
Substituted Triazole-3,5-Diamine Compounds as Novel Human Topoisomerase III Beta Inhibitors

 ,
,  , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Results
2.1. Hits from Virtual Screening of Library of Compounds That Are FDA-Approved or in Clinical Trials
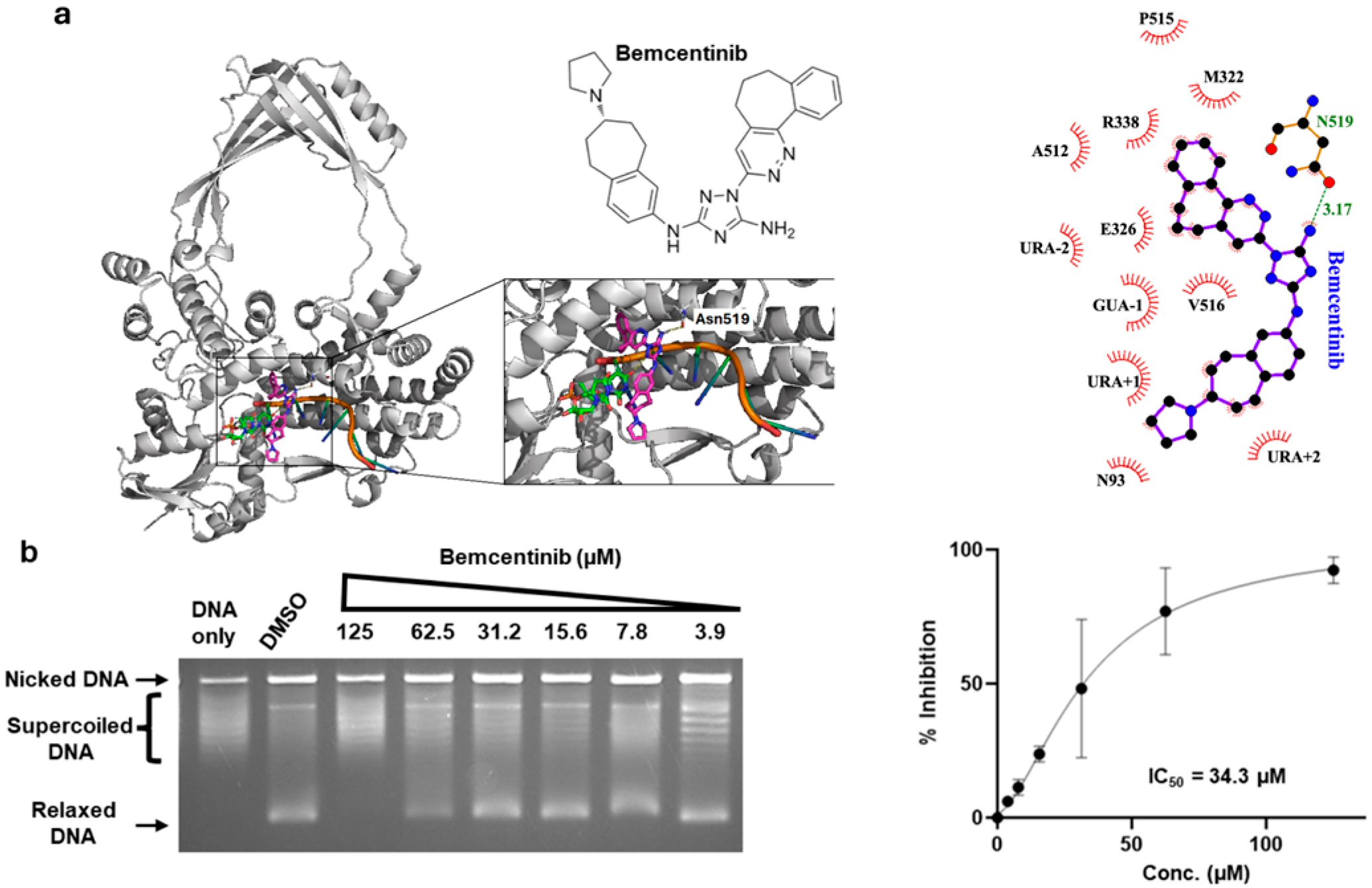
2.2. Inhibition of Human TOP3B Relaxation Activity by Bemcentinib
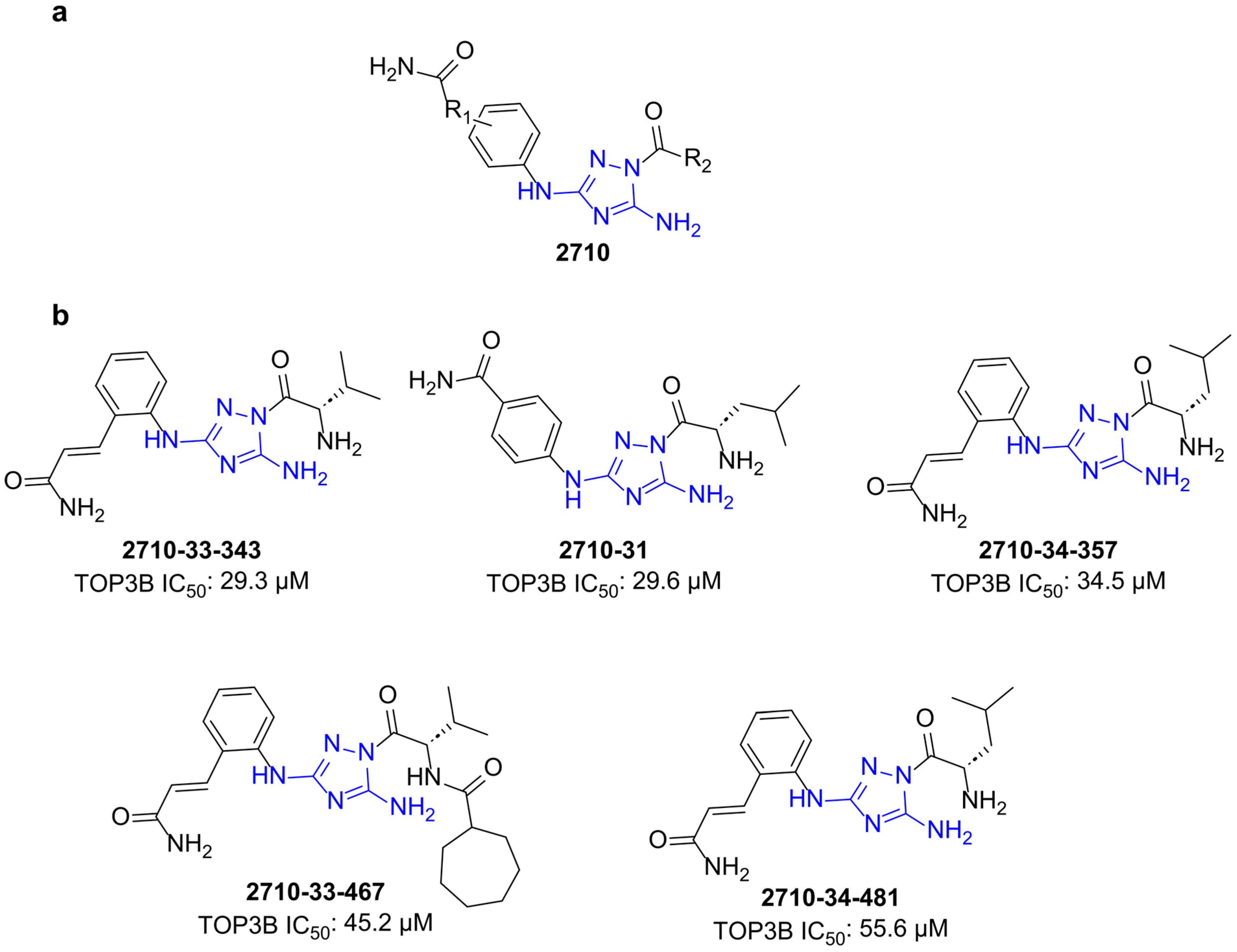
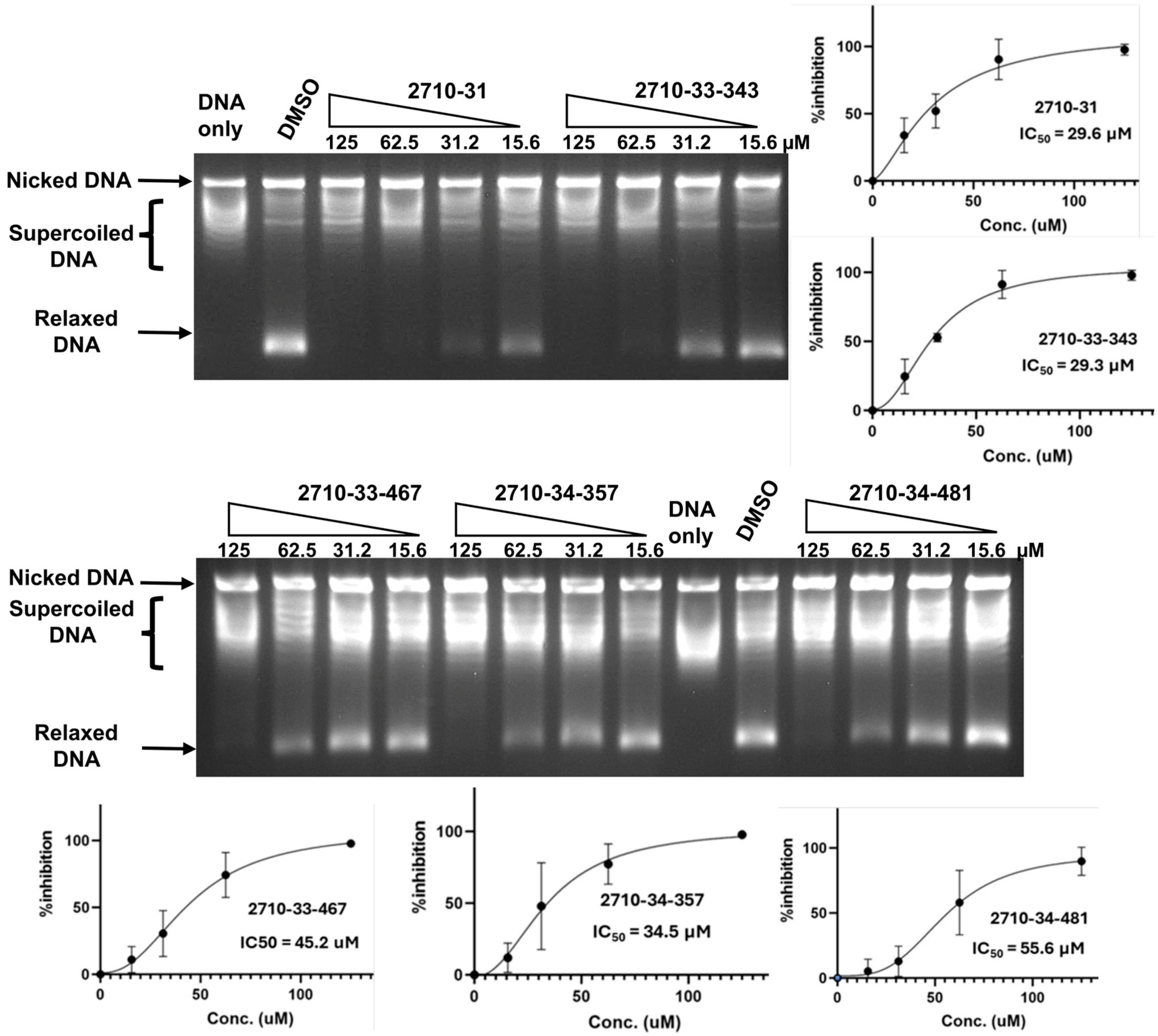
2.3. Testing of Additional Small Molecules for Inhibition of Human TOP3B Relaxation Activity
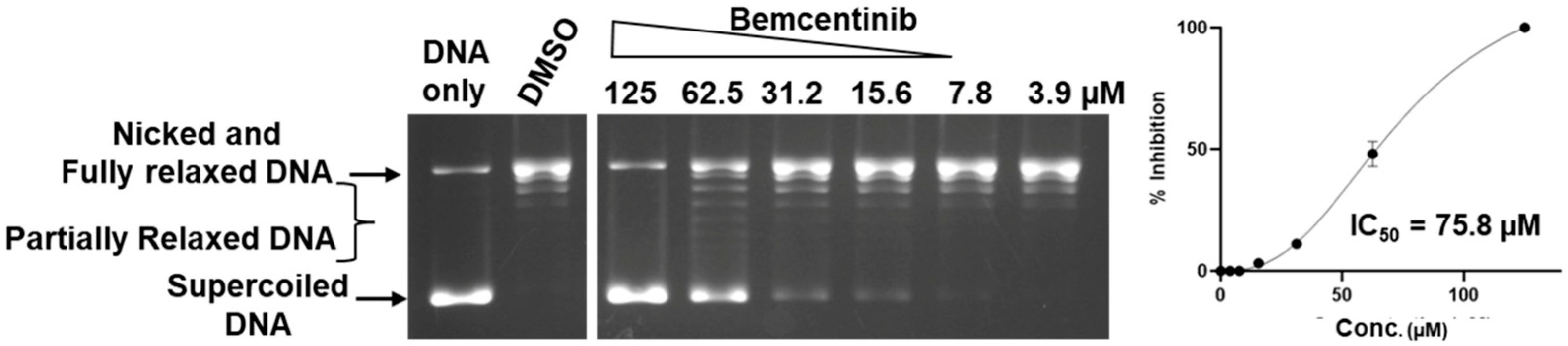
2.4. Comparison of Selectivity for Inhibition of Human TOP3B Versus TOP1
3. Discussion
4. Materials and Methods
4.1. Molecular Dynamics Simulation
4.2. Molecular Docking of Compounds
4.3. Assay of TOP3B Activity Inhibition
4.4. Assay of Inhibition of Human TOP1 Relaxation Activity
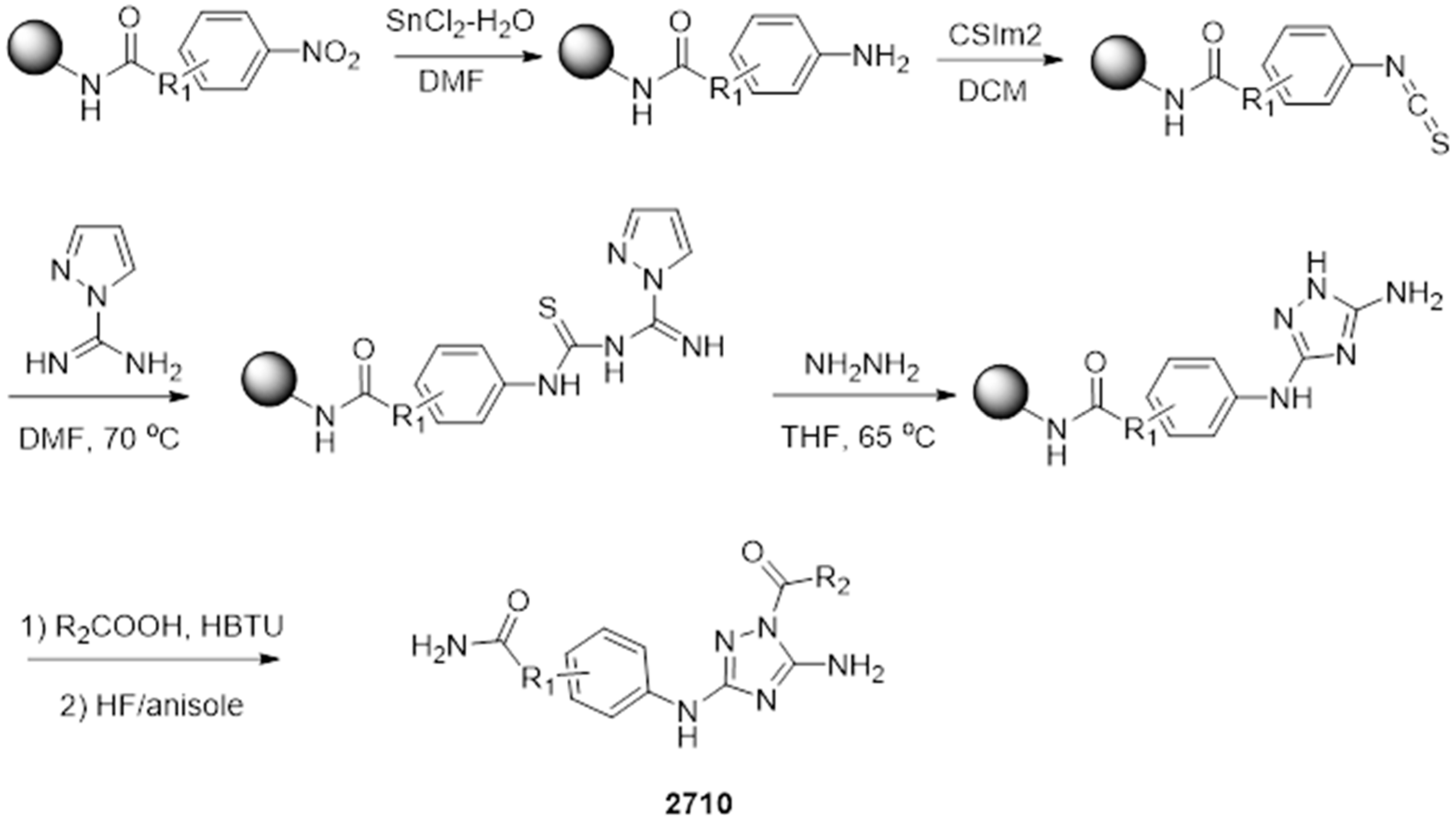
4.5. Synthesis of Small Molecules with Disubstituted N5,N3-1H-1,2,4-Triazole-3,5-Diamine
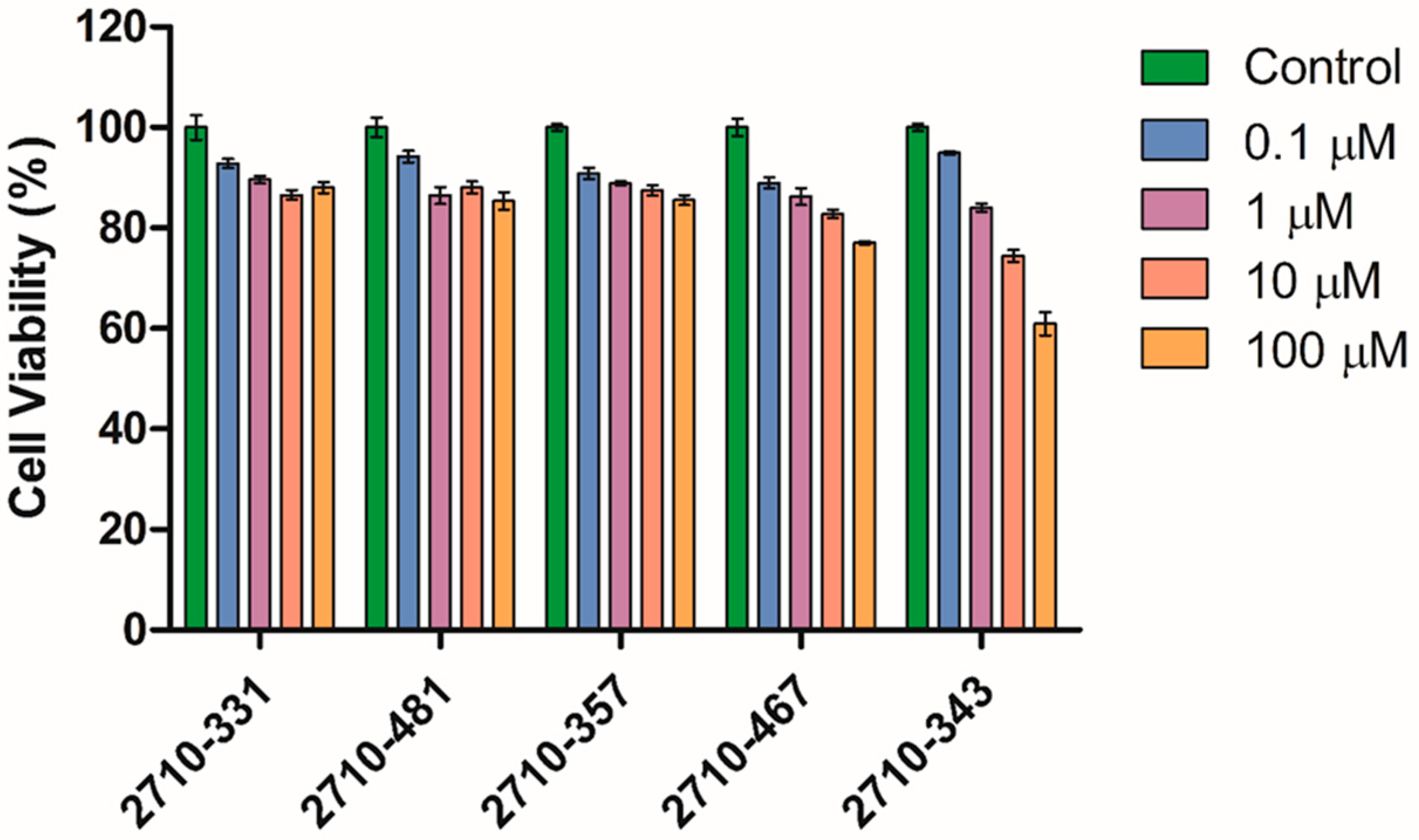
4.6. Dose Response of 2710-Inhibitors on Cell Viability of Human Dermal Fibroblasts (HDFa)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McKie, S.J.; Neuman, K.C.; Maxwell, A. DNA topoisomerases: Advances in understanding of cellular roles and multi-protein complexes via structure-function analysis. Bioessays 2021, 43, e2000286. [Google Scholar] [CrossRef] [PubMed]
- Vos, S.M.; Tretter, E.M.; Schmidt, B.H.; Berger, J.M. All tangled up: How cells direct, manage and exploit topoisomerase function. Nat. Rev. Mol. Cell Biol. 2011, 12, 827–841. [Google Scholar] [CrossRef]
- Wang, J.C. Cellular roles of DNA topoisomerases: A molecular perspective. Nat. Rev. Mol. Cell Biol. 2002, 3, 430–440. [Google Scholar] [CrossRef]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans-Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Baker, N.M.; Rajan, R.; Mondragón, A. Structural studies of type I topoisomerases. Nucleic Acids Res. 2009, 37, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Corbett, K.D.; Berger, J.M. Structure, molecular mechanisms, and evolutionary relationships in DNA topoisomerases. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 95–118. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Shen, W.; Guo, R.; Xue, Y.; Peng, W.; Sima, J.; Yang, J.; Sharov, A.; Srikantan, S.; Yang, J.; et al. Top3beta is an RNA topoisomerase that works with fragile X syndrome protein to promote synapse formation. Nat. Neurosci. 2013, 16, 1238–1247. [Google Scholar] [CrossRef]
- Stoll, G.; Pietilainen, O.P.H.; Linder, B.; Suvisaari, J.; Brosi, C.; Hennah, W.; Leppa, V.; Torniainen, M.; Ripatti, S.; Ala-Mello, S.; et al. Deletion of TOP3beta, a component of FMRP-containing mRNPs, contributes to neurodevelopmental disorders. Nat. Neurosci. 2013, 16, 1228–1237. [Google Scholar] [CrossRef]
- Zhang, T.; Wallis, M.; Petrovic, V.; Challis, J.; Kalitsis, P.; Hudson, D.F. Loss of TOP3B leads to increased R-loop formation and genome instability. Open Biol. 2019, 9, 190222. [Google Scholar] [CrossRef]
- Su, S.; Xue, Y.; Sharov, A.; Zhang, Y.; Lee, S.K.; Martindale, J.L.; Li, W.; Ku, W.L.; Zhao, K.; De, S.; et al. A dual-activity topoisomerase complex regulates mRNA translation and turnover. Nucleic Acids Res. 2022, 50, 7013–7033. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Xue, Y.; Lee, S.K.; Zhang, Y.; Fan, J.; De, S.; Sharov, A.; Wang, W. A dual-activity topoisomerase complex promotes both transcriptional activation and repression in response to starvation. Nucleic Acids Res. 2023, 51, 2415–2433. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Tse-Dinh, Y.C. Variation of Structure and Cellular Functions of Type IA Topoisomerases across the Tree of Life. Cells 2024, 13, 553. [Google Scholar] [CrossRef] [PubMed]
- Prasanth, K.R.; Hirano, M.; Fagg, W.S.; McAnarney, E.T.; Shan, C.; Xie, X.; Hage, A.; Pietzsch, C.A.; Bukreyev, A.; Rajsbaum, R.; et al. Topoisomerase III-ss is required for efficient replication of positive-sense RNA viruses. Antiviral Res. 2020, 182, 104874. [Google Scholar] [CrossRef]
- Sansone, N.M.S.; Marques, L.F.A.; Boschiero, M.N.; Mello, L.S.; Marson, F.A.L. Epidemic after pandemic: Dengue surpasses COVID-19 in number of deaths. Pulmonology 2025, 31, 2448364. [Google Scholar] [CrossRef]
- Dasgupta, T.; Ferdous, S.; Tse-Dinh, Y.C. Mechanism of Type IA Topoisomerases. Molecules 2020, 25, 4769. [Google Scholar] [CrossRef]
- Ahmad, M.; Xu, D.; Wang, W. Type IA topoisomerases can be “magicians” for both DNA and RNA in all domains of life. RNA Biol. 2017, 14, 854–864. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, L.; Zhang, Q.; Lyu, S.; Zhu, D.; Shen, M.; Ke, X.; Qu, Y. Small molecule targeting topoisomerase 3β for cancer therapy. Pharmacol. Res. 2021, 174, 105927. [Google Scholar] [CrossRef]
- Thomas, A.; Pommier, Y. Targeting Topoisomerase I in the Era of Precision Medicine. Clin. Cancer Res. 2019, 25, 6581–6589. [Google Scholar] [CrossRef]
- Bjornsti, M.A.; Kaufmann, S.H. Topoisomerases and cancer chemotherapy: Recent advances and unanswered questions. F1000Research 2019, 8, F1000 Faculty Rev-1704. [Google Scholar] [CrossRef]
- Delgado, J.L.; Hsieh, C.M.; Chan, N.L.; Hiasa, H. Topoisomerases as anticancer targets. Biochem. J. 2018, 475, 373–398. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Saha, S.; Yang, X.; Pommier, Y.; Huang, S.N. Identification and characterization of topoisomerase III beta poisons. Proc. Natl. Acad. Sci. USA 2023, 120, e2218483120. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Pan, A.; Franci, C.; Hu, Y.; Chang, B.; Li, W.; Duan, M.; Torneros, A.; Yu, J.; Heckrodt, T.J.; et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res. 2010, 70, 1544–1554. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.; Sharma, A.; Patne, K.; Tabasum, S.; Suryavanshi, J.; Rawat, L.; Machaalani, M.; Eid, M.; Singh, R.P.; Choueiri, T.K.; et al. AXL signaling in cancer: From molecular insights to targeted therapies. Signal Transduct. Target. Ther. 2025, 10, 37. [Google Scholar] [CrossRef]
- Cohen, P.; Cross, D.; Jänne, P.A. Kinase drug discovery 20 years after imatinib: Progress and future directions. Nat. Rev. Drug Discov. 2021, 20, 551–569. [Google Scholar] [CrossRef]
- Tse-Dinh, Y.C.; Wong, T.W.; Goldberg, A.R. Virus- and cell-encoded tyrosine protein kinases inactivate DNA topoisomerases in vitro. Nature 1984, 312, 785–786. [Google Scholar] [CrossRef]
- Abdelgawad, M.A.; Mohamed, F.E.A.; Lamie, P.F.; Bukhari, S.N.A.; Al-Sanea, M.M.; Musa, A.; Elmowafy, M.; Nayl, A.A.; Karam Farag, A.; Ali, S.M.; et al. Design, synthesis, and biological evaluation of novel pyrido-dipyrimidines as dual topoisomerase II/FLT3 inhibitors in leukemia cells. Bioorg. Chem. 2022, 122, 105752. [Google Scholar] [CrossRef]
- Goto-Ito, S.; Yamagata, A.; Takahashi, T.S.; Sato, Y.; Fukai, S. Structural basis of the interaction between Topoisomerase IIIbeta and the TDRD3 auxiliary factor. Sci. Rep. 2017, 7, 42123. [Google Scholar] [CrossRef]
- Yang, X.; Saha, S.; Yang, W.; Neuman, K.C.; Pommier, Y. Structural and biochemical basis for DNA and RNA catalysis by human Topoisomerase 3β. Nat. Commun. 2022, 13, 4656. [Google Scholar] [CrossRef]
- Cinelli, M.A. Topoisomerase 1B poisons: Over a half-century of drug leads, clinical candidates, and serendipitous discoveries. Med. Res. Rev. 2019, 39, 1294–1337. [Google Scholar] [CrossRef]
- Rialdi, A.; Campisi, L.; Zhao, N.; Lagda, A.C.; Pietzsch, C.; Ho, J.S.Y.; Martinez-Gil, L.; Fenouil, R.; Chen, X.; Edwards, M.; et al. Topoisomerase 1 inhibition suppresses inflammatory genes and protects from death by inflammation. Science 2016, 352, aad7993. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.S.Y.; Mok, B.W.; Campisi, L.; Jordan, T.; Yildiz, S.; Parameswaran, S.; Wayman, J.A.; Gaudreault, N.N.; Meekins, D.A.; Indran, S.V.; et al. TOP1 inhibition therapy protects against SARS-CoV-2-induced lethal inflammation. Cell 2021, 184, 2618–2632.e17. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, P.B.; Chapagain, P.P.; Seddek, A.; Annamalai, T.; Uren, A.; Tse-Dinh, Y.C. Covalent Complex of DNA and Bacterial Topoisomerase: Implications in Antibacterial Drug Development. ChemMedChem 2020, 15, 623–631. [Google Scholar] [CrossRef]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; de Groot, B.L.; Grubmüller, H.; MacKerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Nosé, S.; Klein, M.L. Constant pressure molecular dynamics for molecular systems. Mol. Phys. 1983, 50, 1055–1076. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A major update to the DrugBank database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Swindells, M.B. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Yang, Y.; Lu, Y.; Espejo, A.; Wu, J.; Xu, W.; Liang, S.; Bedford, M.T. TDRD3 is an effector molecule for arginine-methylated histone marks. Mol. Cell 2010, 40, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Seddek, A.; Madeira, C.; Annamalai, T.; Mederos, C.; Tiwari, P.B.; Welch, A.Z.; Tse-Dinh, Y.-C. A Yeast-Based Screening System for Differential Identification of Poisons and Suppressors of Human Topoisomerase I. Front. Biosci. 2022, 27, 93. [Google Scholar] [CrossRef] [PubMed]
- Nefzi, A.; Ostresh, J.M.; Yu, Y.; Houghten, R.A. Combinatorial chemistry: Libraries from libraries, the art of the diversity-oriented transformation of resin-bound peptides and chiral polyamides to low molecular weight acyclic and heterocyclic compounds. J. Org. Chem. 2004, 69, 3603–3609. [Google Scholar] [CrossRef]
- Tantak, M.P.; Rayala, R.; Chaudhari, P.; Danta, C.C.; Nefzi, A. Synthesis of Diazacyclic and Triazacyclic Small-Molecule Libraries Using Vicinal Chiral Diamines Generated from Modified Short Peptides and Their Application for Drug Discovery. Pharmaceuticals 2024, 17, 1566. [Google Scholar] [CrossRef]
- Lin, R.; Connolly, P.J.; Huang, S.; Wetter, S.K.; Lu, Y.; Murray, W.V.; Emanuel, S.L.; Gruninger, R.H.; Fuentes-Pesquera, A.R.; Rugg, C.A.; et al. 1-Acyl-1H-[1,2,4]triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: Synthesis and evaluation of biological activities. J. Med. Chem. 2005, 48, 4208–4211. [Google Scholar] [CrossRef]
- Voytik-Harbin, S.L.; Brightman, A.O.; Waisner, B.; Lamar, C.H.; Badylak, S.F. Application and evaluation of the alamarBlue assay for cell growth and survival of fibroblasts. In Vitro Cell. Dev. Biol. Anim. 1998, 34, 239–246. [Google Scholar] [CrossRef]
- O’Brien, J.; Wilson, I.; Orton, T.; Pognan, F. Investigation of the Alamar Blue (resazurin) fluorescent dye for the assessment of mammalian cell cytotoxicity. Eur. J. Biochem. 2000, 267, 5421–5426. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affinity Score (kcal/mol) | Compound | Known Activities (Drug Bank Description) |
|---|---|---|
| −14.0 | Golvatinib | Dual kinase inhibitor of c-Met (hepatocyte growth factor receptor) and VEGFR-2 (vascular endothelial growth factor receptor). |
| −13.8 | Bemcentinib | Orally available and selective inhibitor of the AXL receptor tyrosine kinase (UFO), with potential antineoplastic activity. |
| −13.6 | Anatibant | Selective and potent Bradykinin B2 receptor antagonist developed for the treatment of traumatic brain injury (TBI). |
| −13.4 | Tucatinib | Tyrosine kinase inhibitor that targets the epidermal growth factor receptor 2 (HER2), used in combination therapy against refractory, advanced or metastatic HER2-positive breast and colorectal cancer. |
| −13.4 | Phthalocyanine | A dye currently under investigation for treating patients with actinic keratosis, Bowen’s disease, skin cancer, or stage I or stage II mycosis fungoides. |
| −13.3 | Conivaptan | Arginine vasopressin (AVP) receptor antagonist with affinity for AVP receptor subtypes V1A and V2, approved for hyponatremia (low blood sodium levels) caused by the syndrome of inappropriate antidiuretic hormone (SIADH). |
| −13.1 | Nilotinib | Tyrosine kinase inhibitor under investigation as a possible treatment for chronic myelogenous leukemia (CML). |
| −13.0 | Rebastinib | Orally bioavailable inhibitor of multiple tyrosine kinases with potential antineoplastic activity. |
| −12.9 | Radotinib | Second-generation tyrosine kinase inhibitor of Bcr-Abl fusion protein and the platelet-derived growth factor receptor (PDGFR), with potential antineoplastic activity. |
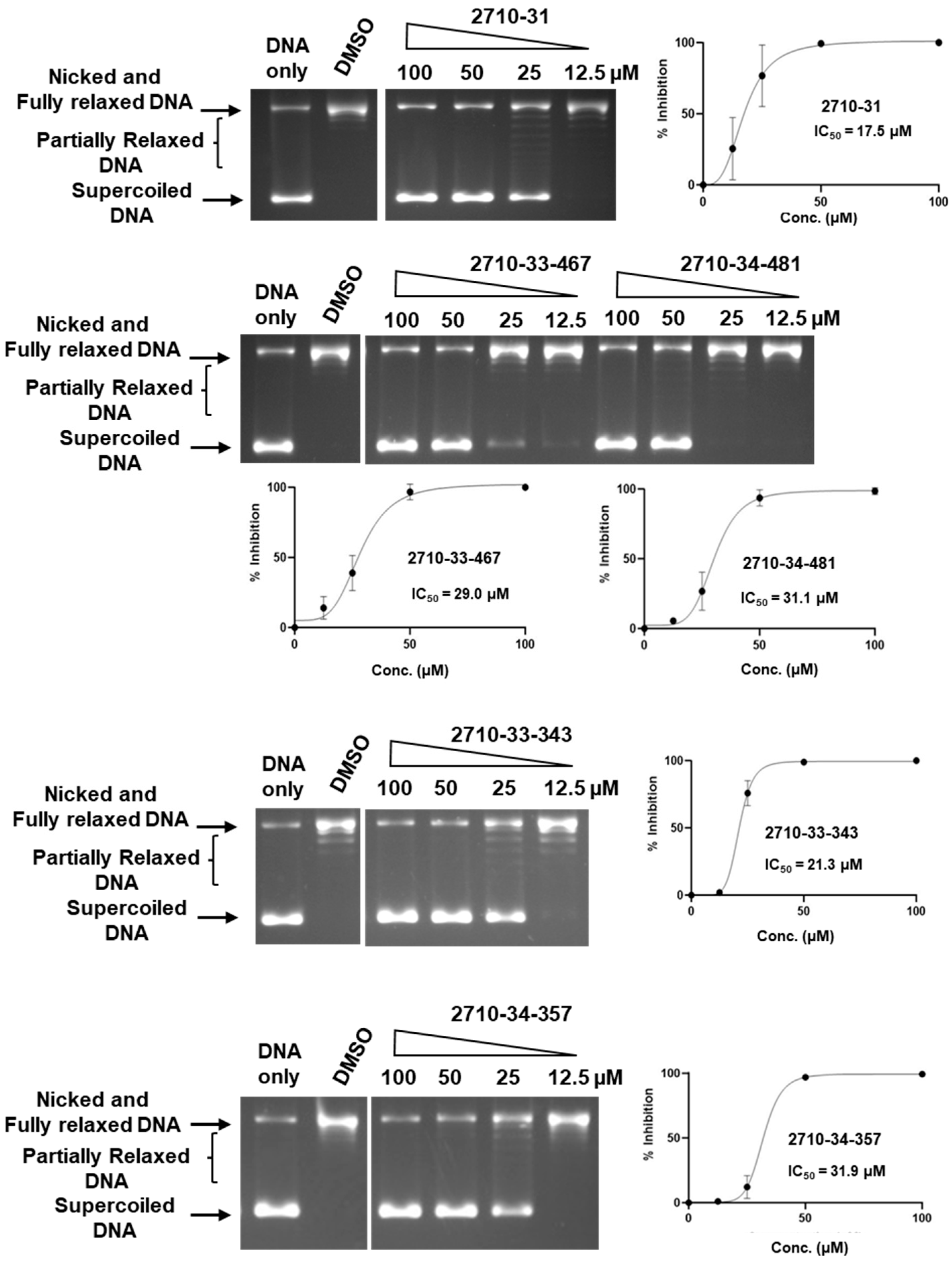
| Compound | TOP3B IC50 µM | 95% CI µM | TOP1 IC50 µM | 95% CI µM |
|---|---|---|---|---|
| Bemcentinib | 30.2 | 21.6–65.1 | 75.8 | 68.9–88.9 |
| 2710-31 | 29.6 | 7.8–41.2 | 17.5 | 13.6–23.1 |
| 2710-33-343 | 29.3 | 23.3–39.0 | 21.3 | 19.5–24.9 |
| 2710-33-467 | 45.2 | 32.8–59.8 | 29.0 | 25.0–33.2 |
| 2710-34-357 | 34.5 | 22.2–49.6 | 31.9 | 27.4–35.4 |
| 2710-34-481 | 55.6 | 40.1–74.2 | 31.1 | 22.89–34.62 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamun, Y.; Chadni, S.H.; Rayala, R.; Shafi, H.; Ferdous, S.; Pokhrel, R.; Nefzi, A.; Chapagain, P.; Tse-Dinh, Y.-C. Substituted Triazole-3,5-Diamine Compounds as Novel Human Topoisomerase III Beta Inhibitors. Int. J. Mol. Sci. 2025, 26, 6193. https://doi.org/10.3390/ijms26136193
Mamun Y, Chadni SH, Rayala R, Shafi H, Ferdous S, Pokhrel R, Nefzi A, Chapagain P, Tse-Dinh Y-C. Substituted Triazole-3,5-Diamine Compounds as Novel Human Topoisomerase III Beta Inhibitors. International Journal of Molecular Sciences. 2025; 26(13):6193. https://doi.org/10.3390/ijms26136193
Chicago/Turabian StyleMamun, Yasir, Somaia Haque Chadni, Ramanjaneyulu Rayala, Hasham Shafi, Shomita Ferdous, Rudramani Pokhrel, Adel Nefzi, Prem Chapagain, and Yuk-Ching Tse-Dinh. 2025. "Substituted Triazole-3,5-Diamine Compounds as Novel Human Topoisomerase III Beta Inhibitors" International Journal of Molecular Sciences 26, no. 13: 6193. https://doi.org/10.3390/ijms26136193
APA StyleMamun, Y., Chadni, S. H., Rayala, R., Shafi, H., Ferdous, S., Pokhrel, R., Nefzi, A., Chapagain, P., & Tse-Dinh, Y.-C. (2025). Substituted Triazole-3,5-Diamine Compounds as Novel Human Topoisomerase III Beta Inhibitors. International Journal of Molecular Sciences, 26(13), 6193. https://doi.org/10.3390/ijms26136193