
Molecular Study from the Signaling Pathways of Four Potential asthma triggers: AKT1, MAPK13, STAT1, and TLR4

 , , , ,
, , , ,  , , , , and
, , , , and
Abstract

1. Introduction
2. Results
2.1. Subjects
2.2. The Four Potential Gene Triggers Show Different Gene and Protein Expression in Asthma Patients than in the Healthy Subjects
2.3. A Deeper Analysis Showed Differences in the Results Obtained for Each Clinical Group When Patients Were Segregated by Severity
2.4. The Expression of the Four Potential Triggers Showed a Good Gene Correlation Between Them, but Worse at a Protein Level, with Differences Among Clinical Groups
2.5. The Four Potential Triggers Showed Different Correlation with Lung Functional Parameters Depending of the Group Studied
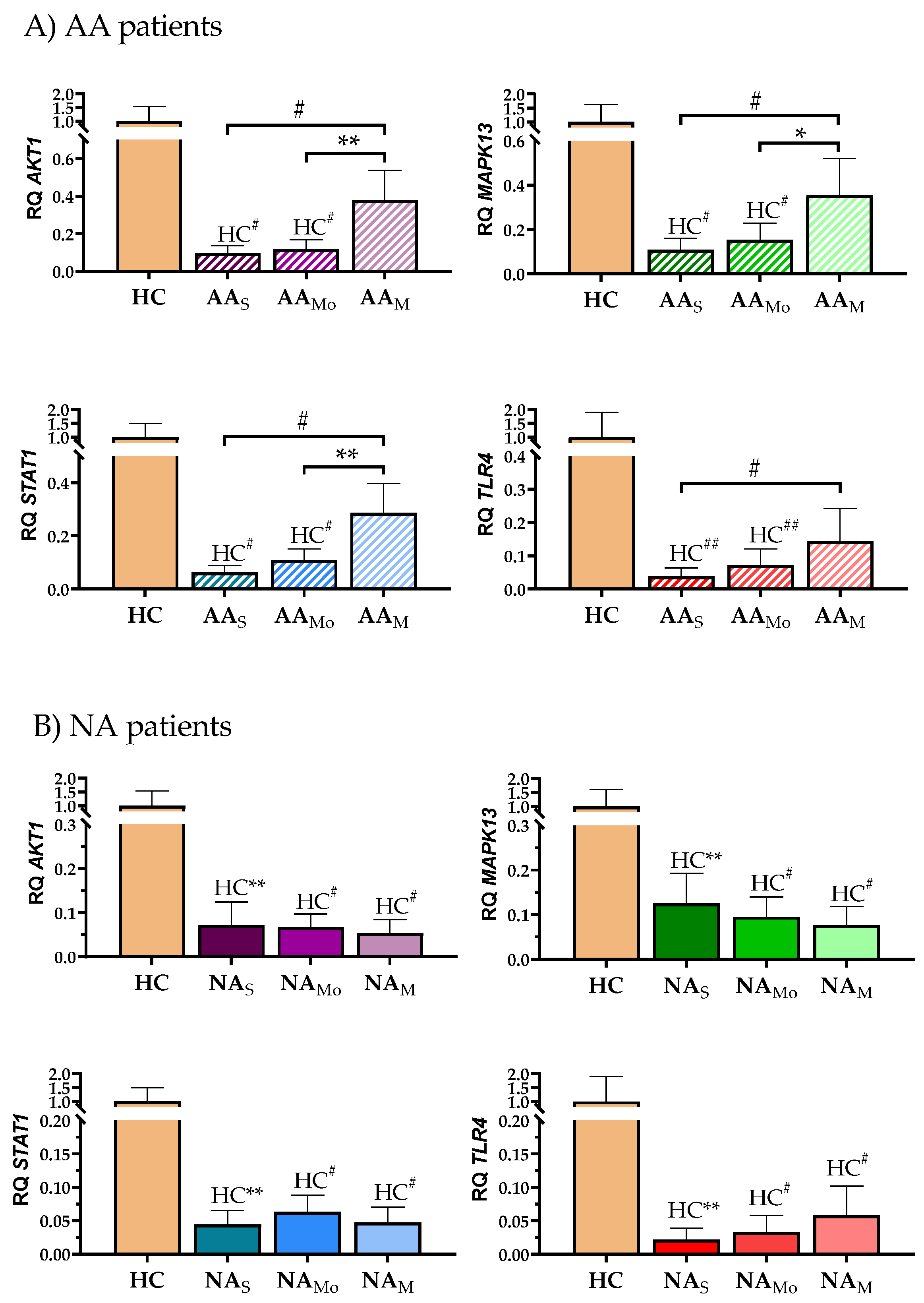
2.6. Analysis of the Signaling Pathways of the Triggers Proteins Show a Significant Gene Expression Decrease in Asthma Groups Compared to HC Subjects
2.7. NA Patients Have Lower Gene Expression than AA Patients on the Common Genes to Both Signaling Pathways
3. Discussion
4. Materials and Methods
4.1. Study Design: Subjects
4.2. Isolation of Peripheral Blood Mononuclear Cells, RNA, and Protein Extraction
4.3. Gene Selection and Differential Expression Analysis by RT-qPCR
4.4. Differential Protein Expression Analysis by Western Blot
4.5. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GINA. Global Initiative for Asthma. Global Strategy for Asthma Management and Prevention. 2021. Available online: http://www.ginasthma.org/ (accessed on 28 May 2024).
- Dubin, S.; Patak, P.; Jung, D. Update on Asthma Management Guidelines. Mo Med. 2024, 121, 364–367. [Google Scholar] [PubMed]
- Pelaia, C.; Crimi, C.; Vatrella, A.; Tinello, C.; Terracciano, R.; Pelaia, G. Molecular Targets for Biological Therapies of Severe Asthma. Front. Immunol. 2020, 11, 603312. [Google Scholar] [CrossRef] [PubMed]
- Vatrella, A.; Maglio, A.; Pelaia, C.; Ciampo, L.; Pelaia, G.; Vitale, C. Eosinophilic inflammation: An Appealing Target for Pharmacologic Treatments in Severe Asthma. Biomedicines 2022, 10, 2181. [Google Scholar] [CrossRef]
- Couillard, S.; Pavord, I.D.; Heaney, L.G.; Petousi, N.; Hinks, T.S.C. Sub-stratification of type-2 high airway disease for therapeutic decision-making: A ‘bomb’ (blood eosinophils) meets the ‘magnet’ (FeNO) framework. Respirology 2022, 27, 573–577. [Google Scholar] [CrossRef]
- Peters, M.C.; Ringel, L.; Dyjack, N.; Herrin, R.; Woodruff, P.G.; Rios, C.; O’cOnnor, B.; Fahy, J.V.; Seibold, M.A. A transcriptomic method to determine airway immune dysfunction in T2-High and T2-Low asthma. Am. J. Respir. Crit. Care Med. 2019, 199, 465–477. [Google Scholar] [CrossRef]
- Berry, A.; Busse, W.W. Biomarkers in asthmatic patients: Has their time come to direct treatment? J. Allergy Clin. Immunol. 2016, 137, 1317–1324. [Google Scholar] [CrossRef]
- Parulekar, A.D.; Diamant, Z.; Nicola, A.; Hanania, N.A. Role of biologics targeting type 2 airway inflammation in asthma: What have we learned so far? Curr. Opin. Pulm. Med. 2017, 23, 3–11. [Google Scholar] [CrossRef]
- Chung, K.F. New treatments for severe treatment-resistant asthma: Targeting the right patient. Lancet Respir. Med. 2013, 1, 639–652. [Google Scholar] [CrossRef]
- Muraro, A.; Lemanske, R.F., Jr.; Hellings, P.W.; Akdis, C.A.; Bieber, T.; Casale, T.B.; Jutel, M.; Ong, P.Y.; Poulsen, L.K.; Schmid-Grendelmeier, P.; et al. Precision medicine in patients with allergic diseases: Airway diseases and atopic dermatitis-PRACTALL document of the European Academy of Allergy and Clinical Immunology and the American Academy of Allergy, Asthma & Immunology. J. Allergy Clin. Immunol. 2016, 137, 1347–1358. [Google Scholar] [CrossRef]
- Tyler, S.R.; Bunyavanich, S. Leveraging -omics for asthma endotyping. J. Allergy Clin. Immunol. 2019, 144, 13–23. [Google Scholar] [CrossRef]
- Cremades-Jimeno, L.; de Pedro, M.Á.; López-Ramos, M.; Sastre, J.; Mínguez, P.; Fernández, I.M.; Baos, S.; Cárdaba, B. Prioritizing Molecular Biomarkers in Asthma and Respiratory Allergy Using Systems Biology. Front. Immunol. 2021, 12, 640791. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Brown, M.; Kogut, P.; Serban, K.; Li, X.; McConville, J.; Chen, B.; Bentley, J.K.; Hershenson, M.B.; Dulin, N.; et al. Akt activation induces hypertrophy without contractile phenotypic maturation in airway smooth muscle. Am. J. Physiol. Cell. Mol. Physiol. 2011, 300, L701–L709. [Google Scholar] [CrossRef]
- Hoshino, M.; Takahashi, M.; Aoike, N. Expression of vascular endothelial growth factor, basic fibroblast growth factor, and angiogenin immunoreactivity in asthmatic airways and its relationship to angiogenesis. J. Allergy Clin. Immunol. 2001, 107, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.F. p38 mitogen-activated protein kinase pathways in asthma and COPD. Chest 2011, 139, 1470–1479. [Google Scholar] [CrossRef]
- Khorasanizadeh, M.; Eskian, M.; Gelfand, E.; Rezaei, N. Mitogen-activated protein kinases as therapeutic targets for asthma. Pharmacol. Ther. 2017, 174, 112–126. [Google Scholar] [CrossRef]
- Wang, J.; Zhou, Y.; Zhang, H.; Hu, L.; Liu, J.; Wang, L.; Wang, T.; Zhang, H.; Cong, L.; Wang, Q. Pathogenesis of allergic diseases and implications for therapeutic interventions. Signal Transduct. Target. Ther. 2023, 8, 138. [Google Scholar] [CrossRef]
- Karvonen, A.M.; Hyvärinen, A.; Rintala, H.; Korppi, M.; Täubel, M.; Doekes, G.; Gehring, U.; Renz, H.; Pfefferle, P.I.; Genuneit, J.; et al. Quantity and diversity of environmental microbial exposure and development of asthma: A birth cohort study. Allergy 2014, 69, 1092–1101. [Google Scholar] [CrossRef]
- Remot, A.; Descamps, D.; Noordine, M.-L.; Boukadiri, A.; Mathieu, E.; Robert, V.; Riffault, S.; Lambrecht, B.; Langella, P.; Hammad, H.; et al. Bacteria isolated from lung modulate asthma susceptibility in mice. ISME J. 2017, 11, 1061–1074. [Google Scholar] [CrossRef]
- Eisenbarth, S.C.; Piggott, D.A.; Huleatt, J.W.; Visintin, I.; Herrick, C.A.; Bottomly, K. Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 2002, 196, 1645–1651. [Google Scholar] [CrossRef]
- Plaza, V.; Blanco, M.; García, G.; Korta, J.; Molina, J.; Quirce, S. Highlights of the Spanish Asthma Guidelines (GEMA), version 5.0. Arch. Bronconeumol 2021, 57, 11–12. [Google Scholar] [CrossRef]
- Balenga, N.A.; Klichinsky, M.; Xie, Z.; Chan, E.C.; Zhao, M.; Jude, J.; Laviolette, M.; Panettieri, R.A., Jr.; Druey, K.M. A fungal protease allergen provokes airway hyper-responsiveness in asthma. Nat. Commun. 2015, 6, 6763. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.D. Critical role of actin-associated proteins in smooth muscle contraction, cell proliferation, airway hyperresponsiveness and airway remodeling. Respir. Res. 2015, 16, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, R.; Gannon, O.J.; Rezey, A.C.; Jiang, S.; Gerlach, B.D.; Liao, G.; Tang, D.D. Polo-like Kinase 1 Regulates Vimentin Phosphorylation at Ser-56 and Contraction in Smooth Muscle. J. Biol. Chem. 2016, 291, 23693–23703. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Wang, Y.; Liao, G.; Chen, B.; Panettieri, R.A., Jr.; Penn, R.B.; Tang, D.D. Abi1 mediates airway smooth muscle cell proliferation and airway remodeling via Jak2/STAT3 signaling. iScience 2022, 25, 103833. [Google Scholar] [CrossRef]
- Liao, G.; Wang, R.; Tang, D.D. Plk1 Regulates Caspase-9 Phosphorylation at Ser-196 and Apoptosis of Human Airway Smooth Muscle Cells. Am. J. Respir. Cell Mol. Biol. 2022, 66, 223–234. [Google Scholar] [CrossRef]
- Hanada, M.; Feng, J.; Hemmings, B.A. Structure, regulation and function of PKB/AKT—A major therapeutic target. Biochim. Biophys. Acta 2004, 1697, 3–16. [Google Scholar] [CrossRef]
- Risco, A.; Miguel A Martin-Serrano, M.A.; Barber, D.F.; Cuenda, A. p38γ and p38d are involved in T lymphocyte development. Front. Immunol. 2018, 9, 65. [Google Scholar] [CrossRef]
- Barrio, L.; Román-García, S.; Díaz-Mora, E.; Risco, A.; Jiménez-Saiz, R.; Carrasco, Y.R.; Cuenda, A. B Cell Development and T-Dependent Antibody Response Are Regulated by p38γ and p38δ. Front. Cell Dev. Biol. 2020, 8, 189. [Google Scholar] [CrossRef]
- Shuai, K.; Liu, B. Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 2003, 11, 900–911. [Google Scholar] [CrossRef]
- Wang, W.; Lopez McDonald, M.C.; Kim, C.; Ma, M.; Pan, Z.T.; Kaufmann, C.; Frank, D.A. The complementary roles of STAT3 and STAT1 in cancer biology: Insights into tumor pathogenesis and therapeutic strategies. Front. Immunol. 2023, 14, 1265818. [Google Scholar] [CrossRef]
- Chiba, Y.; Todoroki, M.; Misawa, M. Antigen exposure causes activations of signal transducer and activator of transcription 6 (STAT6) and STAT1, but not STAT3, in lungs of sensitized mice. Immunopharmacol. Immunotoxicol. 2011, 33, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Quarcoo, D.; Weixler, S.; Groneberg, D.; Joachim, R.; Ahrens, B.; Wagner, A.H.; Hecker, M.; Hamelmann, E. Inhibition of signal transducer and activator of transcription 1 attenuates allergen-induced airway inflammation and hyperreactivity. J. Allergy Clin. Immunol. 2004, 114, 288–295. [Google Scholar] [CrossRef] [PubMed]
- Rao, G.; Mack, C.D.; Nguyen, T.; Wong, N.; Payne, K.; Worley, L.; Gray, P.E.; Wong, M.; Hsu, P.; Stormon, M.O.; et al. Inborn errors of immunity reveal molecular requirements for generation and maintenance of human CD4+ IL-9-expressing cells. J. Allergy Clin. Immunol. 2025, 155, 1161–1178. [Google Scholar] [CrossRef] [PubMed]
- Gangloff, M. Different dimerisation mode for TLR4 upon endosomal acidification? Trends Biochem. Sci. 2012, 37, 92–98. [Google Scholar] [CrossRef]
- Ren, Y.; Ichinose, T.; He, M.; Youshida, S.; Nishikawa, M.; Sun, G. Co-exposure to lipopolysaccharide and desert dust causes exacerbation of ovalbumin-induced allergic lung inflammation in mice via TLR4/MyD88-dependent and -independent pathways. Allergy Asthma Clin. Immunol. 2019, 15, 82. [Google Scholar] [CrossRef]
- Tan, A.M.; Chen, H.C.; Pochard, P.; Eisenbarth, S.C.; Herrick, C.A.; Bottomly, H.K. TLR4 Signaling in Stromal Cells Is Critical for the Initiation of Allergic Th2 Responses to Inhaled Antigen. J. Immunol. 2010, 184, 3535–3544. [Google Scholar] [CrossRef]
- Siwiec, J.; Zaborowski, T.; Jankowska, O.; Wojas-Krawczyk, K.; Krawczyk, P.; Milanowski, J. Evaluation of Th1/Th2 lymphocyte balance and lipopolysaccharide receptor expression in asthma patients. Pneumonol. Alergol. Pol. 2009, 77, 123–130. [Google Scholar]
- Kenny, E.F.; O’Neill, L.A.J. Signalling adaptors used by Toll-like receptors: An update. Cytokine 2008, 43, 342–349. [Google Scholar] [CrossRef]
- Laird, M.H.W.; Rhee, S.H.; Perkins, D.J.; Medvedev, A.E.; Piao, W.; Fenton, M.J.; Vogel, S.N. TLR4/MyD88/PI3K interactions regulate TLR4 signaling. J. Leukoc. Biol. 2009, 85, 966–977. [Google Scholar] [CrossRef]
- Bauerfeld, C.P.; Rastogi, R.; Pirockinaite, G.; Lee, I.; Hüttemann, M.; Monks, B.; Birnbaum, M.J.; Franchi, L.; Nuñez, G.; Samavati, L. TLR4-Mediated AKT Activation Is MyD88/TRIF Dependent and Critical for Induction of Oxidative Phosphorylation and Mitochondrial Transcription Factor A in Murine Macrophages. J. Immunol. 2012, 188, 2847–2857. [Google Scholar] [CrossRef]
- Risco, A.; del Fresno, C.; Mambol, A.; Alsina-Beauchamp, D.; MacKenzie, K.F.; Yang, H.-T.; Barber, D.F.; Morcelle, C.; Arthur, J.S.C.; Ley, S.C.; et al. P38γ and p38δ kinases regulate the Toll-like receptor 4 (TLR4)-induced cytokine production by controlling ERK1/2 protein kinase pathway activation. Proc. Natl. Acad. Sci. USA 2012, 109, 11200–11205. [Google Scholar] [CrossRef] [PubMed]
- Sikorski, K.; Chmielewski, S.; Olejnik, A.; Wesoly, J.Z.; Heemann, U.; Baumann, M.; Bluyssen, H. STAT1 as a central mediator of IFNγ and TLR4 signal integration in vascular dysfunction. JAK-STAT 2012, 1, 241–249. [Google Scholar] [CrossRef] [PubMed]
- Luu, K.; Greenhill, C.J.; Majoros, A.; Decker, T.; Jenkins, B.J.; Mansell, A. STAT1 plays a role in TLR signal transduction and inflammatory responses. Immunol. Cell Biol. 2014, 92, 761–769. [Google Scholar] [CrossRef]
- Nguyen, H.; Ramana, C.V.; Bayes, J.; Stark, G.R. Roles of Phosphatidylinositol 3-Kinase in Interferon-γ-dependent Phosphorylation of STAT1 on Serine 727 and Activation of Gene Expression. J. Biol. Chem. 2001, 276, 33361–33368. [Google Scholar] [CrossRef]
- Chen, K.; Xiang, Y.; Yao, X.; Liu, Y.; Gong, W.; Yoshimura, T.; Wang, J.M. The active contribution of Toll-like receptors to allergic airway inflammation. Int. Immunopharmacol. 2011, 11, 1391–1398. [Google Scholar] [CrossRef]
- Tang, L.; Chen, Q.; Meng, Z.; Sun, L.; Zhu, L.; Liu, J.; Hu, J.; Ni, Z.; Wang, X. Suppression of Sirtuin-1 Increases IL-6 Expression by Activation of the Akt Pathway during Allergic Asthma. Cell. Physiol. Biochem. 2017, 43, 1950–1960. [Google Scholar] [CrossRef]
- Fang, R.; Cui, Q.; Sun, J.; Duan, X.; Ma, X.; Wang, W.; Cheng, B.; Liu, Y.; Hou, Y.; Bai, G. PDK1/Akt/PDE4D axis identified as a target for asthma remedy synergistic with β2AR agonists by a natural agent arctigenin. Allergy 2015, 70, 1622–1632. [Google Scholar] [CrossRef]
- Dahlin, A.; Qiu, W.; Litonjua, A.A.; Lima, J.J.; Tamari, M.; Kubo, M.; Irvin, C.G.; Peters, S.P.; Wu, A.C.; Weiss, S.T.; et al. The phosphatidylinositide 3-kinase (PI3K) signaling pathway is a determinant of zileuton response in adults with asthma. Pharmacogenom J. 2018, 18, 665–677. [Google Scholar] [CrossRef]
- Zou, W.; Ding, F.; Niu, C.; Fu, Z.; Liu, S. Brg1 aggravates airway inflammation in asthma via inhibition of the PI3K/Akt/mTOR pathway. Biochem. Biophys. Res. Commun. 2018, 503, 3212–3218. [Google Scholar] [CrossRef]
- Cui, H.; Cheng, Y.; He, Y.; Cheng, W.; Zhao, W.; Zhao, H.; Zhou, F.H.; Wang, L.; Dong, J.; Cai, S. The AKT inhibitor MK2206 suppresses airway inflammation and the pro-remodeling pathway in a TDI-induced asthma mouse model. Mol. Med. Rep. 2020, 22, 3723–3734. [Google Scholar] [CrossRef]
- Gauthier, M.; Chakraborty, K.; Oriss, T.B.; Raundhal, M.; Das, S.; Chen, J.; Huff, R.; Sinha, A.; Fajt, M.; Ray, P.; et al. Severe asthma in humans and mouse models suggest a CXCL10 signature underlies corticosteroid-resistant Th1 bias. J. Clin. Investig. 2017, 2, e94580. [Google Scholar] [CrossRef] [PubMed]
- Lewis, B.W.; Jackson, D.; Amici, S.A.; Walum, J.; Guessas, M.; Guessas, S.; Coneglio, E.; Boda, A.V.; Guerau-De-Arellano, M.; Grayson, M.H.; et al. Corticosteroid Insensitivity Persists in the Absence of STAT1 Signaling in Severe Allergic Airway Inflammation. Am. J. Physiol. Cell. Mol. Physiol. 2021, 321, L1194–L1205. [Google Scholar] [CrossRef] [PubMed]
- McAlees, J.W.; Whitehead, G.S.; Harley, I.T.; Cappelletti, M.; Rewerts, C.L.; Holdcroft, A.M.; Divanovic, S.; Wills-Karp, M.; Finkelman, F.D.; Karp, C.L.; et al. Distinct Tlr4-expressing cell compartments control neutrophilic and eosinophilic airway inflammation. Mucosal Immunol. 2015, 8, 863–873. [Google Scholar] [CrossRef]
- Wan, B.; Xu, W.-J.; Chen, M.-Z.; Sun, S.-S.; Jin, J.-J.; Lv, Y.-L.; Zhan, P.; Zhu, S.-H.; Wang, X.-X.; Lv, T.-F.; et al. Geranylgeranyl diphosphate synthase 1 knockout ameliorates ventilator-induced lung injury via regulation of TLR2/4-AP-1 signaling. Free Radic Biol. Med. 2020, 147, 159–166. [Google Scholar] [CrossRef]
- Zakeri, A.; Russo, M. Dual role of toll-like receptors in human and experimental asthma models. Front. Immunol. 2018, 9, 1027. [Google Scholar] [CrossRef]
- Lim, J.O.; Kim, W.I.; Pak, S.W.; Lee, S.J.; Park, S.H.; Shin, I.S.; Kim, J.C. Toll-like receptor 4 is a key regulator of asthma exacerbation caused by aluminum oxide nanoparticles via regulation of NF-κB phosphorylation. J. Hazard. Mater. 2023, 448, 130884. [Google Scholar] [CrossRef]
- Ferreira, D.S.; Annoni, R.; Silva, L.F.F.; Buttignol, M.; Santos, A.B.G.; Medeiros, M.C.R.; Andrade, L.N.S.; Yick, C.Y.; Sterk, P.J.; Sampaio, J.L.M.; et al. Toll-like receptors 2, 3 and 4 and thymic stromal lymphopoietin expression in fatal asthma. Clin. Exp. Allergy 2012, 42, 1459–1471. [Google Scholar] [CrossRef]
- Shi, H.L.; Liu, J.B.; Lu, A.P. Expression profiles of PI3K, NF-κB, and STAT1 in peripheral blood mononuclear cells in children with bronchial asthma. Chin. J. Cont. Pediatr. 2016, 18, 614–617. [Google Scholar] [CrossRef]
- Alhetheel, A.; Albarrag, A.; Hakami, A.; Shakoor, Z.; Alswat, K.; Abdo, A.; Al-Hamoudi, W.; Alomar, S. In the peripheral blood mononuclear cells (PBMCs) of HCV infected patients the expression of STAT1 and IRF-1 is downregulated while that of caspase-3 upregulated. Acta Virol. 2020, 64, 352–358. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, P.; Zhao, Y.; Zhuang, Z.; Wang, Z.; Song, R.; Zhang, J.; Liu, C.; Gao, Q.; Xu, Q.; et al. Single-Cell Sequencing of Peripheral Mononuclear Cells Reveals Distinct Immune Response Landscapes of COVID-19 and Influenza Patients. Immunity 2020, 53, 685–696.e3. [Google Scholar] [CrossRef]
- Ehrlich, M.; Sanchez, C.; Shao, C.; Nishiyama, R.; Kehrl, J.; Kuick, R.; Kubota, T.; Hanash, S.M. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity 2008, 41, 253–271. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Smit, M.A.; Oord, J.J.v.D.; Goeman, J.J.; Verdegaal, E.M.E.; van der Burg, S.H.; Stas, M.; Beck, S.; Gruis, N.A.; Tensen, C.P.; et al. Genome-wide promoter methylation analysis identifies epigenetic silencing of MAPK13 in primary cutaneous melanoma. Pigment. Cell Melanoma Res. 2013, 26, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Lun, S.W.M.; Wong, C.K.; Ko, F.W.S.; Hui, D.S.C.; Lam, C.W.K. Expression and functional analysis of toll-like receptors of peripheral blood cells in asthmatic patients: Implication for immunopathological mechanism in asthma. J. Clin. Immunol. 2009, 29, 330–342. [Google Scholar] [CrossRef] [PubMed]
- Bruscia, E.M.; Zhang, P.-X.; Satoh, A.; Caputo, C.; Medzhitov, R.; Shenoy, A.; E Egan, M.; Krause, D.S. Abnormal Trafficking and Degradation of TLR4 Underlie the Elevated Inflammatory Response in Cystic Fibrosis. J. Immunol. 2011, 186, 6990–6998. [Google Scholar] [CrossRef]
- Ciesielska, A.; Matyjek, M.; Kwiatkowska, K. TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell. Mol. Life Sci. 2021, 78, 1233–1261. [Google Scholar] [CrossRef]
- Knobloch, J.; Jungck, D.; Kronsbein, J.; Stoelben, E.; Ito, K.; Koch, A. LABAs and p38MAPK Inhibitors Reverse the Corticosteroid-Insensitivity of IL-8 in Airway Smooth Muscle Cells of COPD. J. Clin. Med. 2019, 8, 2058. [Google Scholar] [CrossRef]
- Zhai, Y.; Zheng, P.; Sun, B.; Li, J.; Wang, B. Allergen-specific immunotherapy with Alutard SQ improves allergic inflammation in house-dust mites-induced allergic asthma rats through inactivation of the HMGB1/TLR4/NF-κB pathway. J. Thorac. Dis. 2023, 15, 77–89. [Google Scholar] [CrossRef]
- Tian, B.; Widen, S.G.; Yang, J.; Wood, T.G.; Kudlicki, A.; Zhao, Y.; Brasier, A.R. The NFκB subunit RELA is a master transcriptional regulator of the committed epithelial-mesenchymal transition in airway epithelial cells. J. Biol. Chem. 2018, 293, 16528–16545. [Google Scholar] [CrossRef]
- Gu, W.; Lei, J.; Zhu, H.; Xiao, Y.; Zhang, Z.; Zhao, L. Effect of the BMPR-II-SMAD3/MRTF pathway on proliferation and migration of ASMCs and the mechanism in asthma. Mol. Biol. Rep. 2022, 49, 9283–9296. [Google Scholar] [CrossRef]
- Yang, Z.; Krammer, S.; Mitländer, H.; Grund, J.C.; Zirlik, S.; Wirtz, S.; Rauh, M.; Shermeh, A.S.; Finotto, S. NFATc1 in CD4+ T cells and CD11c+ dendritic cells drives TH2-mediated eosinophilic inflammation in allergic asthma. J. Allergy Clin. Immunol. Glob. 2024, 4, 100355. [Google Scholar] [CrossRef]
- Drazen, J.M.; Yandava, C.N.; Dubé, L.; Szczerback, N.; Hippensteel, R.; Pillari, A.; Israel, E.; Schork, N.; Silverman, E.S.; Katz, D.A. Pharmacogenetic association between ALOX5 promoter genotype and the response to anti-asthma treatment. Nat. Genet. 1999, 22, 168–170. Available online: http://genetics.nature.com (accessed on 16 June 2025). [CrossRef] [PubMed]
- Mougey, E.; Lang, J.E.; Allayee, H.; Teague, W.G.; Dozor, A.J.; Wise, R.A.; Lima, J.J. ALOX5 Polymorphism associates with increased leukotriene production and reduced lung function and asthma control in children with poorly controlled asthma. Clin. Exp. Allergy 2013, 43, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Alhamdan, F.; Marsh, L.M.; Pedersen, F.; Alhamwe, B.A.; Thölken, C.; Pfefferle, P.I.; Bahmer, T.; Greulich, T.; Potaczek, D.P.; Garn, H. Differential Regulation of Interferon Signaling Pathways in CD4+ T Cells of the Low Type-2 Obesity-Associated Asthma Phenotype. Int. J. Mol. Sci. 2021, 22, 10144. [Google Scholar] [CrossRef] [PubMed]
- Bantulà, M.; Arismendi, E.; Tubita, V.; Roca-Ferrer, J.; Mullol, J.; de Hollanda, A.; Sastre, J.; Valero, A.; Baos, S.; Cremades-Jimeno, L.; et al. Effect of Obesity on the Expression of Genes Associated with Severe Asthma—A Pilot Study. J. Clin. Med. 2023, 12, 4398. [Google Scholar] [CrossRef]
- Muñoz, X.; Álvarez-Puebla, M.J.; Arismendi, E.; Arochena, L.; Ausín, M.d.P.; Barranco, P.; Bobolea, I.; Cañas, J.A.; Cardaba, B.; Crespo, A.; et al. The MEGA Project: A Study of the Mechanisms Involved in the Genesis and Disease Course of Asthma. Asthma Cohort Creation and Long-Term Follow-Up. Arch. Bronconeumol. 2018, 54, 378–385. [Google Scholar] [CrossRef]
- Baos, S.; Calzada, D.; Cremades, L.; Sastre, J.; Quiralte, J.; Florido, F.; Lahoz, C.; Cárdaba, B. Biomarkers associated with disease severity in allergic and nonallergic asthma. Mol. Immunol. 2017, 82, 34–45. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| HC | AA | NA | |
|---|---|---|---|
| N | 15 | 45 | 17 |
| Age (y.o), mean ± SD | 32.87 ± 10.8 | 48.02 ± 12 ## | 54.88 ± 11 ## |
| Gender, N (%) | |||
| Male | 7 (46.7) | 12 (26.7) | 7 (41.2) |
| Female | 8 (53.3) | 33 (73.3) | 10 (58.8) |
| Smoking habit, N (%) | |||
| Smoker | 5 (11.1) | 2 (11.8) | |
| Ex- Smoker | 13 (28.9) | 9 (52.9) | |
| No Smoker | 25 (55.6) | 6 (35.3) | |
| ND | 15 (100) | 2 (4.4) | 0 (0) |
| BMI (Kg/m2), mean ± SD | 23.6 ± 1.3 | 25.87 ± 4.5 | 27.5 ± 4.8 * |
| Asthma severity, N (%) | |||
| Severe | - | 17 (37.8) | 4 (23.5) |
| Moderate | - | 14 (31.1) | 8 (47) |
| Mild | - | 14 (31.1) | 5 (29.5) |
| Spirometry, mean ± SD | |||
| FEV1 (%) | 100.82 ± 7.9 | 87.74 ± 20 | 96.89 ± 21.8 |
| FVC (%) | 104 ± 6.9 | 98.88 ± 18.7 | 110.1 ± 20.6 |
| FEV1/FVC | 82.53 ± 2.2 | 77.09 ± 9.8 | 81.78 ± 14.4 |
| FeNO (ppb), mean ± SD | 11.1 ± 5.9 | 38.4 ± 30.7 ** | 32.38 ± 27 |
| Total IgE (IU/mL), mean ± SD | 63.55 ± 94.7 | 436.71 ± 493.1 #,† | 159.95 ± 173.2 * |
| Blood cells | |||
| Eosinophils (cells/mm3), mean ± SD | 280 ± 168.7 | 324.67 ± 206.1 | 424.8 ± 205 |
| Neutrophils (cells/mm3), mean ± SD | ND | 3823 ± 1574.6 | 4438.33 ± 1172.6 |
| Sputum cells | |||
| Eosinophils (%), mean ± SD | ND | 9.6 ± 17.4 | 16 ± 27.9 |
| ≥3% eosinophils, N (%) | ND | 10 (22.2) | 5 (29.4) |
| Neutrophils (%), mean ± SD | ND | 29.4 ± 24 | 40 ± 30.4 |
| ≥70% neutrophils, N (%) | ND | 4 (8.8) | 3 (17.6) |
| Cellular profile, N (%) | |||
| Eosinophilic | ND | 9 (20) | 5 (29.4) |
| Neutrophilic | ND | 2 (4.4) | 3 (17.7) |
| Mixed | ND | 1 (2.2) | 0 (0) |
| Paucigranulocyte | ND | 13 (28.9) | 4 (23.5) |
| ND | ND | 19 (42.2) | 5 (29.4) |
| Treatment, N (%) | |||
| Inhaled Corticosteroids (ICS) | - | 43 (95.5) | 17 (100) |
| Systemic Corticosteroids | - | 1 (2.2) | 2 (11.8) |
| Long-term β2-Agonists (LABA) | - | 39 (86.7) | 17 (100) |
| Short-term anticholinergics | - | 1 (2.2) | 1 (5.9) |
| Long-term anticholinergics | - | 9 (2) | 2 (11.8) |
| Leukotriens receptor agonists | - | 12 (26.7) | 3 (17.7) |
| Short-term bronchodilators | - | 18 (40) | 7 (41.2) |
| Specific immunotherapy | - | 12 (26.7) | 0 (0) |
| Biologics (Omaluzimab, Mepaluzimab) | - | 14 (31.1), 4 (8.8) | 2 (11.8), 1(5.9) |
| ||||
| AKT1 | MAPK13 | STAT1 | TLR4 | |
| AKT1 | Rs = 0.97; p < 0.001 | Rs = 0.73; p = 0.002 | Rs = 0.60; p = 0.02 | |
| MAPK13 | Rs = 0.82; p = 0.004 | Rs = 0.69; p = 0.005 | Rs = 0.64; p = 0.01 | |
| STAT1 | Rs = 0.51; p = ns | Rs = 0.57; p = ns | Rs = 0.55; p = 0.04 | |
| TLR4 | Rs = 0.89; p = 0.001 | Rs = −0.19; p = ns | Rs = 0.74; p = 0.02 | |
| ||||
| AKT1 | MAPK13 | STAT1 | TLR4 | |
| AKT1 | Rs = 0.86; p < 0.001 | Rs = 0.92; p < 0.001 | Rs = 0.71; p < 0.001 | |
| MAPK13 | Rs = −0.41; p = ns | Rs = 0.83; p < 0.001 | Rs = 0.60; p < 0.001 | |
| STAT1 | Rs = 0.10; p = ns | Rs = 0.74; p = 0.01 | Rs = 0.75; p < 0.001 | |
| TLR4 | Rs = −0.47; p = ns | Rs = 0.59; p = ns | Rs = 0.84; p < 0.001 | |
| ||||
| AKT1 | MAPK13 | STAT1 | TLR4 | |
| AKT1 | Rs = 0.92; p < 0.001 | Rs = 0.94; p < 0.001 | Rs = 0.53; p = 0.04 | |
| MAPK13 | Rs = −0.53; p = ns | Rs = 0.83; p < 0.001 | Rs = 0.53; p = 0.04 | |
| STAT1 | Rs = −0.35; p = ns | Rs = −0.36; p = ns | Rs = 0.53; p = 0.04 | |
| TLR4 | Rs = 0.24; p = ns | Rs = −0.62; p = 0.05 | Rs = 0.55; p = ns | |
| |||||
| FEV1 | FVC | FEV/FVC | PBE | ||
| AKT1 | Gene | Rs = −0.28; p = ns | Rs = −0.30; p = ns | Rs = −0.15; p = ns | Rs = 0.28; p = ns |
| Protein | Rs = −0.59; p = ns | Rs = −0.62; p = ns | Rs = −0.16; p = ns | Rs = −0.16; p = ns | |
| MAPK13 | Gene | Rs = −0.39; p = 0.009 | Rs = −0.36; p = 0.02 | Rs = −0.13; p = ns | Rs = 0.07; p = ns |
| Protein | Rs = 0.27; p = ns | Rs = 0.36; p = ns | Rs = −0.38; p = ns | Rs = −0.19; p = ns | |
| STAT1 | Gene | Rs = −0.41; p = 0.006 | Rs = −0.41; p = 0.005 | Rs = −0.25; p = ns | Rs = 0.30; p = 0.04 |
| Protein | Rs = −0.63; p = 0.01 | Rs = −0.41; p = ns | Rs = −0.14; p = ns | Rs = 0.15; p = ns | |
| TLR4 | Gene | Rs = −0.19; p = ns | Rs = −0.23; p = ns | Rs = −0.41; p = 0.009 | Rs = 0.15; p = ns |
| Protein | Rs = −0.62; p = 0.02 | Rs = −0.62; p = 0.01 | Rs = 0.24; p = ns | Rs = 0.15; p = ns | |
| |||||
| FEV1 | FVC | FEV/FVC | PBE | ||
| AKT1 | Gene | Rs = 0.24; p = ns | Rs = 0.45; p = ns | Rs = 0.39; p = ns | Rs = 0.06; p = ns |
| Protein | Rs = −0.08; p = ns | Rs = 0.53; p = ns | Rs = −0.25; p = ns | Rs = 0.02; p = ns | |
| MAPK13 | Gene | Rs = 0.17; p = ns | Rs = 0.36; p = ns | Rs = 0.49; p = 0.04 | Rs = 0.06; p = ns |
| Protein | Rs = 0.04; p = ns | Rs = 0.57; p = ns | Rs = 0.19; p = ns | Rs = −0.24; p = ns | |
| STAT1 | Gene | Rs = 0.25; p = ns | Rs = 0.46; p = ns | Rs = 0.36; p = ns | Rs = 0.09; p = ns |
| Protein | Rs = 0.43; p = ns | Rs = 0.58; p = 0.04 | Rs = −0.06; p = ns | Rs = −0.56; p = 0.04 | |
| TLR4 | Gene | Rs = −0.22; p = ns | Rs = 0.16; p = ns | Rs = −0.15; p = ns | Rs = −0.11; p = ns |
| Protein | Rs = 0.65; p = 0.03 | Rs = 0.85; p = 0.002 | Rs = 0.20; p = ns | Rs = −0.13; p = ns | |
| ||||||||||
| Gene | Gene Classification | Total AA | Severe AA | Moderate AA | Mild AA | |||||
| RQ | Adj P | RQ | Adj P | RQ | Adj P | RQ | Adj P | |||
| AKT1 pathway | AKT1 | Trigger | 0.177 | <0.001 | 0.109 | <0.001 | 0.154 | <0.001 | 0.354 | ns |
| COL5A1 | Link Node | 0.177 | 0.004 | 0.067 | <0.001 | 0.203 | ns | 0.467 | ns | |
| MMP9 | 0.041 | <0.001 | 0.027 | <0.001 | 0.033 | 0.006 | 0.082 | ns | ||
| RELA | 0.165 | <0.001 | 0.088 | <0.001 | 0.154 | <0.001 | 0.379 | ns | ||
| SMAD3 | 0.25 | <0.001 | 0.134 | <0.001 | 0.218 | <0.001 | 0.616 | ns | ||
| TLR2 | 0.102 | <0.001 | 0.051 | <0.001 | 0.117 | <0.001 | 0.177 | ns | ||
| MAPK13 pathway | MAPK13 | Trigger | 0.177 | <0.001 | 0.109 | <0.001 | 0.154 | <0.001 | 0.354 | ns |
| IL12A | Link Node | 0.218 | 0.011 | 0.154 | <0.001 | 0.165 | 0.018 | 0.435 | ns | |
| IL13RA1 | 0.117 | <0.001 | 0.072 | <0.001 | 0.125 | <0.001 | 0.177 | 0.029 | ||
| NFATC1 | 0.144 | <0.001 | 0.067 | <0.001 | 0.117 | <0.001 | 0.5 | ns | ||
| RELA | 0.165 | <0.001 | 0.088 | <0.001 | 0.154 | <0.001 | 0.379 | ns | ||
| STAT1 pathway | STAT1 | Trigger | 0.117 | <0.001 | 0.063 | <0.001 | 0.109 | <0.001 | 0.287 | ns |
| CCL5 | Link Node | 0.145 | <0.001 | 0.067 | <0.001 | 0.127 | <0.001 | 0.412 | ns | |
| COL5A1 | 0.177 | 0.004 | 0.067 | <0.001 | 0.203 | ns | 0.467 | ns | ||
| IL10 | 0.067 | <0.001 | 0.038 | <0.001 | 0.088 | 0.008 | 0.117 | ns | ||
| IL12A | 0.218 | 0.011 | 0.154 | 0.001 | 0.165 | 0.018 | 0.435 | ns | ||
| MMP9 | 0.041 | <0.001 | 0.027 | <0.001 | 0.033 | 0.006 | 0.082 | ns | ||
| RELA | 0.165 | <0.001 | 0.088 | <0.001 | 0.154 | <0.001 | 0.379 | ns | ||
| TLR4 pathway | TLR4 | Trigger | 0.072 | <0.001 | 0.038 | <0.001 | 0.072 | <0.001 | 0.144 | ns |
| MMP9 | Link Node | 0.041 | <0.001 | 0.027 | <0.001 | 0.033 | 0.006 | 0.082 | ns | |
| RELA | 0.165 | <0.001 | 0.088 | <0.001 | 0.154 | <0.001 | 0.379 | ns | ||
| SMAD3 | 0.250 | <0.001 | 0.134 | <0.001 | 0.218 | <0.001 | 0.616 | ns | ||
| Common | ALOX5 | Mechanistic | 0.102 | <0.001 | 0.044 | <0.001 | 0.088 | <0.001 | 0.287 | ns |
| BAX | 0.233 | <0.001 | 0.154 | <0.001 | 0.203 | <0.001 | 0.5 | ns | ||
| IL4R | 0.134 | <0.001 | 0.063 | <0.001 | 0.117 | <0.001 | 0.435 | ns | ||
| TGFB1 | 0.180 | <0.001 | 0.11 | <0.001 | 0.157 | <0.001 | 0.337 | ns | ||
| ||||||||||
| Gene | Gene classification | Total NA | Severe NA | Moderate NA | Mild NA | |||||
| RQ | Adj P | RQ | Adj P | RQ | Adj P | RQ | Adj P | |||
| AKT1 pathway | AKT1 | Trigger | 0.067 | <0.001 | 0.072 | 0.006 | 0.067 | <0.001 | 0.054 | <0.001 |
| CDH1 | Link Node | 0.044 | <0.001 | 0.077 | ns | 0.038 | <0.001 | 0.036 | <0.001 | |
| RELA | 0.062 | <0.001 | 0.054 | 0.002 | 0.072 | <0.001 | 0.058 | <0.001 | ||
| SMAD3 | 0.125 | <0.001 | 0.117 | 0.002 | 0.134 | <0.001 | 0.109 | <0.001 | ||
| TLR2 | 0.044 | <0.001 | 0.027 | 0.002 | 0.036 | <0.001 | 0.088 | 0.004 | ||
| MAPK13 pathway | MAPK13 | Trigger | 0.036 | <0.001 | 0.022 | 0.004 | 0.033 | <0.001 | 0.058 | <0.001 |
| CXCL8 | Link Node | 0.058 | ns | 0.054 | ns | 0.054 | ns | 0.072 | ns | |
| NFATC1 | 0.067 | <0.001 | 0.063 | 0.002 | 0.067 | <0.001 | 0.077 | <0.001 | ||
| RELA | 0.062 | <0.001 | 0.054 | 0.002 | 0.072 | <0.001 | 0.058 | <0.001 | ||
| STAT1 pathway | STAT1 | Trigger | 0.051 | <0.001 | 0.044 | 0.002 | 0.063 | <0.001 | 0.047 | <0.001 |
| CXCR3 | Link Node | 0.077 | <0.001 | 0.088 | 0.019 | 0.077 | <0.001 | 0.082 | 0.001 | |
| RELA | 0.062 | <0.001 | 0.054 | 0.002 | 0.072 | <0.001 | 0.058 | <0.001 | ||
| VCAN | 0.058 | <0.001 | 0.041 | 0.002 | 0.054 | <0.001 | 0.095 | <0.001 | ||
| TLR4 pathway | TLR4 | Trigger | 0.036 | <0.001 | 0.022 | 0.004 | 0.033 | <0.001 | 0.058 | <0.001 |
| CDH1 | Link Node | 0.044 | <0.001 | 0.077 | ns | 0.038 | <0.001 | 0.036 | <0.001 | |
| RELA | 0.062 | <0.001 | 0.054 | 0.002 | 0.072 | <0.001 | 0.058 | <0.001 | ||
| SMAD3 | 0.125 | <0.001 | 0.117 | 0.002 | 0.134 | <0.001 | 0.109 | <0.001 | ||
| TLR5 | 0.051 | <0.001 | 0.102 | ns | 0.038 | <0.001 | 0.054 | 0.002 | ||
| Common | ALOX5 | Mechanistic | 0.038 | <0.001 | 0.038 | 0.002 | 0.038 | <0.001 | 0.036 | <0.001 |
| CCL5 | 0.058 | <0.001 | 0.051 | 0.002 | 0.063 | <0.001 | 0.054 | <0.001 | ||
| PTGER2 | 0.067 | <0.001 | 0.036 | 0.002 | 0.077 | <0.001 | 0.077 | <0.001 | ||
| Gene | Gene Classification | AA RQ | NA RQ | Adjusted P | |
|---|---|---|---|---|---|
| AKT1 Pathway | AKT1 | Trigger | 0.177 | 0.067 | 0.008 |
| RELA | Link Node | 0.165 | 0.062 | <0.001 | |
| SMAD3 | 0.250 | 0.125 | 0.021 | ||
| TLR2 | 0.102 | 0.044 | 0.022 | ||
| MAPK13 Pathway | MAPK13 | Trigger | 0.177 | 0.036 | 0.021 |
| NFATC1 | Link Node | 0.144 | 0.067 | 0.028 | |
| RELA | 0.165 | 0.062 | <0.001 | ||
| STAT1 pathway | STAT1 | Trigger | 0.117 | 0.051 | 0.007 |
| RELA | Link Node | 0.165 | 0.062 | <0.001 | |
| TLR4 pathway | TLR4 | Trigger | 0.072 | 0.036 | ns |
| RELA | Link Node | 0.165 | 0.062 | <0.001 | |
| SMAD3 | 0.250 | 0.125 | 0.021 | ||
| Commons | ALOX5 | Mechanistic | 0.102 | 0.038 | 0.011 |
| Gene | Definition of the Gene According to Biology Systems [12] | Chromosome Location (GRCh38/hg38) | Primer Reference |
|---|---|---|---|
| AKT1 | Trigger | Chr.14: 104769349–104795743 | Hs00178289_m1 |
| ALOX5 | Mechanistic in NA and AA | Chr.10: 45374166–45446121 | Hs01095330_m1 |
| BAX | Mechanistic in AA | Chr.19: 48954825–48961798 | Hs00180269_m1 |
| CCL5 | Mechanistic in NA, link node in AA | Chr.17: 35871491–35880373 | Hs00982282_m1 |
| CCL11 | Mechanistic in NA | Chr.17: 34285668–34288180 | Hs00237013_m1 |
| CCL17 | Link node in NA | Chr.16: 57396076–57416063 | Hs00171074_m1 |
| CDH1 | Link node in NA | Chr.16: 68737290–68835542 | Hs01023895_m1 |
| COL5A1 | Link node in AA | Chr.9: 134641790–134844843 | Hs00609088_m1 |
| CXCR3 | Link node in NA | Chr.X: 71615913–71618517 | Hs01847760_s1 |
| IL4R | Mechanistic in AA | Chr.16: 27313668–27364778 | Hs00166237_m1 |
| IL8 (CXCL8) | Link node in NA | Chr.4: 73740506–73743716 | Hs00174103_m1 |
| IL10 | Link node in AA | Chr.1: 206767603–206772494 | Hs00961622_m1 |
| IL12A | Link node in AA | Chr.3: 159988836–159996019 | Hs01073447_m1 |
| IL12B | Link node in AA | Chr.5: 159314783–159330473 | Hs01011518_m1 |
| IL13RA1 | Link node in AA | Chr.X: 118726954–118794533 | Hs00609817_m1 |
| IL17A | Mechanistic in NA and AA | Chr.6: 52186387–52190638 | Hs00174383_m1 |
| MAPK13 | Trigger | Chr.6: 36130484–36144524 | Hs00559623_m1 |
| MUC5B | Mechanistic in NA and AA | Chr.11: 1223065–1262176 | Hs00861595_m1 |
| MMP9 | Link node in AA | Chr.20: 46008908–46016561 | Hs00957562_m1 |
| NFATC1 | Link node in NA and AA | Chr.18: 79395772–79529323 | Hs00542678_m1 |
| PTGER2 | Mechanistic in NA | Chr.14: 52314298–52328606 | Hs04183523_m1 |
| RELA | Link node in NA and AA | Chr.11: 65653596–65662972 | Hs00153294_m1 |
| SMAD3 | Link node in NA and AA | Chr.15: 67065698–67195195 | Hs00969210_m1 |
| STAT1 | Trigger | Chr.2: 190969036–191014250 | Hs01013996_m1 |
| TGFB1 | Mechanistic in AA | Chr.19: 41330531–41353933 | Hs00998133_m1 |
| TLR2 | Link node in NA and AA | Chr.4: 153684080–153710643 | Hs00152932_m1 |
| TLR4 | Trigger | Chr.9: 117704175–117717491 | Hs00152939_m1 |
| TLR5 | Link node in NA | Chr.1: 223108401–223143282 | Hs01920773_s1 |
| VCAN | Link node in NA | Chr.5: 83471674–83582303 | Hs00171642_m1 |
| 18S | Endogenous | - | Hs99999901_s1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cremades-Jimeno, L.; López-Ramos, M.; Fernández-Santamaría, R.; De Pedro, M.Á.; Mahillo, I.; Rosales-Ariza, C.; Olaguibel, J.M.; Pozo, V.d.; Caballero, M.L.; Luna-Porta, J.A.; et al. Molecular Study from the Signaling Pathways of Four Potential asthma triggers: AKT1, MAPK13, STAT1, and TLR4. Int. J. Mol. Sci. 2025, 26, 6240. https://doi.org/10.3390/ijms26136240
Cremades-Jimeno L, López-Ramos M, Fernández-Santamaría R, De Pedro MÁ, Mahillo I, Rosales-Ariza C, Olaguibel JM, Pozo Vd, Caballero ML, Luna-Porta JA, et al. Molecular Study from the Signaling Pathways of Four Potential asthma triggers: AKT1, MAPK13, STAT1, and TLR4. International Journal of Molecular Sciences. 2025; 26(13):6240. https://doi.org/10.3390/ijms26136240
Chicago/Turabian StyleCremades-Jimeno, Lucía, María López-Ramos, Rubén Fernández-Santamaría, María Ángeles De Pedro, Ignacio Mahillo, Cristina Rosales-Ariza, José María Olaguibel, Victoria del Pozo, María Luisa Caballero, Juan Alberto Luna-Porta, and et al. 2025. "Molecular Study from the Signaling Pathways of Four Potential asthma triggers: AKT1, MAPK13, STAT1, and TLR4" International Journal of Molecular Sciences 26, no. 13: 6240. https://doi.org/10.3390/ijms26136240
APA StyleCremades-Jimeno, L., López-Ramos, M., Fernández-Santamaría, R., De Pedro, M. Á., Mahillo, I., Rosales-Ariza, C., Olaguibel, J. M., Pozo, V. d., Caballero, M. L., Luna-Porta, J. A., Quirce, S., Barroso, B., Betancor, D., Valverde-Monge, M., Sastre, J., Baos, S., & Cárdaba, B. (2025). Molecular Study from the Signaling Pathways of Four Potential asthma triggers: AKT1, MAPK13, STAT1, and TLR4. International Journal of Molecular Sciences, 26(13), 6240. https://doi.org/10.3390/ijms26136240