Waardenburg Syndrome Type 4 in Mongolian Children: Genetic and Clinical Characterization

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
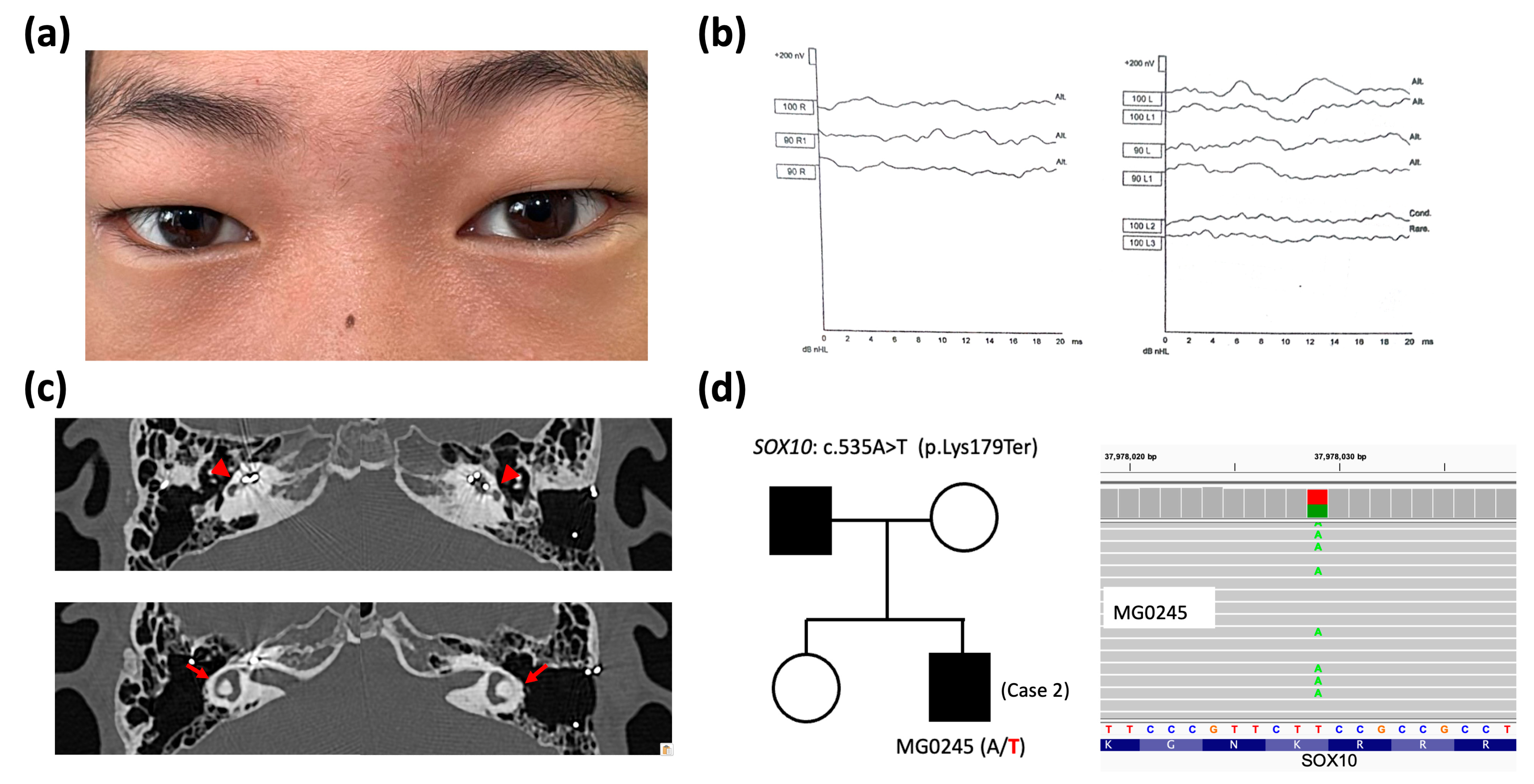
2.1. Clinical and Genetic Findings
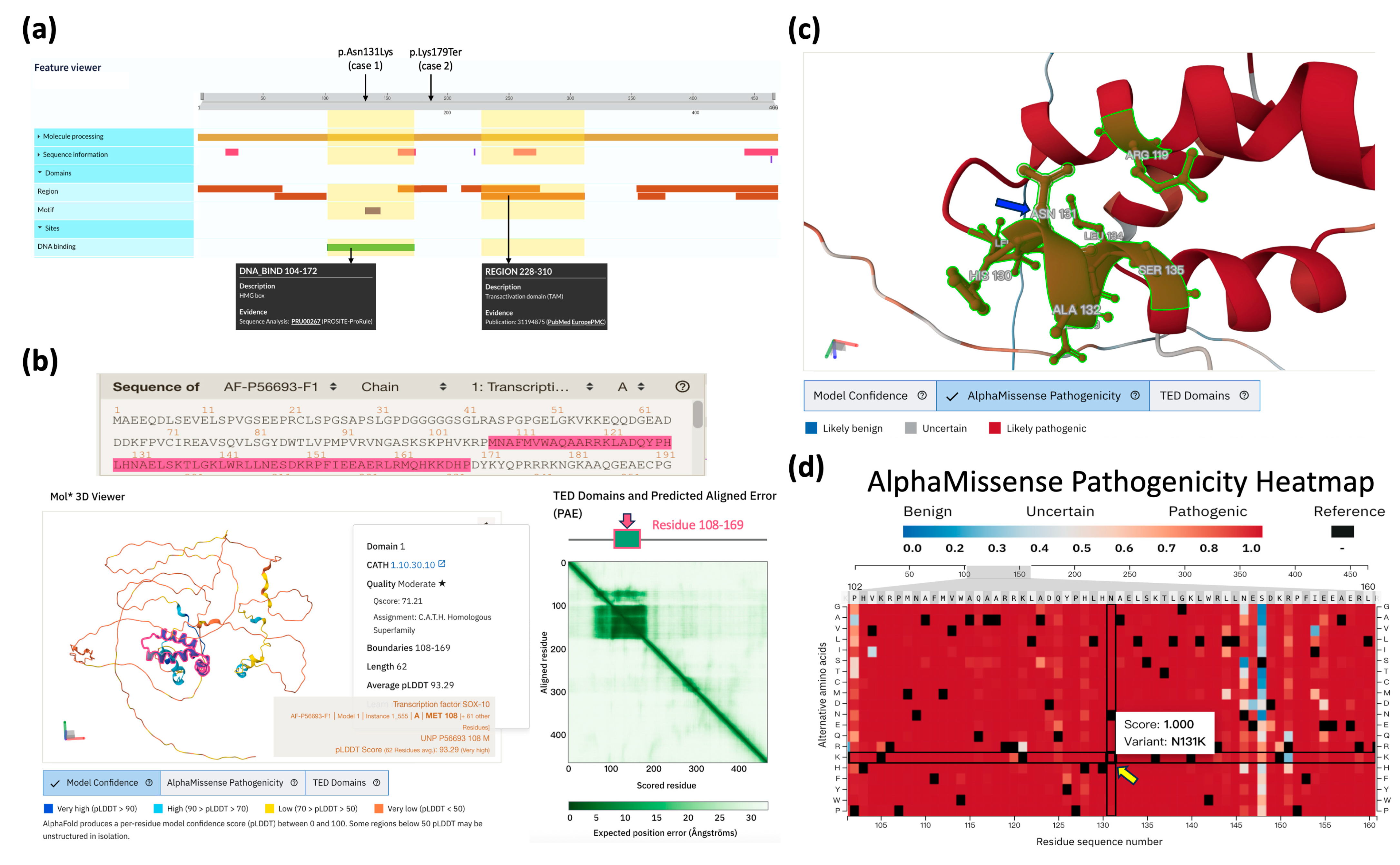
2.2. Structural and Pathogenic Analysis of SOX10 Variants
3. Discussion
4. Materials and Methods
4.1. Subjects and Clinical Assessments
4.2. Genetic Examination with Multi-Database Assessment for Pathogenicity
4.3. Homologous Sequence Analyses and Structural Characterizations
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Winters, R.; Masood, S. “Waardenburg Syndrome” in StatPearls [Internet]; StatPearls: Treasure Island, FL, USA, 2025. [Google Scholar]
- Read, A.P.; Newton, V.E. Waardenburg syndrome. J. Med. Genet. 1997, 34, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Feng, Y.; Acke, F.; Coucke, P.; Vleminckx, K.; Dhooge, I. Hearing loss in Waardenburg syndrome: A systematic review. Clin. Genet. 2016, 89, 416–425. [Google Scholar] [CrossRef] [PubMed]
- Boubekri, A.; Atmani, W.; Bensghir, M. [Waardenburg syndrome]. Rev Prat 2023, 73, 1106. Available online: https://www.ncbi.nlm.nih.gov/pubmed/38294479 (accessed on 16 May 2025). [PubMed]
- Ramakrishnan, I.L.; Taksande, A. Waardenburg syndrome. Pan Afr. Med. J. 2023, 45, 23. [Google Scholar] [CrossRef]
- Ringer, J. Identification of Waardenburg syndrome and the management of hearing loss and associated sequelae: A review for the pediatric nurse practitioner. J. Pediatr. Health Care 2019, 33, 694–701. [Google Scholar] [CrossRef]
- Winsbip, I.; Brighton, P. Phenotypic discriminants in the Waardenburg syndrome. Clin. Genet. 1992, 41, 181–188. [Google Scholar] [CrossRef]
- Milunsky, J.M. “Waardenburg Syndrome Type I” in GeneReviews® [Internet]; University of Washington: Seattle, WA, USA, 2001. [Google Scholar]
- Kumar, S.; Natraj, R.; Dutta, A. Waardenburg Syndrome Type 2 in Paediatrics: A Case Highlighting Diagnostic Complexities and the Efficacy of Cochlear Implantation. Indian J. Otolaryngol. Head Neck Surg. 2024, 76, 2100–2103. [Google Scholar] [CrossRef]
- Sil, A.; Panigrahi, A. Visual dermatology: Waardenburg syndrome type II. J. Cutan. Med. Surg. 2020, 24, 305. [Google Scholar] [CrossRef]
- Shaw, S.C.; Neema, S.; Devgan, A.; Maggon, R. Waardenburg syndrome type 2. Med. J. Armed Forces India 2018, 74, 380–382. [Google Scholar] [CrossRef]
- Tekin, M.; Bodurtha, J.; Nance, W.; Pandya, A. Waardenburg syndrome type 3 (Klein–Waardenburg syndrome) segregating with a heterozygous deletion in the paired box domain of PAX3: A simple variant or a true syndrome? Clin. Genet. 2001, 60, 301–304. [Google Scholar] [CrossRef]
- Zhang, L.; Wan, Y.; Wang, N. Waardenburg syndrome type 4 coexisting with open-angle glaucoma: A case report. J. Med. Case Rep. 2022, 16, 264. [Google Scholar] [CrossRef] [PubMed]
- Appak, Y.C.; Tinastepe, T.; Aksoy, B.; Turhan, S.; Tugmen, C.; Sert, I.; Baran, M. Outcome of Children With Intestinal Failure Due to Waardenburg Syndrome From an Intestinal Transplant Center: A Case Series. Exp. Clin. Transplant. Off. J. Middle East Soc. Organ Transplant. 2020. [Google Scholar] [CrossRef]
- Nusrat, M.; Tariq, M.A.; Aslam, S.; Zil-E-Ali, A.; Shahid, M.; Mahmood, S. A case of Waardenburg-Shah syndrome type 4 presenting with bilateral homochromatic blue Irises from Pakistan. Cureus 2018, 10, e3143. [Google Scholar] [CrossRef] [PubMed]
- Apaydin, F.; Bereketoglu, M.; Turan, O.; Hribar, K.; Maassen, M.; Günhan, Ö.; Zenner, H.-P.; Pfister, M. Waardenburg syndrome: A heterogenic disorder with variable penetrance. Hno 2004, 52, 533–537. [Google Scholar] [CrossRef]
- Huang, S.; Song, J.; He, C.; Cai, X.; Yuan, K.; Mei, L.; Feng, Y. Genetic insights, disease mechanisms, and biological therapeutics for Waardenburg syndrome. Gene Ther. 2022, 29, 479–497. [Google Scholar] [CrossRef]
- Pingault, V.; Bondurand, N.; Kuhlbrodt, K.; Goerich, D.E.; Préhu, M.-O.; Puliti, A.; Herbarth, B.; Hermans-Borgmeyer, I.; Legius, E.; Matthijs, G. SOX10 mutations in patients with Waardenburg-Hirschsprung disease. Nat. Genet. 1998, 18, 171–173. [Google Scholar] [CrossRef]
- Touraine, R.L.; Attié-Bitach, T.; Manceau, E.; Korsch, E.; Sarda, P.; Pingault, V.; Encha-Razavi, F.; Pelet, A.; Augé, J.; Nivelon-Chevallier, A. Neurological phenotype in Waardenburg syndrome type 4 correlates with novel SOX10 truncating mutations and expression in developing brain. Am. J. Hum. Genet. 2000, 66, 1496–1503. [Google Scholar] [CrossRef]
- Bondurand, N.; Dastot-Le Moal, F.; Stanchina, L.; Collot, N.; Baral, V.; Marlin, S.; Attie-Bitach, T.; Giurgea, I.; Skopinski, L.; Reardon, W. Deletions at the SOX10 gene locus cause Waardenburg syndrome types 2 and 4. Am. J. Hum. Genet. 2007, 81, 1169–1185. [Google Scholar] [CrossRef]
- Gombojav, B.; Erdenechuluun, J.; Makhbal, Z.; Danshiitsoodol, N.; Purevdorj, E.; Jargalmaa, M.; Batsaikhan, T.; Lin, P.-H.; Lu, Y.-S.; Lo, M.-Y. Genetic Basis of Hearing Loss in Mongolian Patients: A Next-Generation Sequencing Study. Genes 2024, 15, 1227. [Google Scholar] [CrossRef]
- Pingault, V.; Zerad, L.; Bertani-Torres, W.; Bondurand, N. SOX10: 20 years of phenotypic plurality and current understanding of its developmental function. J. Med. Genet. 2022, 59, 105–114. [Google Scholar] [CrossRef]
- Wegner, M. From head to toes: The multiple facets of Sox proteins. Nucleic Acids Res. 1999, 27, 1409–1420. [Google Scholar] [CrossRef] [PubMed]
- Schepers, G.E.; Teasdale, R.D.; Koopman, P. Twenty pairs of sox: Extent, homology, and nomenclature of the mouse and human sox transcription factor gene families. Dev. Cell 2002, 3, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Nguengang Wakap, S.; Lambert, D.M.; Olry, A.; Rodwell, C.; Gueydan, C.; Lanneau, V.; Murphy, D.; Le Cam, Y.; Rath, A. Estimating cumulative point prevalence of rare diseases: Analysis of the Orphanet database. Eur. J. Hum. Genet. 2020, 28, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Khan, T.A.; Safdar, C.A.; Zameer, S.; Khushdil, A. Waardenburg-Shah syndrome (WS type IV): A rare case from Pakistan. Perioper. Med. 2020, 9, 4. [Google Scholar] [CrossRef]
- Buonfiglio, P.I.; Izquierdo, A.; Pace, M.V.; Grinberg, S.; Lotersztein, V.; Brun, P.; Bruque, C.D.; Elgoyhen, A.B.; Dalamón, V. Comprehensive approach for the genetic diagnosis of patients with Waardenburg syndrome. J. Pers. Med. 2024, 14, 906. [Google Scholar] [CrossRef]
- Kankipati, S.M.; Mahalingam, A.; Reshie, A.; Fayaz, F.; Nimal, S.; Duggineni, D. Clinical insights into Waardenburg-Shah syndrome: A case series and literature review. Cureus 2024, 16, e59858. [Google Scholar] [CrossRef]
- Bertani-Torres, W.; Lezirovitz, K.; Alencar-Coutinho, D.; Pardono, E.; da Costa, S.S.; Antunes, L.d.N.; de Oliveira, J.; Otto, P.A.; Pingault, V.; Mingroni-Netto, R.C. Waardenburg syndrome: The contribution of next-generation sequencing to the identification of novel causative variants. Audiol. Res. 2023, 14, 9–25. [Google Scholar] [CrossRef]
- Tawfik, C.A.; Essawi, M.L.; Nowara, M.; Mohsen, R.; Elbagoury, N.M. Concurrent novel mutations in PAX3 and CFAP410 in a patient with Waardenburg syndrome type 1 associated with Retinitis Pigmentosa. Ophthalmic Genet. 2025, 46, 1–8. [Google Scholar] [CrossRef]
- Somashekar, P.H.; Upadhyai, P.; Narayanan, D.L.; Kamath, N.; Bajaj, S.; Girisha, K.M.; Shukla, A. Phenotypic diversity and genetic complexity of PAX3-related Waardenburg syndrome. Am. J. Med. Genet. Part A 2020, 182, 2951–2958. [Google Scholar] [CrossRef]
- Choi, E.Y.; Choi, W.; Lee, C.S. A novel PAX3 mutation in a Korean patient with Waardenburg syndrome type 1 and unilateral branch retinal vein and artery occlusion: A case report. BMC Ophthalmol. 2018, 18, 266. [Google Scholar] [CrossRef]
- Nasirshalal, M.; Panahi, M.; Javanshir, N.; Salmani, H. Identification of a novel heterozygous mutation in the MITF gene in an Iranian family with Waardenburg syndrome type II using next-generation sequencing. J. Clin. Lab. Anal. 2021, 35, e23792. [Google Scholar] [CrossRef] [PubMed]
- Albarry, M.A.; Latif, M.; Alreheli, A.Q.; Awadh, M.A.; Almatrafi, A.M.; Albalawi, A.M.; Basit, S. Frameshift variant in MITF gene in a large family with Waardenburg syndrome type II and a co-segregation of a C2orf74 variant. PLoS ONE 2021, 16, e0246607. [Google Scholar] [CrossRef] [PubMed]
- Mohan, S.L.C. Case of Waardenburg Shah syndrome in a family with review of literature. J. Otol. 2018, 13, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhu, Y.; Shen, N.; Peng, J.; Wang, C.; Liu, H.; Lu, Y. A de novo deletion mutation in SOX10 in a Chinese family with Waardenburg syndrome type 4. Sci. Rep. 2017, 7, 41513. [Google Scholar] [CrossRef]
- Loupe, J.; Sampath, S.; Lacassie, Y. Familial co-segregation of Coffin–Lowry syndrome inherited from the mother and autosomal dominant Waardenburg type IV syndrome due to deletion of EDNRB inherited from the father. Eur. J. Med. Genet. 2014, 57, 562–566. [Google Scholar] [CrossRef]
- Jiang, L.; Chen, H.; Jiang, W.; Hu, Z.; Mei, L.; Xue, J.; He, C.; Liu, Y.; Xia, K.; Feng, Y. Novel mutations in the SOX10 gene in the first two Chinese cases of type IV Waardenburg syndrome. Biochem. Biophys. Res. Commun. 2011, 408, 620–624. [Google Scholar] [CrossRef]
- Pang, X.; Zheng, X.; Kong, X.; Chai, Y.; Wang, Y.; Qian, H.; Yang, B.; Wu, C.; Chu, J.; Yang, T. A homozygous MITF mutation leads to familial Waardenburg syndrome type 4. Am. J. Med. Genet. Part A 2019, 179, 243–248. [Google Scholar] [CrossRef]
- Sangkhathat, S.; Chiengkriwate, P.; Kusafuka, T.; Patrapinyokul, S.; Fukuzawa, M. Novel mutation of endothelin-B receptor gene in Waardenburg–Hirschsprung disease. Pediatr. Surg. Int. 2005, 21, 960–963. [Google Scholar] [CrossRef]
- Syrris, P.; Carter, N.D.; Patton, M.A. Novel nonsense mutation of the endothelin-B receptor gene in a family with Waardenburg-Hirschsprung disease. Am. J. Med. Genet. 1999, 87, 69–71. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, W.; Chen, M.; Yang, Y.; Yang, Y.; Hong, E.; Lu, J.; Zheng, J.; Ni, X.; Guo, Y. Two novel mutations of PAX3 and SOX10 were characterized as genetic causes of Waardenburg Syndrome. Mol. Genet. Genom. Med. 2020, 8, e1217. [Google Scholar] [CrossRef]
- Verheij, J.B.; Sival, D.A.; van der Hoeven, J.H.; Vos, Y.J.; Meiners, L.C.; Brouwer, O.F.; van Essen, A.J. Shah-Waardenburg syndrome and PCWH associated with SOX10 mutations: A case report and review of the literature. Eur. J. Paediatr. Neurol. 2006, 10, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Fernández, R.M.; Núñez-Ramos, R.; Enguix-Riego, M.V.; Román-Rodríguez, F.J.; Galán-Gómez, E.; Blesa-Sánchez, E.; Antinolo, G.; Núñez-Núñez, R.; Borrego, S. Waardenburg syndrome type 4: Report of two new cases caused by SOX10 mutations in Spain. Am. J. Med. Genet. Part A 2014, 164, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, K.-F.; Nguyen, C.M.; Cardinal, T.; Charrier, B.; Silversides, D.W.; Pilon, N. Upregulation of the Nr2f1-A830082K12Rik gene pair in murine neural crest cells results in a complex phenotype reminiscent of Waardenburg syndrome type 4. Dis. Models Mech. 2016, 9, 1283–1293. [Google Scholar] [CrossRef]
- Lovett, A.; Eastwood, M.; Metcalfe, C.; Muzaffar, J.; Monksfield, P.; Bance, M. Outcomes of Cochlear implantation in early-deafened patients with Waardenburg syndrome: A systematic review and narrative synthesis. Laryngoscope Investig. Otolaryngol. 2023, 8, 1094–1107. [Google Scholar] [CrossRef]
- Eppsteiner, R.W.; Shearer, A.E.; Hildebrand, M.S.; DeLuca, A.P.; Ji, H.; Dunn, C.C.; Black-Ziegelbein, E.A.; Casavant, T.L.; Braun, T.A.; Scheetz, T.E. Prediction of cochlear implant performance by genetic mutation: The spiral ganglion hypothesis. Hear. Res. 2012, 292, 51–58. [Google Scholar] [CrossRef]
- Wu, C.-C.; Lin, Y.-H.; Liu, T.-C.; Lin, K.-N.; Yang, W.-S.; Hsu, C.-J.; Chen, P.-L.; Wu, C.-M. Identifying children with poor cochlear implantation outcomes using massively parallel sequencing. Medicine 2015, 94, e1073. [Google Scholar] [CrossRef]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Aguilera, M.A.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef]
- Cheng, J.; Novati, G.; Pan, J.; Bycroft, C.; Žemgulytė, A.; Applebaum, T.; Pritzel, A.; Wong, L.H.; Zielinski, M.; Sargeant, T. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science 2023, 381, eadg7492. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, N.M.; Rothstein, J.H.; Pejaver, V.; Middha, S.; McDonnell, S.K.; Baheti, S.; Musolf, A.; Li, Q.; Holzinger, E.; Karyadi, D. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 2016, 99, 877–885. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.R.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Jang, W. ClinVar: Improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 2018, 46, D1062–D1067. [Google Scholar] [CrossRef]
- Azaiez, H.; Booth, K.T.; Ephraim, S.S.; Crone, B.; Black-Ziegelbein, E.A.; Marini, R.J.; Shearer, A.E.; Sloan-Heggen, C.M.; Kolbe, D.; Casavant, T. Genomic landscape and mutational signatures of deafness-associated genes. Am. J. Hum. Genet. 2018, 103, 484–497. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosenbloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- Navarro Gonzalez, J.; Zweig, A.S.; Speir, M.L.; Schmelter, D.; Rosenbloom, K.R.; Raney, B.J.; Powell, C.C.; Nassar, L.R.; Maulding, N.D.; Lee, C.M. The UCSC genome browser database: 2021 update. Nucleic Acids Res. 2021, 49, D1046–D1057. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- UniProt_Consortium, UniProt: The Universal protein knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [CrossRef]
- Varadi, M.; Bertoni, D.; Magana, P.; Paramval, U.; Pidruchna, I.; Radhakrishnan, M.; Tsenkov, M.; Nair, S.; Mirdita, M.; Yeo, J. AlphaFold Protein Structure Database in 2024: Providing structure coverage for over 214 million protein sequences. Nucleic Acids Res. 2024, 52, D368–D375. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patients | Loci (hg38) and HGVS ‡ of Variants | Grpmax Allele Frequency § | Predictive Scores ¶ | Database Assertions | ACMG Criteria (Classification) | Ref. |
|---|---|---|---|---|---|---|
| Case 1 (MG0252) | chr22-37983392-G-C SOX10: c.393C>G (p.Asn131Lys) | N.A. | CADD: 27.8 AlphaMissense: 1.0 (PS) SIFT: 0.001 (D) POL2: 0.998 (PD) REVEL: 0.917 (PS) | ClinVar: P DVD: N/A | PS1, PS2, PM2, PP3, PP4 (Pathogenic) | [21] |
| Case 2 (MG0245) | chr22-37978029-T-A SOX10: c.535A>T (p.Lys179Ter) | N.A. | CADD: 37 BayesDel: 0.994 (PS) | ClinVar: N/A DVD: N/A | PVS1, PM2, PP4 (Pathogenic) | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gombojav, B.; Erdenechuluun, J.; Batsaikhan, T.; Danshiitsoodol, N.; Makhbal, Z.; Jargalmaa, M.; Jargalkhuu, T.; Lu, Y.-S.; Lin, P.-H.; Hsu, J.S.-J.; et al. Waardenburg Syndrome Type 4 in Mongolian Children: Genetic and Clinical Characterization. Int. J. Mol. Sci. 2025, 26, 6258. https://doi.org/10.3390/ijms26136258
Gombojav B, Erdenechuluun J, Batsaikhan T, Danshiitsoodol N, Makhbal Z, Jargalmaa M, Jargalkhuu T, Lu Y-S, Lin P-H, Hsu JS-J, et al. Waardenburg Syndrome Type 4 in Mongolian Children: Genetic and Clinical Characterization. International Journal of Molecular Sciences. 2025; 26(13):6258. https://doi.org/10.3390/ijms26136258
Chicago/Turabian StyleGombojav, Bayasgalan, Jargalkhuu Erdenechuluun, Tserendulam Batsaikhan, Narandalai Danshiitsoodol, Zaya Makhbal, Maralgoo Jargalmaa, Tuvshinbayar Jargalkhuu, Yue-Sheng Lu, Pei-Hsuan Lin, Jacob Shu-Jui Hsu, and et al. 2025. "Waardenburg Syndrome Type 4 in Mongolian Children: Genetic and Clinical Characterization" International Journal of Molecular Sciences 26, no. 13: 6258. https://doi.org/10.3390/ijms26136258
APA StyleGombojav, B., Erdenechuluun, J., Batsaikhan, T., Danshiitsoodol, N., Makhbal, Z., Jargalmaa, M., Jargalkhuu, T., Lu, Y.-S., Lin, P.-H., Hsu, J. S.-J., Tsai, C.-Y., & Wu, C.-C. (2025). Waardenburg Syndrome Type 4 in Mongolian Children: Genetic and Clinical Characterization. International Journal of Molecular Sciences, 26(13), 6258. https://doi.org/10.3390/ijms26136258