p domain-containing protein essential to ribosome biogenesis and seed development in maize. New Phytol. 2024, 241, 1662–1675. [Google Scholar] [CrossRef] [PubMed]
- Bernal-Vasquez, A.-M.; Utz, H.F.; Piepho, H.-P. Outlier detection methods for generalized lattices: A case study on the transition from ANOVA to REML. Theor. Appl. Genet. 2016, 129, 787–804. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.; Maechler, M.; Bolker, B.M.; Walker, S.C. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Covarrubias-Pazaran, G. Genome-Assisted Prediction of Quantitative Traits Using the R Package sommer. PLoS ONE 2016, 11, e0156744. [Google Scholar] [CrossRef]
- Cullis, B.R.; Smith, A.B.; Coombes, N.E. On the design of early generation variety trials with correlated data. J. Agric. Biol. Environ. Stat. 2006, 11, 381–393. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Lian, L.; DeLosCampos, G. FW: An R Package for Finlay–Wilkinson Regression that Incorporates Genomic/Pedigree Information and Covariance Structures Between Environments. G3 Genes Genomes Genet. 2016, 6, 589–597. [Google Scholar] [CrossRef]
- Browning, B.L.; Tian, X.; Zhou, Y.; Browning, S.R. Fast two-stage phasing of large-scale sequence data. Am. J. Hum. Genet. 2021, 108, 1880–1890. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: Software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Z. GAPIT Version 3: Boosting Power and Accuracy for Genomic Association and Prediction. Genom. Proteom. Bioinform. 2021, 19, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Zhang, H.; Tang, Z.; Xu, J.; Yin, D.; Zhang, Z.; Yuan, X.; Zhu, M.; Zhao, S.; Li, X.; et al. rMVP: A Memory-efficient, Visualization-enhanced, and Parallel-accelerated Tool for Genome-wide Association Study. Genom. Proteom. Bioinform. 2021, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Zhang, H.; Long, Y.; Shu, Y.; Zhai, J. Plant Public RNA-seq Database: A comprehensive online database for expression analysis of ∼45 000 plant public RNA-Seq libraries. Plant Biotechnol. J. 2022, 20, 806–808. [Google Scholar] [CrossRef]
- Thatcher, S.R.; Danilevskaya, O.N.; Meng, X.; Beatty, M.; Zastrow-Hayes, G.; Harris, C.; Van Allen, B.; Habben, J.; Li, B. Genome-Wide Analysis of Alternative Splicing during Development and Drought Stress in Maize. Plant Physiol. 2016, 170, 586–599. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Treatment | Min | Max | Mean | SD | Ratio | H2 | ||
|---|---|---|---|---|---|---|---|---|---|
| PH | WW | 142.0 | 261.9 | 197.8 | 21.9 | 424.6 *** | 15.4 ** | 0.04 | 0.88 |
| PH | WD | 121.7 | 222.7 | 172.0 | 20.1 | 277.1 *** | 85.2 *** | 0.31 | 0.71 |
| EH | WW | 32.9 | 126.0 | 66.2 | 13.4 | 146.9 *** | 22.7 *** | 0.15 | 0.81 |
| EH | WD | 29.5 | 104.6 | 57.6 | 10.7 | 90.4 *** | 9.3 *** | 0.10 | 0.80 |
| EL | WW | 10.0 | 21.1 | 15.2 | 1.9 | 3.0 *** | 0.38 *** | 0.13 | 0.82 |
| EL | WD | 9.1 | 18.3 | 13.4 | 1.8 | 2.2 *** | 0.67 *** | 0.30 | 0.69 |
| ED | WW | 3.3 | 5.3 | 4.3 | 0.33 | 0.07 *** | 0.04 *** | 0.57 | 0.58 |
| ED | WD | 2.7 | 4.8 | 4.0 | 0.35 | 0.08 *** | 0.04 *** | 0.50 | 0.64 |
| RNPE | WW | 11.4 | 19.2 | 15.2 | 1.5 | 1.6 *** | 0.57 *** | 0.35 | 0.72 |
| RNPE | WD | 10.9 | 18.7 | 14.6 | 1.5 | 1.5 *** | 0.50 *** | 0.33 | 0.69 |
| KNPR | WW | 11.9 | 42.1 | 26.9 | 4.0 | 12.8 *** | 1.5 ** | 0.12 | 0.79 |
| KNPR | WD | 11.4 | 36.4 | 23.9 | 4.2 | 12.4 *** | 0.78 * | 0.06 | 0.72 |
| GYPE | WW | 29.9 | 253.0 | 108.5 | 26.4 | 555.9 *** | 122.6 *** | 0.22 | 0.76 |
| GYPE | WD | 24.4 | 142.0 | 75.7 | 18.4 | 179.1 *** | 65.9 ** | 0.37 | 0.53 |
| Gene | Description | Class | Traits | QTL | Homology in Rice | Reference |
|---|---|---|---|---|---|---|
| Zm00001d023262 | Brick3 | LP | RNPE | qLP65 | OsDS8 | [50] |
| Zm00001d028574 | Protein phosphatase homolog5 | LP | EL | qLP7 | OsPP2C09 | [51] |
| Zm00001d046501 | AP2-EREBP-transcription factor 237 | LP | KNPR | qLP62 | OsWR1 | [52] |
| Zm00001d052847 | WRKY-transcription factor 87 | LP | GYPE | qLP26 | OsWRKY30 | [53] |
| Zm00001d016105 | Pyrabactin resistance-like protein10 | DRI | PH | qDRI59 | OsPYL10 | [54] |
| Zm00001d002545 | MYB-related-transcription factor 20 | DRI | EL | qDRI24 | OsMYBR1 | [55] |
| Zm00001d026501 | Glutamine synthetase1 | DRI | EL | qDRI19 | OsGS2 | [56] |
| WD | EH | qWD13 | ||||
| Zm00001d052738 | HSF transcription factor 7 | STI | EL | qSTI42 | OsHsfB2b | [57] |
| WD | EL | qWD31 | ||||
| Zm00001d007962 | G2-like-transcription factor 27 | STI | GYPE | qSTI66 | OsHHO3 | [58] |
| STI | KNPR | qSTI81 | ||||
| Zm00001d051804 | Glutamine synthetase5 | WD | EL | qWD30 | OsGS1;1 | [59] |
| Zm00001d017294 | Gibberellin 2-oxidase4 | WD | EL | qWD35 | GA2ox6 | [60] |
| Zm00001d011765 | Methylsterol monooxygenase 2-2 | WW | GYPE | qWW46 | OsGL1-8 | [61] |
| Class | Traits | Description | Measuring Unit |
|---|---|---|---|
| Phenological | PH | The height from ground to the tassel tip | centimeters (cm) |
| EH | The height from ground to the node of the highest ear | centimeters (cm) | |
| Yield | EL | The length of maize ear from base to top | centimeters (cm) |
| composition | ED | The diameter of the middle ear | centimeters (cm) |
| RNPE | The number of rows of complete ear | count (rows/ear) | |
| KNPR | The kernel number per row | count (grains/row) | |
| GYPE | The yield per ear as the mean value of the effective plants in the plot | grams (g) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Li, D.; Guo, Y.; Wang, Y.; Zhang, Y.; Li, L.; Yang, X.; Chen, S.; Würschum, T.; Liu, W. Genetic Dissection of Drought Tolerance in Maize Through GWAS of Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity. Int. J. Mol. Sci. 2025, 26, 6285. https://doi.org/10.3390/ijms26136285
Li R, Li D, Guo Y, Wang Y, Zhang Y, Li L, Yang X, Chen S, Würschum T, Liu W. Genetic Dissection of Drought Tolerance in Maize Through GWAS of Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity. International Journal of Molecular Sciences. 2025; 26(13):6285. https://doi.org/10.3390/ijms26136285
Chicago/Turabian StyleLi, Ronglan, Dongdong Li, Yuhang Guo, Yueli Wang, Yufeng Zhang, Le Li, Xiaosong Yang, Shaojiang Chen, Tobias Würschum, and Wenxin Liu. 2025. "Genetic Dissection of Drought Tolerance in Maize Through GWAS of Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity" International Journal of Molecular Sciences 26, no. 13: 6285. https://doi.org/10.3390/ijms26136285
APA StyleLi, R., Li, D., Guo, Y., Wang, Y., Zhang, Y., Li, L., Yang, X., Chen, S., Würschum, T., & Liu, W. (2025). Genetic Dissection of Drought Tolerance in Maize Through GWAS of Agronomic Traits, Stress Tolerance Indices, and Phenotypic Plasticity. International Journal of Molecular Sciences, 26(13), 6285. https://doi.org/10.3390/ijms26136285