
Immuno-Oncology at the Crossroads: Confronting Challenges in the Quest for Effective Cancer Therapies

 and
and 
Abstract
1. Introduction
2. Immune Evasion Mechanisms in Cancer
2.1. Mechanisms Directly Involving T Cells
2.2. Mechanisms Involving Other Immune Cells
3. Immunotherapy Approaches
3.1. Immune Checkpoint Inhibitors (ICIs)
3.2. Cancer Vaccines
3.3. Synthetic Long Peptides (SLPs)
3.4. Adoptive Cell Transfer (ACT) Therapy
3.5. Cytokine-Based Therapies
3.6. Resistance to Immunotherapy

{kind=link}
{kind=link}
| Immunotherapy Type | Examples | Mechanism of Resistance | References |
|---|---|---|---|
| Immune Checkpoint Inhibitors (ICIs) | Anti-PD-1 (e.g., nivolumab) Anti-PD-L1 (e.g., atezolizumab) Anti-CTLA-4 (e.g., ipilimumab) | Loss of tumor antigen expression Mutations in IFN-γ/JAK/STAT pathway (e.g., JAK1/2 mutations) Upregulation of alternative immune checkpoints (e.g., LAG-3, TIM-3, TIGIT) Immunosuppressive tumor microenvironment (e.g., Tregs, MDSCs, TAMs) Activation of WNT/β-catenin signaling leading to T-cell exclusion | [88,89,90] |
| Adoptive Cell Therapies (ACTs) | CAR-T cells, CAR-NK cells | Antigen loss or downregulation on tumor cells Immunosuppressive cytokines in the tumor microenvironment Physical barriers preventing T-cell infiltration Exhaustion of transferred T cells | [88] |
| Cancer Vaccines | Peptide-based vaccines, dendritic cell vaccines | Low immunogenicity of tumor antigens Tumor-induced immunosuppression Antigenic variation leading to immune escape | [88] |
| Cytokine Therapies | Interleukin-2 (IL-2), Interferon-alpha (IFN-α) | Activation of regulatory T cells leading to immunosuppression Systemic toxicity limiting therapeutic doses Short half-life requiring frequent administration | [88] |
4. Biomarkers Associated with Efficacy of ICI Therapy
4.1. Programmed Death Ligand 1 (PD-L1)
4.2. Tumor Mutational Burden (TMB) and Microsatellite Instability (MSI)
4.3. Tumor Microenvironment (TME)
Tumor-Infiltrating Immune Cells
4.4. Circulating Biomarkers
4.5. Transcriptome Signatures
4.6. Metabolic Products
| Metabolite | Biomarker Role | Cancer Type | Impact for Immunotherapy | References |
|---|---|---|---|---|
| Kynurenine/IDO1 | Immune suppression marker | Metastatic renal cell carcinoma Acute myeloid leukemia Glioblastoma Hepatocellular carcinoma. | Predicts poor response | [182,183,184,185] |
| Lactate | T-cell suppression in TME | Pan-cancer Pancreatic cancer | Associated with resistance | [186,187,188] |
| Arginine | T-cell proliferation and activation | Liver cancer | Low levels = reduced efficacy | [189] |
| SCFAs (e.g., butyrate) | Immune modulation via gut microbiome | Solid tumor | Correlates with better outcomes | [190] |
| Acylcarnitines | Lipid metabolism dysregulation | Acute myeloid leukemia Hepatocellular carcinoma | Linked to immune dysfunction | [191,192] |
| Polyamines | Tumor-promoting, immunosuppressive | Colorectal cancer | Elevated in non-responders | [193] |
| Glutamine | Supports tumor and T-cell metabolism | Lung adenocarcinoma | Metabolic imbalance affects response | [194] |
4.7. Microbiome
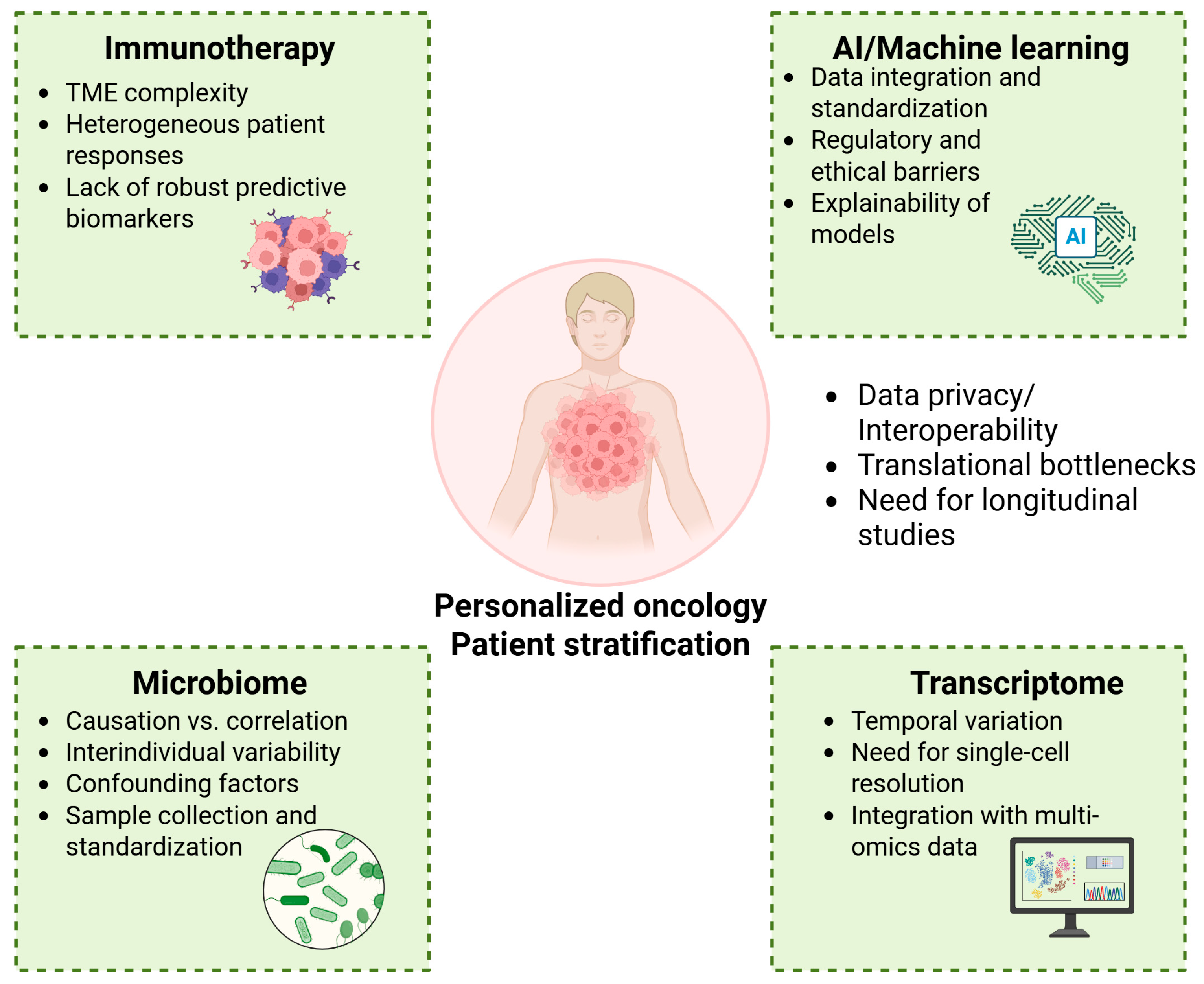
5. Discussion
6. Future Directions
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Global Cancer Burden Growing Amidst Mounting Need for Services; WHO: Geneva, Switzerland, 2024; Available online: https://www.who.int/news/item/01-02-2024-global-cancer-burden-growing--amidst-mounting-need-for-services (accessed on 24 May 2025).
- National Cancer Institute. Cancer Statistics; NCI: Bethesda, MD, USA, 2025. Available online: https://www.cancer.gov/about-cancer/understanding/statistics (accessed on 24 May 2025).
- Zafar, A.; Khatoon, S.; Khan, M.J.; Abu, J.; Naeem, A. Advancements and Limitations in Traditional Anti-Cancer Therapies: A Comprehensive Review of Surgery, Chemotherapy, Radiation Therapy, and Hormonal Therapy. Discov. Oncol. 2025, 16, 607. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zhou, H.; Tan, L.; Siu, K.T.; Guan, X.Y. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct. Target. Ther. 2024, 9, 175. [Google Scholar] [CrossRef]
- Marei, H.E.; Hasan, A.; Pozzoli, G.; Cenciarelli, C. Cancer immunotherapy with immune checkpoint inhibitors (ICIs): Potential, mechanisms of resistance, and strategies for reinvigorating T cell responsiveness when resistance is acquired. Cancer Cell Int. 2023, 23, 64. [Google Scholar] [CrossRef]
- Kong, X.; Zhang, J.; Chen, S.; Wang, X.; Xi, Q.; Shen, H.; Zhang, R. Immune Checkpoint Inhibitors: Breakthroughs in Cancer Treatment. Cancer Biol. Med. 2024, 21, 451–472. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, C.; Tu, J.; Tang, M.; Ashrafizadeh, M.; Nabavi, N.; Sethi, G.; Zhao, P.; Liu, S. Advances in cancer immunotherapy: Historical perspectives, current developments, and future directions. Mol. Cancer 2025, 24, 1–48. [Google Scholar] [CrossRef]
- Pilard, C.; Ancion, M.; Delvenne, P.; Jerusalem, G.; Hubert, P.; Herfs, M. Cancer Immunotherapy: It’s Time to Better Predict Patients’ Response. Br. J. Cancer 2021, 125, 927–938. [Google Scholar] [CrossRef]
- Desai, S.A.; Patel, V.P.; Bhosle, K.P.; Nagare, S.D.; Thombare, K.C. The Tumor Microenvironment: Shaping Cancer Progression and Treatment Response. J. Chemother. 2025, 37, 15–44. [Google Scholar] [CrossRef]
- Fang, J.; Lu, Y.; Zheng, J.; Jiang, X.; Shen, H.; Shang, X.; Lu, Y.; Fu, P. Exploring the crosstalk between endothelial cells, immune cells, and immune checkpoints in the tumor microenvironment: New insights and therapeutic implications. Cell Death Dis. 2023, 14, 586. [Google Scholar] [CrossRef]
- Yang, Y.; Li, S.; To, K.K.; Zhu, S.; Wang, F.; Fu, L. Tumor-associated macrophages remodel the suppressive tumor immune microenvironment and targeted therapy for immunotherapy. J. Exp. Clin. Cancer Res. 2025, 44, 1–28. [Google Scholar] [CrossRef]
- Liu, Y.; Sinjab, A.; Min, J.; Han, G.; Paradiso, F.; Zhang, Y.; Wang, R.; Pei, G.; Dai, Y.; Liu, Y.; et al. Conserved Spatial Subtypes and Cellular Neighborhoods of Cancer-Associated Fibroblasts Revealed by Single-Cell Spatial Multi-Omics. Cancer Cell 2025, 43, 905–924.e6. [Google Scholar] [CrossRef]
- Liu, C.; Cao, J. The Pivotal Role of Tertiary Lymphoid Structures in the Tumor Immune Microenvironment. Front. Oncol. 2025, 15, 1616904. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Yu, X.; Han, X.; Lian, C.; Wang, Z.; Shao, S.; Shao, F.; Wang, H.; Ma, S.; Liu, J. Innate Immune Cells in Tumor Microenvironment: A New Frontier in Cancer Immunotherapy. iScience 2024, 27, 110750. [Google Scholar] [CrossRef] [PubMed]
- Derosa, L.; Iebba, V.; Alves Costa Silva, C.; Piccinno, G.; Wu, G.; Lordello, L.; Routy, B.; Zhao, N.; Thelemaque, C.; Birebent, R.; et al. Custom Scoring Based on Ecological Topology of Gut Microbiota Associated with Cancer Immunotherapy Outcome. Cell 2024, 187, 3373–3389.e16. [Google Scholar] [CrossRef]
- Ghorani, E.; Swanton, C.; Quezada, S.A. Cancer cell-intrinsic mechanisms driving acquired immune tolerance. Immunity 2023, 56, 2270–2295. [Google Scholar] [CrossRef]
- Dunn, G.P.; Old, L.J.; Schreiber, R.D. The three Es of cancer immunoediting. Annu. Rev. Immunol. 2004, 22, 329–360. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef]
- van Weverwijk, A.; de Visser, K.E. Mechanisms driving the immunoregulatory function of cancer cells. Nat. Rev. Cancer 2023, 23, 193–215. [Google Scholar] [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: Insights into tumour immunogenicity and immune evasion. Nat. Rev. Cancer 2021, 21, 298–312. [Google Scholar] [CrossRef]
- Vinay, D.S.; Ryan, E.P.; Pawelec, G.; Talib, W.H.; Stagg, J.; Elkord, E.; Lichtor, T.; Decker, W.K.; Whelan, R.L.; Kumara, H.S.; et al. Immune evasion in cancer: Mechanistic basis and therapeutic strategies. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2015; Volume 35, pp. S185–S198. [Google Scholar]
- Piali, L.; Fichtel, A.; Terpe, H.J.; Imhof, B.A.; Gisler, R.H. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J. Exp. Med. 1995, 181, 811–816. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Damen, C.A.; Martinotti, S.; Blijham, G.H.; Groenewegen, G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: The role of angiogenic factors. Cancer Res. 1996, 56, 1111–1117. [Google Scholar] [PubMed]
- Motz, G.T.; Santoro, S.P.; Wang, L.P.; Garrabrant, T.; Lastra, R.R.; Hagemann, I.S.; Lal, P.; Feldman, M.D.; Benencia, F.; Coukos, G. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 2014, 20, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Akimoto, N.; Väyrynen, J.P.; Zhao, M.; Ugai, T.; Fujiyoshi, K.; Borowsky, J.; Zhong, R.; Haruki, K.; Arima, K.; Lau, M.C.; et al. Desmoplastic reaction, immune cell response, and prognosis in colorectal cancer. Front. Immunol. 2022, 13, 840198. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti–PD-L1 immunotherapy in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef]
- Li, H.; Courtois, E.T.; Sengupta, D.; Tan, Y.; Chen, K.H.; Goh, J.J.; Kong, S.L.; Chua, C.; Hon, L.K.; Tan, W.S.; et al. Reference component analysis of single-cell transcriptomes elucidates cellular heterogeneity in human colorectal tumors. Nat. Genet. 2017, 49, 708–718. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Pentcheva-Hoang, T.; Carstens, J.L.; Zheng, X.; Wu, C.C.; Simpson, T.R.; Laklai, H.; Sugimoto, H.; Kahlert, C.; Novitskiy, S.V.; et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell 2014, 25, 719–734. [Google Scholar] [CrossRef]
- Chakravarthy, A.; Khan, L.; Bensler, N.P.; Bose, P.; De Carvalho, D.D. TGF-β-associated extracellular matrix genes link cancer-associated fibroblasts to immune evasion and immunotherapy failure. Nat. Commun. 2018, 9, 4692. [Google Scholar] [CrossRef]
- Harlin, H.; Meng, Y.; Peterson, A.C.; Zha, Y.; Tretiakova, M.; Slingluff, C.; McKee, M.; Gajewski, T.F. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009, 69, 3077–3085. [Google Scholar] [CrossRef]
- Abou El Hassan, M.; Huang, K.; Eswara, M.B.; Zhao, M.; Song, L.; Yu, T.; Liu, Y.; Liu, J.C.; McCurdy, S.; Ma, A.; et al. Cancer cells hijack PRC2 to modify multiple cytokine pathways. PLoS ONE 2015, 10, e0126466. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, E.; Togashi, Y.; Takeuchi, Y.; Shinya, S.; Tada, Y.; Kataoka, K.; Tane, K.; Sato, E.; Ishii, G.; Goto, K.; et al. Blockade of EGFR improves responsiveness to PD-1 blockade in EGFR-mutated non–small cell lung cancer. Sci. Immunol. 2020, 5, eaav3937. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Puyang, X.; Wu, Z.J.; Seiler, R.; Furman, C.; Oo, H.Z.; Seiler, M.; Irwin, S.; Subramanian, V.; Julie Joshi, J.; et al. Evasion of immunosurveillance by genomic alterations of PPARγ/RXRα in bladder cancer. Nat. Commun. 2017, 8, 103. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef]
- Dubrot, J.; Du, P.P.; Lane-Reticker, S.K.; Kessler, E.A.; Muscato, A.J.; Mehta, A.; Freeman, S.S.; Allen, P.M.; Olander, K.E.; Ockerman, K.M.; et al. In vivo CRISPR screens reveal the landscape of immune evasion pathways across cancer. Nat. Immunol. 2022, 23, 1495–1506. [Google Scholar] [CrossRef]
- Forloni, M.; Albini, S.; Limongi, M.Z.; Cifaldi, L.; Boldrini, R.; Nicotra, M.R.; Giannini, G.; Natali, P.G.; Giacomini, P.; Fruci, D. NF-κB, and not MYCN, regulates MHC class I and endoplasmic reticulum aminopeptidases in human neuroblastoma cells. Cancer Res. 2010, 70, 916–924. [Google Scholar] [CrossRef]
- Matsushita, H.; Vesely, M.D.; Koboldt, D.C.; Rickert, C.G.; Uppaluri, R.; Magrini, V.J.; Arthur, C.D.; White, J.M.; Chen, Y.S.; Shea, L.K.; et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012, 482, 400–404. [Google Scholar] [CrossRef]
- Martínez-Ruiz, C.; Black, J.R.; Puttick, C.; Hill, M.S.; Demeulemeester, J.; Larose Cadieux, E.; Thol, K.; Jones, T.P.; Veeriah, S.; Naceur-Lombardelli, C.; et al. Genomic–transcriptomic evolution in lung cancer and metastasis. Nature 2023, 616, 543–552. [Google Scholar] [CrossRef]
- Ruiz de Galarreta, M.; Bresnahan, E.; Molina-Sánchez, P.; Lindblad, K.E.; Maier, B.; Sia, D.; Puigvehi, M.; Miguela, V.; Casanova-Acebes, M.; Dhainaut, M.; et al. β-catenin activation promotes immune escape and resistance to anti–PD-1 therapy in hepatocellular carcinoma. Cancer Discov. 2019, 9, 1124–1141. [Google Scholar] [CrossRef]
- Pan, Y.; Yu, Y.; Wang, X.; Zhang, T. Tumor-associated macrophages in tumor immunity. Front. Immunol. 2020, 11, 583084. [Google Scholar] [CrossRef]
- Chen, P.; Huang, Y.; Bong, R.; Ding, Y.; Song, N.; Wang, X.; Song, X.; Luo, Y. Tumor-associated macrophages promote angiogenesis and melanoma growth via adrenomedullin in a paracrine and autocrine manner. Clin. Cancer Res. 2011, 17, 7230–7239. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.P.; Cheng, X.; Liu, Z.D.; Xu, S.F. Comparative beneficiary effects of immunotherapy against chemotherapy in patients with advanced NSCLC: Meta-analysis and systematic review. Oncol. Lett. 2017, 14, 1568–1580. [Google Scholar] [CrossRef] [PubMed]
- Sanmamed, M.F.; Chen, L. A paradigm shift in cancer immunotherapy: From enhancement to normalization. Cell 2018, 175, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Cha, J.H.; Chan, L.C.; Song, M.S.; Hung, M.C. New approaches on cancer immunotherapy. Cold Spring Harb. Perspect. Med. 2020, 10, a036863. [Google Scholar] [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune checkpoint blockade: A common denominator approach to cancer therapy. Cancer Cell 2015, 27, 450–461. [Google Scholar] [CrossRef]
- Gavrielatou, N.; Doumas, S.; Economopoulou, P.; Foukas, P.G.; Psyrri, A. Biomarkers for immunotherapy response in head and neck cancer. Cancer Treat. Rev. 2020, 84, 101977. [Google Scholar] [CrossRef]
- Esfahani, K.; Roudaia, L.; Buhlaiga, N.A.; Del Rincon, S.V.; Papneja, N.; Miller, W.H. A review of cancer immunotherapy: From the past, to the present, to the future. Curr. Oncol. 2020, 27, 87–97. [Google Scholar] [CrossRef]
- Galon, J.; Bruni, D. Approaches to treat immune hot, altered and cold tumours with combination immunotherapies. Nat. Rev. Drug Discov. 2019, 18, 197–218. [Google Scholar] [CrossRef]
- Hu, Z.; Ott, P.A.; Wu, C.J. Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol. 2018, 18, 168–182. [Google Scholar] [CrossRef]
- Hollingsworth, R.E.; Jansen, K. Turning the corner on therapeutic cancer vaccines. npj Vaccines 2019, 4, 7. [Google Scholar] [CrossRef] [PubMed]
- Disis, M.L.; Wallace, D.R.; Gooley, T.A.; Dang, Y.; Slota, M.; Lu, H.; Coveler, A.L.; Childs, J.S.; Higgins, D.M.; Fintak, P.A.; et al. Concurrent trastuzumab and HER2/neu-specific vaccination in patients with metastatic breast cancer. J. Clin. Oncol. 2009, 27, 4685–4692. [Google Scholar] [CrossRef] [PubMed]
- Correale, P.; Nieroda, C.; Zaremba, S.; Zhu, M.; Schlom, J.; Tsang, K.Y.; Konstantin, W. In vitro generation of human cytotoxic T lymphocytes specific for peptides derived from prostate-specific antigen. J. Natl. Cancer Inst. 1997, 89, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Finn, O.J. The dawn of vaccines for cancer prevention. Nat. Rev. Immunol. 2018, 18, 183–194. [Google Scholar] [CrossRef]
- De Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef]
- van der Burg, S.H.; Arens, R.; Ossendorp, F.; van Hall, T.; Melief, C.J. Vaccines for established cancer: Overcoming the challenges posed by immune evasion. Nat. Rev. Cancer 2016, 16, 219–233. [Google Scholar] [CrossRef]
- Melief, C.J.; van Hall, T.; Arens, R.; Ossendorp, F.; van der Burg, S.H. Therapeutic cancer vaccines. J. Clin. Investig. 2015, 125, 3401–3412. [Google Scholar] [CrossRef]
- Alloatti, A.; Kotsias, F.; Magalhaes, J.G.; Amigorena, S. Dendritic cell maturation and cross-presentation: Timing matters! Immunol. Rev. 2016, 272, 97–108. [Google Scholar] [CrossRef]
- Melief, C.J.; Welters, M.J.; Vergote, I.; Kroep, J.R.; Kenter, G.G.; Ottevanger, P.B.; Tjalma, W.A.; Denys, H.; van Poelgeest, M.I.; Nijman, H.W.; et al. Strong vaccine responses during chemotherapy are associated with prolonged cancer survival. Sci. Transl. Med. [Internet] 2020, 12, 535. [Google Scholar] [CrossRef]
- Choi, Y.J.; Hur, S.Y.; Kim, T.J.; Hong, S.R.; Lee, J.K.; Cho, C.H.; Park, K.S.; Woo, J.W.; Sung, Y.C.; Suh, Y.S.; et al. A phase II, prospective, randomized, multicenter, open-label study of GX-188E, an HPV DNA vaccine, in patients with cervical intraepithelial neoplasia 3. Clin. Cancer Res. 2020, 26, 1616–1623. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M.; Türeci, Ö.; Sahin, U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu. Rev. Med. 2019, 70, 395–407. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Bijker, M.S.; van den Eeden, S.J.; Franken, K.L.; Melief, C.J.; van der Burg, S.H.; Offringa, R. Superior induction of anti-tumor CTL immunity by extended peptide vaccines involves prolonged, DC-focused antigen presentation. Eur. J. Immunol. 2008, 38, 1033–1042. [Google Scholar] [CrossRef]
- Speetjens, F.M.; Welters, M.J.; Slingerland, M.; van Poelgeest, M.I.; van Steenwijk, P.J.; Roozen, I.; Boekestijn, S.; Loof, N.M.; Zom, G.G.; Valentijn, A.R.; et al. Intradermal vaccination of HPV-16 E6 synthetic peptides conjugated to an optimized Toll-like receptor 2 ligand shows safety and potent T cell immunogenicity in patients with HPV-16 positive (pre-) malignant lesions. J. Immunother. Cancer 2022, 10, e005016. [Google Scholar] [CrossRef]
- Baldin, A.V.; Savvateeva, L.V.; Bazhin, A.V.; Zamyatnin, A.A., Jr. Dendritic cells in anticancer vaccination: Rationale for ex vivo loading or in vivo targeting. Cancers 2020, 12, 590. [Google Scholar] [CrossRef]
- Qu, C.; Brinck-Jensen, N.S.; Zang, M.; Chen, K. Monocyte-derived dendritic cells: Targets as potent antigen-presenting cells for the design of vaccines against infectious diseases. Int. J. Infect. Dis. 2014, 19, 1–5. [Google Scholar] [CrossRef]
- Murphy, T.L.; Murphy, K.M. Dendritic cells in cancer immunology. Cell. Mol. Immunol. 2022, 19, 3–13. [Google Scholar] [CrossRef]
- Del Prete, A.; Salvi, V.; Soriani, A.; Laffranchi, M.; Sozio, F.; Bosisio, D.; Sozzani, S. Dendritic cell subsets in cancer immunity and tumor antigen sensing. Cell. Mol. Immunol. 2023, 20, 432–447. [Google Scholar] [CrossRef]
- Cheever, M.A.; Higano, C.S. PROVENGE (Sipuleucel-T) in prostate cancer: The first FDA-approved therapeutic cancer vaccine. Clin. Cancer Res. 2011, 17, 3520–3526. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, S.A.; Restifo, N.P. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015, 348, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.C.; Sterner, R.M. CAR-T cell therapy: Current limitations and potential strategies. Blood Cancer J. 2021, 11, 69. [Google Scholar] [CrossRef]
- Braendstrup, P.; Levine, B.L.; Ruella, M. The long road to the first FDA-approved gene therapy: Chimeric antigen receptor T cells targeting CD19. Cytotherapy 2020, 22, 57–69. [Google Scholar] [CrossRef]
- Vitale, C.; Strati, P. CAR T-cell therapy for B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia: Clinical trials and real-world experiences. Front. Oncol. 2020, 10, 849. [Google Scholar] [CrossRef]
- Grupp, S.A.; Kalos, M.; Barrett, D.; Aplenc, R.; Porter, D.L.; Rheingold, S.R.; Teachey, D.T.; Chew, A.; Hauck, B.; Wright, J.F.; et al. Chimeric antigen receptor–modified T cells for acute lymphoid leukemia. N. Engl. J. Med. 2013, 368, 1509–1518. [Google Scholar] [CrossRef]
- Li, Y.; Hermanson, D.L.; Moriarity, B.S.; Kaufman, D.S. Human iPSC-derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell 2018, 23, 181–192. [Google Scholar] [CrossRef]
- Muhammad, S.; Fan, T.; Hai, Y.; Gao, Y.; He, J. Reigniting hope in cancer treatment: The promise and pitfalls of IL-2 and IL-2R targeting strategies. Mol. Cancer 2023, 22, 121. [Google Scholar] [CrossRef]
- Payne, R.; Glenn, L.; Hoen, H.; Richards, B.; Smith, J.W.; Lufkin, R.; Crocenzi, T.S.; Urba, W.J.; Curti, B.D. Durable responses and reversible toxicity of high-dose interleukin-2 treatment of melanoma and renal cancer in a Community Hospital Biotherapy Program. J. Immunother. Cancer 2014, 2, 13. [Google Scholar] [CrossRef]
- Zhang, Q.; Hresko, M.E.; Picton, L.K.; Su, L.; Hollander, M.J.; Nunez-Cruz, S.; Zhang, Z.; Assenmacher, C.A.; Sockolosky, J.T.; Garcia, K.C.; et al. A human orthogonal IL-2 and IL-2Rβ system enhances CAR T cell expansion and antitumor activity in a murine model of leukemia. Sci. Transl. Med. 2021, 13, eabg6986. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed]
- van Elsas, M.J.; van Hall, T.; van der Burg, S.H. Future challenges in cancer resistance to immunotherapy. Cancers 2020, 12, 935. [Google Scholar] [CrossRef] [PubMed]
- Bashash, D.; Zandi, Z.; Kashani, B.; Pourbagheri-Sigaroodi, A.; Salari, S.; Ghaffari, S.H. Resistance to immunotherapy in human malignancies: Mechanisms, research progresses, challenges, and opportunities. J. Cell. Physiol. 2022, 237, 346–372. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.; Pich, O.; Thol, K.; Watkins, T.B.; Luebeck, J.; Rowan, A.; Stavrou, G.; Weiser, N.E.; Dameracharla, B.; Bentham, R.; et al. Origins and impact of extrachromosomal DNA. Nature 2024, 635, 193–200. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Mittra, A.; Naqash, A.R.; Takebe, N. A review of mechanisms of resistance to immune checkpoint inhibitors and potential strategies for therapy. Cancer Drug Resist. 2020, 3, 252. [Google Scholar] [CrossRef]
- Kawashima, S.; Togashi, Y. Resistance to immune checkpoint inhibitors and the tumor microenvironment. Exp. Dermatol. 2023, 32, 240–249. [Google Scholar] [CrossRef]
- Wang, B.; Han, Y.; Zhang, Y.; Zhao, Q.; Wang, H.; Wei, J.; Meng, L.; Xin, Y.; Jiang, X. Overcoming acquired resistance to cancer immune checkpoint therapy: Potential strategies based on molecular mechanisms. Cell Biosci. 2023, 13, 120. [Google Scholar] [CrossRef]
- Tan, S.; Day, D.; Nicholls, S.J.; Segelov, E. Immune Checkpoint Inhibitor Therapy in Oncology: Current Uses and Future Directions: JACC: CardioOncology State-of-the-Art Review. JACC CardioOncol. 2022, 4, 579–597. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhao, X.; Bao, Y.; Meng, B.; Xu, Z.; Li, S.; Wang, X.; Hou, R.; Ma, W.; Liu, D.; Zheng, J.; et al. From rough to precise: PD-L1 evaluation for predicting the efficacy of PD-1/PD-L1 blockades. Front. Immunol. 2022, 13, 920021. [Google Scholar] [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, I.I.; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nat. Rev. Clin. Oncol. 2021, 18, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Scheper, W.; Kvistborg, P. Cancer neoantigens. Annu. Rev. Immunol. 2019, 37, 173–200. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Hopkins, A.; Jaffee, E.M. Tumor mutational burden and response rate to PD-1 inhibition. N. Engl. J. Med. 2017, 377, 2500–2501. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M. Tumor mutation burden for predicting immune checkpoint blockade response: The more, the better. J. Immunother. Cancer 2022, 10, e003087. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Strickler, J.H.; Hanks, B.A.; Khasraw, M. Tumor Mutational Burden as a Predictor of Immunotherapy Response: Is More Always Better? Clin. Cancer Res. 2021, 27, 1236–1241. [Google Scholar] [CrossRef]
- Iwahori, K. Cytotoxic CD8+ lymphocytes in the tumor microenvironment. Tumor Microenviron. Hematop. Cells–Part A 2020, 1224, 53–62. [Google Scholar]
- Paul, M.S.; Ohashi, P.S. The roles of CD8+ T cell subsets in antitumor immunity. Trends Cell Biol. 2020, 30, 695–704. [Google Scholar] [CrossRef]
- Peralta, R.M.; Xie, B.; Lontos, K.; Nieves-Rosado, H.; Spahr, K.; Joshi, S.; Ford, B.R.; Quann, K.; Frisch, A.T.; Dean, V.; et al. Dysfunction of exhausted T cells is enforced by MCT11-mediated lactate metabolism. Nat. Immunol. 2024, 25, 2297–2307. [Google Scholar] [CrossRef]
- Lipp, J.J.; Wang, L.; Yang, H.; Yao, F.; Harrer, N.; Müller, S.; Berezowska, S.; Dorn, P.; Marti, T.M.; Schmid, R.A.; et al. Functional and molecular characterization of PD1+ tumor-infiltrating lymphocytes from lung cancer patients. Oncoimmunology 2022, 11, 2019466. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Stanton, S.E.; Disis, M.L. Clinical significance of tumor-infiltrating lymphocytes in breast cancer. J. Immunother. Cancer 2016, 4, 59. [Google Scholar] [CrossRef]
- Tramm, T.; Vinter, H.; Vahl, P.; Özcan, D.; Alsner, J.; Overgaard, J. Tumor-infiltrating lymphocytes predict improved overall survival after post-mastectomy radiotherapy: A study of the randomized DBCG82bc cohort. Acta Oncol. 2022, 61, 153–162. [Google Scholar] [CrossRef]
- Sun, P.; He, J.; Chao, X.; Chen, K.; Xu, Y.; Huang, Q.; Yun, J.; Li, M.; Luo, R.; Kuang, J.; et al. A computational tumor-infiltrating lymphocyte assessment method comparable with visual reporting guidelines for triple-negative breast cancer. EBioMedicine 2021, 70, 103492. [Google Scholar] [CrossRef] [PubMed]
- Albusayli, R.; Graham, J.D.; Pathmanathan, N.; Shaban, M.; Raza, S.E.; Minhas, F.; Armes, J.E.; Rajpoot, N. Artificial intelligence-based digital scores of stromal tumour-infiltrating lymphocytes and tumour-associated stroma predict disease-specific survival in triple-negative breast cancer. J. Pathol. 2023, 260, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Maibach, F.; Sadozai, H.; Seyed Jafari, S.M.; Hunger, R.E.; Schenk, M. Tumor-infiltrating lymphocytes and their prognostic value in cutaneous melanoma. Front. Immunol. 2020, 11, 2105. [Google Scholar] [CrossRef] [PubMed]
- Saberzadeh-Ardestani, B.; Foster, N.R.; Lee, H.E.; Shi, Q.; Alberts, S.R.; Smyrk, T.C.; Sinicrope, F.A. Association of tumor-infiltrating lymphocytes with survival depends on primary tumor sidedness in stage III colon cancers (NCCTG N0147) [Alliance]. Ann. Oncol. 2022, 33, 1159–1167. [Google Scholar] [CrossRef]
- Omura, Y.; Toiyama, Y.; Okugawa, Y.; Yin, C.; Shigemori, T.; Kusunoki, K.; Kusunoki, Y.; Ide, S.; Shimura, T.; Fujikawa, H.; et al. Prognostic impacts of tumoral expression and serum levels of PD-L1 and CTLA-4 in colorectal cancer patients. Cancer Immunol. Immunother. 2020, 69, 2533–2546. [Google Scholar] [CrossRef]
- Herrera, M.; Berral-González, A.; López-Cade, I.; Galindo-Pumariño, C.; Bueno-Fortes, S.; Martín-Merino, M.; Carrato, A.; Ocaña, A.; De La Pinta, C.; López-Alfonso, A.; et al. Cancer-associated fibroblast-derived gene signatures determine prognosis in colon cancer patients. Mol. Cancer 2021, 20, 73. [Google Scholar] [CrossRef]
- Pan, H.; Pan, J.; Wu, J. Development and validation of a cancer-associated fibroblast-derived lncRNA signature for predicting clinical outcomes in colorectal cancer. Front. Immunol. 2022, 13, 934221. [Google Scholar] [CrossRef]
- Gao, Y.; Sun, Z.; Gu, J.; Li, Z.; Xu, X.; Xue, C.; Li, X.; Zhao, L.; Zhou, J.; Bai, C.; et al. Cancer-associated fibroblasts promote the upregulation of PD-L1 expression through Akt phosphorylation in colorectal cancer. Front. Oncol. 2021, 11, 748465. [Google Scholar] [CrossRef]
- Wang, Y.; Dong, J.; Quan, Q.; Liu, S.; Chen, X.; Cai, X.; Qiu, H.; Zhang, B.; Guo, G. Immune Cell Infiltration of the Primary Tumor Microenvironment Predicted the Treatment Outcome of Chemotherapy with or without Bevacizumab in Metastatic Colorectal Cancer Patients. Front. Oncol. 2021, 10, 581051. [Google Scholar] [CrossRef]
- Wang, X.; Barrera, C.; Bera, K.; Viswanathan, V.S.; Azarianpour-Esfahani, S.; Koyuncu, C.; Velu, P.; Feldman, M.D.; Yang, M.; Fu, P.; et al. Spatial interplay patterns of cancer nuclei and tumor-infiltrating lymphocytes (TILs) predict clinical benefit for immune checkpoint inhibitors. Sci. Adv. 2022, 8, eabn3966. [Google Scholar] [CrossRef]
- Rakaee, M.; Adib, E.; Ricciuti, B.; Sholl, L.M.; Shi, W.; Alessi, J.V.; Cortellini, A.; Fulgenzi, C.A.; Viola, P.; Pinato, D.J.; et al. Association of machine learning–based assessment of tumor-infiltrating lymphocytes on standard histologic images with outcomes of immunotherapy in patients with NSCLC. JAMA Oncol. 2023, 9, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ock, C.Y.; Kim, H.; Pereira, S.; Park, S.; Ma, M.; Choi, S.; Kim, S.; Shin, S.; Aum, B.J.; et al. Artificial intelligence–powered spatial analysis of tumor-infiltrating lymphocytes as complementary biomarker for immune checkpoint inhibition in non–small-cell lung cancer. J. Clin. Oncol. 2022, 40, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.H.; Kim, Y.W.; Sefcovicova, N.; Laye, J.P.; Hewitt, L.C.; Irvine, A.F.; Vromen, V.; Janssen, Y.; Davarzani, N.; Fazzi, G.E.; et al. Tumour infiltrating lymphocytes and survival after adjuvant chemotherapy in patients with gastric cancer: Post-hoc analysis of the CLASSIC trial. Br. J. Cancer 2023, 128, 2318–2325. [Google Scholar] [CrossRef]
- Hoesli, R.; Birkeland, A.C.; Rosko, A.J.; Issa, M.; Chow, K.L.; Michmerhuizen, N.L.; Mann, J.E.; Chinn, S.B.; Shuman, A.G.; Prince, M.E.; et al. Proportion of CD4 and CD8 tumor infiltrating lymphocytes predicts survival in persistent/recurrent laryngeal squamous cell carcinoma. Oral Oncol. 2018, 77, 83–89. [Google Scholar] [CrossRef]
- Kuba, K.; Inoue, H.; Matsumura, S.; Enoki, Y.; Kogashiwa, Y.; Ebihara, Y.; Nakahira, M.; Yamazaki, T.; Yasuda, M.; Kaira, K.; et al. A retrospective analysis of tumor infiltrating lymphocytes in head and neck squamous cell carcinoma patients treated with nivolumab. Sci. Rep. 2022, 12, 22557. [Google Scholar] [CrossRef]
- Burandt, E.; Blessin, N.C.; Rolschewski, A.C.; Lutz, F.; Mandelkow, T.; Yang, C.; Bady, E.; Reiswich, V.; Simon, R.; Sauter, G.; et al. T-Cell Density at the Invasive Margin and Immune Phenotypes Predict Outcome in Vulvar Squamous Cell Cancer. Cancers 2022, 14, 4246. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Oshi, M.; Chida, K.; Roy, A.M.; Mann, G.K.; An, N.; Yan, L.; Endo, I.; Takabe, K. Higher inflammatory response in hepatocellular carcinoma is associated with immune cell infiltration and a better outcome. Hepatol. Int. 2024, 18, 1299–1309. [Google Scholar] [CrossRef]
- Hamada, K.; Murakami, R.; Ueda, A.; Kashima, Y.; Miyagawa, C.; Taki, M.; Yamanoi, K.; Yamaguchi, K.; Hamanishi, J.; Minamiguchi, S.; et al. A Deep Learning-Based Assessment Pipeline for Intraepithelial and Stromal Tumor-Infiltrating Lymphocytes in High-Grade Serous Ovarian Carcinoma. Am. J. Pathol. 2024, 194, 1272–1284. [Google Scholar] [CrossRef]
- Hashemi, S.; Fransen, M.F.; Niemeijer, A.; Ben Taleb, N.; Houda, I.; Veltman, J.; Becker-Commissaris, A.; Daniels, H.; Crombag, L.; Radonic, T.; et al. Surprising impact of stromal TIL’s on immunotherapy efficacy in a real-world lung cancer study. Lung Cancer 2021, 153, 81–89. [Google Scholar] [CrossRef]
- Perez, E.A.; Ballman, K.V.; Tenner, K.S.; Thompson, E.A.; Badve, S.S.; Bailey, H.; Baehner, F.L. Association of Stromal Tumor-Infiltrating Lymphocytes with Recurrence-Free Survival in the N9831 Adjuvant Trial in Patients with Early-Stage HER2-Positive Breast Cancer. JAMA Oncol. 2016, 2, 56–64. [Google Scholar] [CrossRef]
- De Angelis, C.; Nagi, C.; Hoyt, C.C.; Liu, L.; Roman, K.; Wang, C.; Zheng, Y.; Veeraraghavan, J.; Sethunath, V.; Nuciforo, P.; et al. Evaluation of the Predictive Role of Tumor Immune Infiltrate in Patients with HER2-Positive Breast Cancer Treated with Neoadjuvant Anti-HER2 Therapy without Chemotherapy. Clin. Cancer Res. 2020, 26, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, R.; Chitre, A.S.; Scherl, A.; Wu, T.D.; Patil, N.S.; de Almeida, P.; Kadel, E.E., III; Madireddi, S.; Au-Yeung, A.; Takahashi, C.; et al. Intratumoral CD103+ CD8+ T cells predict response to PD-L1 blockade. J. Immunother. Cancer 2021, 9, e002231. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Damei, I.; Trickovic, T.; Mami-Chouaib, F.; Corgnac, S. Tumor-resident memory T cells as a biomarker of the response to cancer immunotherapy. Front. Immunol. 2023, 14, 1205984. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Lu, J.; Zhang, G.; Wang, Y.; He, M.; Xu, Q.; Xu, C.; Liu, H. CXCL13 shapes immunoactive tumor microenvironment and enhances the efficacy of PD-1 checkpoint blockade in high-grade serous ovarian cancer. J. Immunother. Cancer 2021, 9, e001136. [Google Scholar] [CrossRef]
- Westwood, A.C.; Wilson, B.I.; Laye, J.; Grabsch, H.I.; Mueller, W.; Magee, D.R.; Quirke, P.; West, N.P. Deep-learning enabled combined measurement of tumour cell density and tumour infiltrating lymphocyte density as a prognostic biomarker in colorectal cancer. BJC Rep. 2025, 3, 12. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, L.; Li, L.; Liu, Y.; Das, B.; Zhai, S.; Tan, J.; Jiang, Y.; Turco, S.; Yao, Y.; et al. Prediction and analysis of tumor infiltrating lymphocytes across 28 cancers by TILScout using deep learning. npj Precis. Oncol. 2025, 9, 76. [Google Scholar] [CrossRef]
- Bang, Y.H.; Lee, C.K.; Bang, K.; Kim, H.D.; Kim, K.P.; Jeong, J.H.; Park, I.; Ryoo, B.Y.; Lee, D.K.; Choi, H.J.; et al. Artificial Intelligence-Powered Spatial Analysis of Tumor-Infiltrating Lymphocytes as a Potential Biomarker for Immune Checkpoint Inhibitors in Patients with Biliary Tract Cancer. Clin. Cancer Res. 2024, 30, 4635–4643. [Google Scholar] [CrossRef] [PubMed]
- Wakiyama, H.; Masuda, T.; Motomura, Y.; Hu, Q.; Tobo, T.; Eguchi, H.; Sakamoto, K.; Hirakawa, M.; Honda, H.; Mimori, K. Cytolytic Activity (CYT) Score Ιs a Prognostic Biomarker Reflecting Host Immune Status in Hepatocellular Carcinoma (HCC). Anticancer Res. 2018, 38, 6631–6638. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Xia, H.; Yan, Y.; Zhu, X.; Sun, F.; Sun, L.; Li, S.; Li, D.; Wang, J.; et al. Tumor microenvironment remodeling after neoadjuvant immunotherapy in non-small cell lung cancer revealed by single-cell RNA sequencing. Genome Med. 2023, 15, 14. [Google Scholar] [CrossRef]
- Narayanan, S.; Kawaguchi, T.; Peng, X.; Qi, Q.; Liu, S.; Yan, L.; Takabe, K. Tumor Infiltrating Lymphocytes and Macrophages Improve Survival in Microsatellite Unstable Colorectal Cancer. Sci. Rep. 2019, 9, 13455. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Park, C.; Na, K.J.; Choi, H.; Ock, C.Y.; Ha, S.; Kim, M.; Park, S.; Keam, B.; Kim, T.M.; Paeng, J.C.; et al. Tumor immune profiles noninvasively estimated by FDG PET with deep learning correlate with immunotherapy response in lung adenocarcinoma. Theranostics 2020, 10, 10838–10848. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Agioti, S.; Zaravinos, A. Immune Cytolytic Activity and Strategies for Therapeutic Treatment. Int. J. Mol. Sci. 2024, 25, 3624. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Helmink, B.A.; Reddy, S.M.; Gao, J.; Zhang, S.; Basar, R.; Thakur, R.; Yizhak, K.; Sade-Feldman, M.; Blando, J.; Han, G.; et al. B cells and tertiary lymphoid structures promote immunotherapy response. Nature 2020, 577, 549–555. [Google Scholar] [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561–565. [Google Scholar] [CrossRef]
- Petitprez, F.; de Reyniès, A.; Keung, E.Z.; Chen, T.W.; Sun, C.M.; Calderaro, J.; Jeng, Y.M.; Hsiao, L.P.; Lacroix, L.; Bougoüin, A.; et al. B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020, 577, 556–560. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, L.; Song, G.; Peng, G.; Chen, Z.; Yuan, Y.; Chen, T.; Zhong, T.; Chen, S.; Luo, Z.; et al. A novel tertiary lymphoid structure-associated signature accurately predicts patient prognosis and facilitates the selection of personalized treatment strategies for HNSCC. Front. Immunol. 2025, 16, 1551844. [Google Scholar] [CrossRef]
- Cascone, T.; Leung, C.H.; Weissferdt, A.; Pataer, A.; Carter, B.W.; Godoy, M.C.; Feldman, H.; William, W.N., Jr.; Xi, Y.; Basu, S.; et al. Neoadjuvant chemotherapy plus nivolumab with or without ipilimumab in operable non-small cell lung cancer: The phase 2 platform NEOSTAR trial. Nat. Med. 2023, 29, 593–604. [Google Scholar] [CrossRef]
- Long, S.; Li, M.; Chen, J.; Zhong, L.; Dai, G.; Pan, D.; Liu, W.; Yi, F.; Ruan, Y.; Zou, B.; et al. Transfer learning radiomic model predicts intratumoral tertiary lymphoid structures in hepatocellular carcinoma: A multicenter study. J. Immunother. Cancer 2025, 13, e011126. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef]
- Chamseddine, A.N.; Assi, T.; Mir, O.; Chouaib, S. Modulating tumor-associated macrophages to enhance the efficacy of immune checkpoint inhibitors: A TAM-pting approach. Pharmacol. Ther. 2022, 231, 107986. [Google Scholar] [CrossRef]
- De Keukeleire, S.J.; Vermassen, T.; Hilgert, E.; Creytens, D.; Ferdinande, L.; Rottey, S. Immuno-Oncological Biomarkers for Squamous Cell Cancer of the Head and Neck: Current State of the Art and Future Perspectives. Cancers 2021, 13, 1714. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jin, H.; He, Y.; Zhao, P.; Hu, Y.; Tao, J.; Chen, J.; Huang, Y. Targeting lipid metabolism to overcome EMT-associated drug resistance via integrin β3/FAK pathway and tumor-associated macrophage repolarization using legumain-activatable delivery. Theranostics 2019, 9, 265. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Rao, L.; Yao, H.; Wang, Z.; Ning, P.; Chen, X. Engineering macrophages for cancer immunotherapy and drug delivery. Adv. Mater. 2020, 32, 2002054. [Google Scholar] [CrossRef] [PubMed]
- Coulton, A.; Murai, J.; Qian, D.; Thakkar, K.; Lewis, C.E.; Litchfield, K. Using a pan-cancer atlas to investigate tumour associated macrophages as regulators of immunotherapy response. Nat. Commun. 2024, 15, 5665. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tharp, K.M.; Kersten, K.; Maller, O.; Timblin, G.A.; Stashko, C.; Canale, F.P.; Menjivar, R.E.; Hayward, M.K.; Berestjuk, I.; Ten Hoeve, J.; et al. Tumor-associated macrophages restrict CD8+ T cell function through collagen deposition and metabolic reprogramming of the breast cancer microenvironment. Nat. Cancer 2024, 5, 1045–1062. [Google Scholar] [CrossRef] [PubMed]
- Lang, R.; Patel, D.; Morris, J.J.; Rutschman, R.L.; Murray, P.J. Shaping gene expression in activated and resting primary macrophages by IL-10. J. Immunol. 2002, 169, 2253–2263. [Google Scholar] [CrossRef]
- Sawant, D.V.; Yano, H.; Chikina, M.; Zhang, Q.; Liao, M.; Liu, C.; Callahan, D.J.; Sun, Z.; Sun, T.; Tabib, T.; et al. Adaptive plasticity of IL-10+ and IL-35+ Treg cells cooperatively promotes tumor T cell exhaustion. Nat. Immunol. 2019, 20, 724–735. [Google Scholar] [CrossRef]
- Dadey, R.E.; Workman, C.J.; Vignali, D.A. Regulatory T cells in the tumor microenvironment. In Tumor Microenvironment: Hematopoietic Cells–Part B; Springer: Cham, Switzerland, 2020; pp. 105–134. [Google Scholar]
- Denize, T.; Jegede, O.A.; Matar, S.; El Ahmar, N.; West, D.J.; Walton, E.; Bagheri, A.S.; Savla, V.; Nabil Laimon, Y.; Gupta, S.; et al. PD-1 Expression on Intratumoral Regulatory T Cells Is Associated with Lack of Benefit from Anti–PD-1 Therapy in Metastatic Clear-Cell Renal Cell Carcinoma Patients. Clin. Cancer Res. 2024, 30, 803–813. [Google Scholar] [CrossRef]
- Lavie, D.; Ben-Shmuel, A.; Erez, N.; Scherz-Shouval, R. Cancer-associated fibroblasts in the single-cell era. Nat. Cancer 2022, 3, 793–807. [Google Scholar] [CrossRef]
- Mishra, P.; Banerjee, D.; Ben-Baruch, A. Chemokines at the crossroads of tumor-fibroblast interactions that promote malignancy. J. Leukoc. Biol. 2011, 89, 31–39. [Google Scholar] [CrossRef]
- Kuzet, S.E.; Gaggioli, C. Fibroblast activation in cancer: When seed fertilizes soil. Cell Tissue Res. 2016, 365, 607–619. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, X.; Zhou, Q.; Li, P.; Yu, B.; Li, J.; Qu, Y.; Yan, J.; Yu, Y.; Yan, M.; et al. Hepatocyte growth factor activates tumor stromal fibroblasts to promote tumorigenesis in gastric cancer. Cancer Lett. 2013, 335, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Rimal, R.; Desai, P.; Daware, R.; Hosseinnejad, A.; Prakash, J.; Lammers, T.; Singh, S. Cancer-associated fibroblasts: Origin, function, imaging, and therapeutic targeting. Adv. Drug Deliv. Rev. 2022, 189, 114504. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Wei, R.; Liu, C.; Zhao, Z.; Liu, X.; Wang, Y.; Liu, F.; Liu, X. Antigen-presenting cancer associated fibroblasts enhance antitumor immunity and predict immunotherapy response. Nat. Commun. 2025, 16, 2175. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, K.; Liu, K.; Hu, B.; Du, G.; Chen, X.; Xiao, L.; Zhang, Y.; Jiang, L.; Jing, N.; Cheng, C.; et al. Iron-loaded cancer-associated fibroblasts induce immunosuppression in prostate cancer. Nat. Commun. 2024, 15, 9050. [Google Scholar] [CrossRef]
- Nikanjam, M.; Kato, S.; Kurzrock, R. Liquid biopsy: Current technology and clinical applications. J. Hematol. Oncol. 2022, 15, 131. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chan, Y.T.; Zhang, C.; Wu, J.; Lu, P.; Xu, L.; Yuan, H.; Feng, Y.; Chen, Z.S.; Wang, N. Biomarkers for diagnosis and therapeutic options in hepatocellular carcinoma. Mol. Cancer 2024, 23, 189. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hwang, J.; Holl, E.; Wu, Y.; Agarwal, A.; Starr, M.D.; Reyes Martinez, M.A.; Wang, A.Z.; Armstrong, A.J.; Harrison, M.R.; George, D.J.; et al. Circulating immune biomarkers correlating with response in patients with metastatic renal cell carcinoma on immunotherapy. JCI Insight 2025, 10, e185963. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rogado, J.; Pozo, F.; Troule, K.; Sánchez-Torres, J.M.; Romero-Laorden, N.; Mondejar, R.; Donnay, O.; Ballesteros, A.; Pacheco-Barcia, V.; Aspa, J.; et al. Peripheral blood mononuclear cells predict therapeutic efficacy of immunotherapy in NSCLC. Cancers. 2022, 14, 2898. [Google Scholar] [CrossRef]
- Kievit, H.; Muntinghe-Wagenaar, M.B.; Abdulahad, W.H.; Rutgers, A.; Hijmering-Kappelle, L.B.M.; Hiddinga, B.I.; Ubbels, J.F.; Wijsman, R.; van der Leij, M.J.; Bijzet, J.; et al. Baseline Blood CD8+ T Cell Activation Potency Discriminates Responders from Non-Responders to Immune Checkpoint Inhibition Combined with Stereotactic Radiotherapy in Non-Small-Cell Lung Cancer. Cancers 2024, 16, 2592. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Koh, J.; Kim, Y.; Lee, K.Y.; Hur, J.Y.; Kim, M.S.; Kim, B.; Cho, H.J.; Lee, Y.C.; Bae, Y.H.; Ku, B.M.; et al. MDSC subtypes and CD39 expression on CD8+ T cells predict the efficacy of anti-PD-1 immunotherapy in patients with advanced NSCLC. Eur. J. Immunol. 2020, 50, 1810–1819. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Li, Y.; Tan, S.; Cheng, T.; Luo, Y.; Zhang, L. Pretreatment neutrophil-to-lymphocyte ratio is associated with immunotherapy efficacy in patients with advanced cancer: A systematic review and meta-analysis. Sci. Rep. 2025, 15, 446. [Google Scholar] [CrossRef] [PubMed]
- Dragu, D.; Necula, L.G.; Bleotu, C.; Diaconu, C.C.; Chivu-Economescu, M. Soluble PD-L1: From Immune Evasion to Cancer Therapy. Life 2025, 15, 626. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Zhang, X.; Li, L.; Shi, X.; Zhao, Y.; Cai, Z.; Ni, N.; Yang, D.; Meng, Z.; Gao, X.; Huang, L.; et al. Development of a tertiary lymphoid structure-based prognostic model for breast cancer: Integrating single-cell sequencing and machine learning to enhance patient outcomes. Front. Immunol. 2025, 16, 1534928. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yang, J.; Liu, Q.; Shyr, Y. A Large-Scale Meta-Analysis Reveals Positive Feedback between Macrophages and T Cells That Sensitizes Tumors to Immunotherapy. Cancer Res. 2024, 84, 626–638. [Google Scholar] [CrossRef]
- Xie, Y.; Guan, S.; Li, Z.; Cai, G.; Liu, Y.; Li, G.; Huang, P.; Lin, M. Identification of a metabolic-immune signature associated with prognosis in colon cancer and exploration of potential predictive efficacy of immunotherapy response. Clin. Exp. Med. 2025, 25, 46. [Google Scholar] [CrossRef]
- Xia, Y.; Huang, C.; Zhong, M.; Zhong, H.; Ruan, R.; Xiong, J.; Yao, Y.; Zhou, J.; Deng, J. Targeting HGF/c-MET signaling to regulate the tumor microenvironment: Implications for counteracting tumor immune evasion. Cell Commun. Signal. 2025, 23, 46. [Google Scholar] [CrossRef]
- Zhang, Y.; Ding, L.; Zhang, Z.; Shen, L.; Guo, Y.; Zhang, W.; Yu, Y.; Gu, Z.; Liu, J.; Kadier, A.; et al. An Integrated Approach Utilizing Single-Cell and Bulk RNA-Sequencing for the Identification of a Mitophagy-Associated Genes Signature: Implications for Prognostication and Therapeutic Stratification in Prostate Cancer. Biomedicines 2025, 13, 311. [Google Scholar] [CrossRef]
- Cao, K.; Wei, S.; Ma, T.; Yang, X.; Wang, Y.; He, X.; Lu, M.; Bai, Y.; Qi, C.; Zhang, L.; et al. Integrating bulk, single-cell, and spatial transcriptomics to identify and functionally validate novel targets to enhance immunotherapy in NSCLC. npj Precis. Oncol. 2025, 9, 112. [Google Scholar] [CrossRef]
- Platten, M.; Nollen, E.A.; Röhrig, U.F.; Fallarino, F.; Opitz, C.A. Tryptophan metabolism as a common therapeutic target in cancer, neurodegeneration and beyond. Nat. Rev. Drug Discov. 2019, 18, 379–401. [Google Scholar] [CrossRef]
- de Jong, C.; Deneer, V.H.M.; Kelder, J.C.; Ruven, H.; Egberts, T.C.G.; Herder, G.J.M. Association between serum biomarkers CEA and LDH and response in advanced non-small cell lung cancer patients treated with platinum-based chemotherapy. Thorac. Cancer 2020, 11, 1790–1800. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, Y.; Yan, X.; Song, Q.; Wang, G.; Hu, Y.; Jiao, S.; Wang, J. Pretreatment lactate dehydrogenase may predict outcome of advanced non small-cell lung cancer patients treated with immune checkpoint inhibitors: A meta-analysis. Cancer Med. 2019, 8, 1467–1473. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Lian, D.; Chen, M.; Liu, Y.; Zhang, M.; Zeng, D.; Zhou, S.K.; Ying, W. Prognostic value of a lactate metabolism gene signature in lung adenocarcinoma and its associations with immune checkpoint blockade therapy response. Medicine 2024, 103, e39371. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Geiger, R.; Rieckmann, J.C.; Wolf, T.; Basso, C.; Feng, Y.; Fuhrer, T.; Kogadeeva, M.; Picotti, P.; Meissner, F.; Mann, M.; et al. L-arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell 2016, 167, 829–842. [Google Scholar] [CrossRef]
- Coutzac, C.; Jouniaux, J.M.; Paci, A.; Schmidt, J.; Mallardo, D.; Seck, A.; Asvatourian, V.; Cassard, L.; Saulnier, P.; Lacroix, L.; et al. Systemic short chain fatty acids limit antitumor effect of CTLA-4 blockade in hosts with cancer. Nat. Commun. 2020, 11, 2168. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.N.; et al. An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef]
- Johnson, M.O.; Wolf, M.M.; Madden, M.Z.; Andrejeva, G.; Sugiura, A.; Contreras, D.C.; Maseda, D.; Liberti, M.V.; Paz, K.; Kishton, R.J.; et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase-dependent metabolism. Cell 2018, 175, 1780–1795. [Google Scholar] [CrossRef]
- Lara, P.N., Jr.; Villanueva, L.; Ibanez, C.; Erman, M.; Lee, J.L.; Heinrich, D.; Lipatov, O.N.; Gedye, C.; Gokmen, E.; Acevedo, A.; et al. A randomized, open-label, phase 3 trial of pembrolizumab plus epacadostat versus sunitinib or pazopanib as first-line treatment for metastatic renal cell carcinoma (KEYNOTE-679/ECHO-302). BMC Cancer 2024, 23 (Suppl. 1), 1253. [Google Scholar] [CrossRef]
- Mabuchi, R.; Hara, T.; Matsumoto, T.; Shibata, Y.; Nakamura, N.; Nakamura, H.; Kitagawa, J.; Kanemura, N.; Goto, N.; Shimizu, M.; et al. High serum concentration of L-kynurenine predicts unfavorable outcomes in patients with acute myeloid leukemia. Leuk. Lymphoma 2016, 57, 92–98. [Google Scholar] [CrossRef]
- Zhai, L.; Dey, M.; Lauing, K.L.; Gritsina, G.; Kaur, R.; Lukas, R.V.; Nicholas, M.K.; Rademaker, A.W.; Dostal, C.R.; McCusker, R.H.; et al. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J. Clin. Neurosci. 2015, 22, 1964–1968. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bekki, S.; Hashimoto, S.; Yamasaki, K.; Komori, A.; Abiru, S.; Nagaoka, S.; Saeki, A.; Suehiro, T.; Kugiyama, Y.; Beppu, A.; et al. Serum kynurenine levels are a novel biomarker to predict the prognosis of patients with hepatocellular carcinoma. PLoS ONE 2020, 15, e0241002. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, P.; Lu, X.; Li, J.; Qi, D.; Zang, L.; Lin, J.; Liu, Y.; Zhai, S.; Fu, D.; et al. Pan-cancer analysis implicates novel insights of lactate metabolism into immunotherapy response prediction and survival prognostication. J. Exp. Clin. Cancer Res. 2024, 43, 125. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.; Sun, F.; Yang, J.C.; Cai, M.H.; Huai, M.X.; Pan, J.X.; Zhang, F.Y.; Xu, L.M. Novel lactylation-related signature to predict prognosis for pancreatic adenocarcinoma. World J. Gastroenterol. 2024, 30, 2575–2602. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Xu, X.; Pan, X.; Fan, Z.; Xia, J.; Ren, X. Lactate dehydrogenase B as a metabolism-related marker for immunotherapy in head and neck squamous cell carcinoma. Cell Signal. 2024, 120, 111200. [Google Scholar] [CrossRef]
- Peyraud, F.; Guégan, J.P.; Bodet, D.; Nafia, I.; Fontan, L.; Auzanneau, C.; Cousin, S.; Roubaud, G.; Cabart, M.; Chomy, F.; et al. Circulating L-arginine predicts the survival of cancer patients treated with immune checkpoint inhibitors. Ann. Oncol. 2022, 33, 1041–1051. [Google Scholar] [CrossRef]
- Nomura, M.; Nagatomo, R.; Doi, K.; Shimizu, J.; Baba, K.; Saito, T.; Matsumoto, S.; Inoue, K.; Muto, M. Association of Short-Chain Fatty Acids in the Gut Microbiome with Clinical Response to Treatment with Nivolumab or Pembrolizumab in Patients with Solid Cancer Tumors. JAMA Netw. Open 2020, 3, e202895. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Siamakpour-Reihani, S.; Cao, F.; Lyu, J.; Ren, Y.; Nixon, A.B.; Xie, J.; Bush, A.T.; Starr, M.D.; Bain, J.R.; Muehlbauer, M.J.; et al. Evaluating immune response and metabolic related biomarkers pre-allogenic hematopoietic stem cell transplant in acute myeloid leukemia. PLoS ONE 2022, 17, e0268963, Erratum in PLoS ONE 2024, 19, e0313701. [Google Scholar] [CrossRef]
- Cheng, X.; Tan, X.; Wang, W.; Zhang, Z.; Zhu, R.; Wu, M.; Li, M.; Chen, Y.; Liang, Z.; Zhu, P.; et al. Long-Chain Acylcarnitines Induce Senescence of Invariant Natural Killer T Cells in Hepatocellular Carcinoma. Cancer Res. 2023, 83, 582–594. [Google Scholar] [CrossRef]
- Zhang, E.; Ding, C.; Li, S.; Aikemu, B.; Zhou, X.; Fan, X.; Sun, J.; Yang, X.; Zheng, M. Polyamine metabolism patterns characterized tumor microenvironment, prognosis, and response to immunotherapy in colorectal cancer. Cancer Cell Int. 2023, 23, 96. [Google Scholar] [CrossRef]
- Du, M.; Meng, X.; Zhou, B.; Song, W.; Shi, J.; Liang, M.; Liang, Y.; Gao, Y. A risk score based on polyamine metabolism and chemotherapy-related genes predicts prognosis and immune cells infiltration of lung adenocarcinoma. J. Cell Mol. Med. 2024, 28, e18387. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Schwabe, R.F.; Jobin, C. The microbiome and cancer. Nat. Rev. Cancer 2013, 13, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Constantin, M.; Chifiriuc, M.C.; Mihaescu, G.; Corcionivoschi, N.; Burlibasa, L.; Bleotu, C.; Tudorache, S.; Mitache, M.M.; Filip, R.; Munteanu, S.G.; et al. Microbiome and Cancer: From Mechanistic Implications in Disease Progression and Treatment to Development of Novel Antitumoral Strategies. Front. Immunol. 2024, 15, 1373504. [Google Scholar] [CrossRef]
- Fernandez, E.; Wargo, J.A.; Helmink, B.A. The Microbiome and Cancer: A Translational Science Review. JAMA 2025, 333, 2188–2196. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef]
- Nejman, D.; Livyatan, I.; Fuks, G.; Gavert, N.; Zwang, Y.; Geller, L.T.; Rotter-Maskowitz, A.; Weiser, R.; Mallel, G.; Gigi, E.; et al. The human tumor microbiome is composed of tumor type–specific intracellular bacteria. Science 2020, 368, 973–980. [Google Scholar] [CrossRef]
- Riquelme, E.; Zhang, Y.; Zhang, L.; Montiel, M.; Zoltan, M.; Dong, W.; Quesada, P.; Sahin, I.; Chandra, V.; San Lucas, F.A.; et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 2019, 178, 795–806.e12. [Google Scholar] [CrossRef]
- Coker, O.O.; Dai, Z.; Nie, Y.; Zhao, G.; Cao, L.; Nakatsu, G.; Wu, W.K.K.; Wong, S.H.; Chen, H.; Fang, J.Y.; et al. Mucosal microbiome dysbiosis in gastric carcinogenesis. Gut 2018, 67, 1024–1032. [Google Scholar] [CrossRef]
- Pushalkar, S.; Hun-deyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The pancreatic cancer microbiome promotes oncogenesis by in-duction of innate and adaptive immune suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Long, J.; Yang, X.; Lin, J.; Song, Y.; Xie, F.; Xun, Z.; Wang, Y.; Wang, Y.; et al. Gut microbiome is associated with the clinical response to anti-PD-1 based immunotherapy in hepatobiliary cancers. J. Immunother. Cancer 2021, 9, e003334. [Google Scholar] [CrossRef]
- Zitvogel, L.; Ma, Y.; Raoult, D.; Kroemer, G.; Gajewski, T.F. The microbiome in cancer immunotherapy: Diagnostic tools and therapeutic strategies. Science 2018, 359, 1366–1370. [Google Scholar] [CrossRef]
- Dean, A.P.; Comito, O.R.; Yusoff, I.; Rao, S.; Johansson, M.; Navadgi, S.; Webber, L.; Watanabe, Y. Potential Effect of the Gut Microbiome on Tumour Response to Anti-Cancer Treatment in Patients Diagnosed with Pancreatic Ductal Adenocarcinoma. J. Clin. Oncol. 2025, 43, 757. [Google Scholar] [CrossRef]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Nat. Med. 2018, 24, 144–153. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut microbiome influences efficacy of PD-1–based immunotherapy against epithelial tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef]
- Matson, V.; Fessler, J.; Bao, R.; Chongsuwat, T.; Zha, Y.; Alegre, M.-L.; Luke, J.J.; Gajewski, T.F. The commensal microbiome is associated with anti–PD-1 efficacy in metastatic melanoma patients. Science 2018, 359, 104–108. [Google Scholar] [CrossRef]
- Frankel, A.E.; Coughlin, L.A.; Kim, J.; Froehlich, T.W.; Xie, Y.; Frenkel, E.P.; Koh, A.Y. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia 2017, 19, 848–855. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Derosa, L.; Routy, B.; Fidelle, M.; Iebba, V.; Alla, L.; Pasolli, E.; Segata, N.; Desnoyer, A.; Pietrantonio, F.; Ferrere, G.; et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur. Urol. 2020, 78, 195–206. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nat. Med. 2019, 25, 377–388. [Google Scholar] [CrossRef]
- Andrews, M.C.; Duong, C.P.; Gopalakrishnan, V.; Iebba, V.; Chen, W.S.; Derosa, L.; Khan, M.A.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut microbiota signatures are associated with toxicity to combined CTLA-4 and PD-1 blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef]
- Vivarelli, S.; Salemi, R.; Candido, S.; Falzone, L.; Santagati, M.; Stefani, S.; Torino, F.; Banna, G.L.; Tonini, G.; Libra, M. Gut microbiota and cancer: From pathogenesis to therapy. Cancers 2019, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Farhadi Rad, H.; Tahmasebi, H.; Javani, S.; Hemati, M.; Zakerhamidi, D.; Hosseini, M.; Aliba-baei, F.; Banihashemian, S.Z.; Oksenych, V.; Eslami, M. Microbiota and Cytokine Modula-tion: Innovations in Enhancing Anticancer Immunity and Personalized Cancer Thera-pies. Biomedicines 2024, 12, 2776. [Google Scholar] [CrossRef] [PubMed]
- Luu, M.; Riester, Z.; Baldrich, A.; Reichardt, N.; Yuille, S.; Busetti, A.; Klein, M.; Wempe, A.; Leister, H.; Raifer, H.; et al. Microbial short-chain fatty acids modulate CD8+ T cell responses and improve adoptive immunotherapy for cancer. Nat. Commun. 2021, 12, 4077. [Google Scholar] [CrossRef] [PubMed]
- Baruch, E.N.; Youngster, I.; Ben-Betzalel, G.; Ortenberg, R.; Lahat, A.; Katz, L.; Adler, A.; Dick-Necula, D.; Fishman, S.; Mandelboim, O.; et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science 2021, 371, 602–609. [Google Scholar] [CrossRef]
- Davar, D.; Dzutsev, A.K.; McCulloch, J.A.; Rodrigues, R.R.; Chauvin, J.M.; Morrison, R.M.; Deblasio, R.N.; Menna, C.; Ding, Q.; Pagliano, O.; et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma. Science 2021, 371, 595–602. [Google Scholar] [CrossRef]
- Sharma, P.; Siddiqui, B.A.; Anandhan, S.; Yadav, S.S.; Subudhi, S.K.; Gao, J.; Goswami, S.; Allison, J.P. The next decade of immune checkpoint therapy. Cancer Discov. 2021, 11, 838–857. [Google Scholar] [CrossRef]
- Hegde, P.S.; Chen, D.S. Top 10 challenges in cancer immunotherapy. Immunity 2023, 58, 312–326. [Google Scholar] [CrossRef]
- Knorr, D.A.; Ni, Z.; Hermanson, D.; Hexum, M.K.; Bendzick, L.; Cooper, L.J.; Lee, D.A.; Kaufman, D.S. Clinical-scale derivation of natural killer cells from human pluripotent stem cells for cancer therapy. Stem Cells Transl. Med. 2013, 2, 274–283. [Google Scholar] [CrossRef]
- Peng, K.; Fu, Y.X.; Liang, Y. Engineering cytokines for tumor-targeting and selective T cell activation. Trends Mol. Med. 2025, 31, 373–387. [Google Scholar] [CrossRef]
- Silk, A.W.; O’Day, S.J.; Kaufman, H.L.; Bryan, J.; Norrell, J.T.; Imbergamo, C.; Portal, D.; Zambrano-Acosta, E.; Palmeri, M.; Fein, S.; et al. A phase 1b single-arm trial of intratumoral oncolytic virus V937 in combination with pembrolizumab in patients with advanced melanoma: Results from the CAPRA study. Cancer Immunol. Immunother. 2023, 72, 1405–1415. [Google Scholar] [CrossRef]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti–PD-L1 efficacy. Science 2023, 381, 1341–1347. [Google Scholar] [CrossRef] [PubMed]
- Sarnaik, A.A.; Hwu, P.; Mulé, J.J.; Pilon-Thomas, S. Tumor-infiltrating lymphocytes: A new hope. Cancer Cell 2024, 42, 1315–1318. [Google Scholar] [CrossRef] [PubMed]
- Bandara, S.; Raveendran, S. Current Landscape and Future Directions in Cancer Immunotherapy: Therapies, Trials, and Challenges. Cancers 2025, 17, 821. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.Y.; Zhang, S. Safety and efficacy of personalized cancer vaccines in combination with immune checkpoint inhibitors in cancer treatment. Front. Oncol. 2021, 11, 663264. [Google Scholar] [CrossRef]
- Wu, D.W.; Jia, S.P.; Xing, S.J.; Ma, H.L.; Wang, X.; Tang, Q.Y.; Li, Z.W.; Wu, Q.; Bai, M.; Zhang, X.Y.; et al. Personalized neoantigen cancer vaccines: Current progression, challenges and a bright future. Clin. Exp. Med. 2024, 24, 229. [Google Scholar] [CrossRef]
- Yan, C.; Yang, J.; Saleh, N.; Chen, S.C.; Ayers, G.D.; Abramson, V.G.; Mayer, I.A.; Richmond, A. Inhibition of the PI3K/mTOR pathway in breast cancer to enhance response to immune checkpoint inhibitors in breast cancer. Int. J. Mol. Sci. 2021, 22, 5207. [Google Scholar] [CrossRef]
- Peng, X.; Huang, X.; Lulu, T.B.; Jia, W.; Zhang, S.; Cohen, L.; Huang, S.; Fan, J.; Chen, X.; Liu, S.; et al. A novel pan-PI3K inhibitor KTC1101 synergizes with anti-PD-1 therapy by targeting tumor suppression and immune activation. Mol. Cancer 2024, 23, 54. [Google Scholar] [CrossRef]
- Isoyama, S.; Mori, S.; Sugiyama, D.; Kojima, Y.; Tada, Y.; Shitara, K.; Hinohara, K.; Dan, S.; Nishikawa, H. Cancer immunotherapy with PI3K and PD-1 dual-blockade via optimal modulation of T cell activation signal. J. Immunother. Cancer 2021, 9, e002279. [Google Scholar] [CrossRef]
- Suleiman, R.; McGarrah, P.; Baral, B.; Owen, D.; Vera Aguilera, J.; Halfdanarson, T.R.; Price, K.A.; Fuentes Bayne, H.E. Alpelisib and Immunotherapy: A Promising Combination for Recurrent and Metastatic Squamous Cell Carcinoma of the Head and Neck. Cancer Rep. 2024, 7, e70023. [Google Scholar] [CrossRef]
- Shalhout, S.Z.; Miller, D.M.; Emerick, K.S.; Kaufman, H.L. Therapy with oncolytic viruses: Progress and challenges. Nat. Rev. Clin. Oncol. 2023, 20, 160–177. [Google Scholar] [CrossRef]
- Shah, D.D.; Chorawala, M.R.; Raghani, N.R.; Patel, R.; Fareed, M.; Kashid, V.A.; Prajapati, B.G. Tumor microenvironment: Recent advances in understanding and its role in modulating cancer therapies. Med. Oncol. 2025, 42, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Spagnolo, C.C.; Pepe, F.; Ciappina, G.; Nucera, F.; Ruggeri, P.; Squeri, A.; Speranza, D.; Silvestris, N.; Malapelle, U.; Santarpia, M. Circulating biomarkers as predictors of response to immune checkpoint inhibitors in NSCLC: Are we on the right path? Crit. Rev. Oncol./Hematol. 2024, 197, 104332. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Guo, H.; Zhao, Y.; Liu, Z.; Wang, C.; Bu, J.; Sun, T.; Wei, J. Liquid biopsy in cancer current: Status, challenges and future prospects. Signal Transduct. Target. Ther. 2024, 9, 336. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; Liu, J.; Subudhi, S.K.; Poon, C.; Gant, K.L.; et al. Immune checkpoint therapy—Current perspectives and future directions. Cell 2023, 186, 1652–1669. [Google Scholar] [CrossRef]
- Goswami, S.; Chen, Y.; Anandhan, S.; Szabo, P.M.; Basu, S.; Blando, J.M.; Liu, W.; Zhang, J.; Natarajan, S.M.; Xiong, L.; et al. ARID1A mutation plus CXCL13 expression act as combinatorial biomarkers to predict responses to immune checkpoint therapy in mUCC. Sci. Transl. Med. 2020, 12, eabc4220. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Chen, Y.; Li, X.; Long, S.; Shi, Y.; Yu, Y.; Wu, W.; Han, L.; Wang, S. The role of PD-1/PD-L1 and application of immune-checkpoint inhibitors in human cancers. Front. Immunol. 2022, 13, 964442. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kato, R.; Obara, W. Persisting challenges in the development of predictive biomarkers for immuno-oncology therapies for renal cell carcinoma. Expert Rev. Anticancer Ther. 2025, 25, 97–103. [Google Scholar] [CrossRef]
- Kim, P.; Joe, S.; Kim, H.; Jeong, H.; Park, S.; Song, J.; Kim, W.; Lee, Y.G. Hidden Partner of Immunity: Microbiome as an Innovative Companion in Immunotherapy. Int. J. Mol. Sci. 2025, 26, 856. [Google Scholar] [CrossRef]
- Fuentes, A.M. The Role of the Microbiome in Skin Cancer Development and Treatment. Curr. Opin. Oncol. 2025, 37, 121–125. [Google Scholar] [CrossRef]
- Lu, Y.; Yuan, X.; Wang, M.; He, Z.; Li, H.; Wang, J.; Li, Q. Gut microbiota influence immunotherapy responses: Mechanisms and therapeutic strategies. J. Hematol. Oncol. 2022, 15, 47. [Google Scholar] [CrossRef]
- Kermany, D.S.; Ahn, J.Y.; Vasquez, M.; Zhang, W.; Wang, L.; Liu, K.; Xu, Z.; Cho, M.S.; Carlos-Alcalde, W.; Lee, H.; et al. Multiscale 3D spatial analysis of the tumor microenvironment using whole-tissue digital histopathology. Cancer Commun. 2025, 45, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Lehr, K.; Oosterlinck, B.; Then, C.K.; Gemmell, M.R.; Gedgaudas, R.; Bornschein, J.; Kupcinskas, J.; Smet, A.; Hold, G.; Link, A. Comparison of different microbiome analysis pipe-lines to validate their reproducibility of gastric mucosal microbiome composition. mSystems 2025, 10, e01358-24. [Google Scholar] [CrossRef]
- Ghaddar, B.C.; Blaser, M.J.; De, S. Revisiting the Cancer Microbiome Using PRISM. bioRxiv 2025. [Google Scholar] [CrossRef]
- Simonsen, M.; Mendoza López, R.V.; Maistro, S.; Ikeoka, L.T.; Pereira, G.F.L.; Lugão, A.B.; Sadalla, J.C.; Katayama, M.L.H.; Folgueira, M.A.A.K. Peritoneal Chemotherapy Delivery Systems for Ovarian Cancer Treatment: Systematic Review of Animal Models. Front. Oncol. 2025, 14, 1487376. [Google Scholar] [CrossRef]
- Gamrath, L.; Pedersen, T.B.; Møller, M.V.; Volmer, L.M.; Holst-Christensen, L.; Vestermark, L.W.; Donskov, F. Role of the Microbiome and Diet for Response to Cancer Checkpoint Immunotherapy: A Narrative Review of Clinical Trials. Curr. Oncol. Rep. 2025, 27, 45–58. [Google Scholar] [CrossRef]
- Monette, A.; Aguilar-Mahecha, A.; Altinmakas, E.; Angelos, M.G.; Assad, N.; Batist, G.; Bommareddy, P.K.; Bonilla, D.L.; Borchers, C.H.; Church, S.E.; et al. The Society for Immunotherapy of Cancer Perspective on Tissue-Based Technologies for Immuno-Oncology Biomarker Discovery and Application. Clin. Cancer Res. 2025, 31, 439–456. [Google Scholar] [CrossRef] [PubMed]
- Taube, J.M.; Sunshine, J.C.; Angelo, M.; Akturk, G.; Eminizer, M.; Engle, L.L.; Ferreira, C.S.; Gnjatic, S.; Green, B.; Greenbaum, S.; et al. Society for Immunotherapy of Cancer: Updates and best practices for multiplex immunohistochemistry (IHC) and immunofluorescence (IF) image analysis and data sharing. J. Immunother. Cancer 2025, 13, e008875. [Google Scholar] [CrossRef]
- Li, T.; Li, Y.; Zhu, X.; He, Y.; Wu, Y.; Ying, T.; Xie, Z. Artificial intelligence in cancer immunotherapy: Applications in neoantigen recognition, antibody design and immunotherapy response prediction. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2023; Volume 91, pp. 50–69. [Google Scholar]
- Yin, X.; Liao, H.; Yun, H.; Lin, N.; Li, S.; Xiang, Y.; Ma, X. Artificial intelligence-based prediction of clinical outcome in immunotherapy and targeted therapy of lung cancer. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2022; Volume 86, pp. 146–159. [Google Scholar]
- Painuli, D.; Bhardwaj, S. Recent advancement in cancer diagnosis using machine learning and deep learning techniques: A comprehensive review. Comput. Biol. Med. 2022, 146, 105580. [Google Scholar] [CrossRef]
- Li, K.; Luo, H.; Huang, L.; Luo, H.; Zhu, X. Microsatellite instability: A review of what the oncologist should know. Cancer Cell Int. 2020, 20, 1–3. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, X.; Tang, Q.; Yang, K.; Guo, X.; Wang, A.; Zhang, Y.; Hao, Y.; Pei, Z.; Wang, D.; et al. Gene expression-based machine learning approach to predict immunotherapy response and survival in patients with advanced cancer. JCO 2023, 41, 1550. [Google Scholar] [CrossRef]
- Tao, W.; Sun, Q.; Xu, B.; Wang, R. Towards the Prediction of Responses to Cancer Immunotherapy: A Multi-Omics Review. Life 2025, 15, 283. [Google Scholar] [CrossRef]

| Multiplex Tissue Staining | Advantages | Limiting Factors | Perspectives |
|---|---|---|---|
| mIHC/IF technologies | In-depth research on functional cellular states and spatial dynamics related to cell-to-cell interactions within intricate tumor microenvironments (TMEs). | The workflow presents several challenges, including pre-analytical issues such as staining variability, as well as analytical complexity and difficulties in the interpretation and querying of post-analytical data. | The whole-slide AI-based “AstroPath” platform has identified predictive features in the pre-treatment of melanoma tissue samples that are associated with the response to anti–PD-1 therapy. |
| PhenoCycler-Fusion: single-cell phenotypes and spatial relationships via DNA-conjugated antibodies | Employs DNA-conjugated antibodies along with the cyclic addition and removal of complementary fluorescently labeled DNA probes to enable the simultaneous visualization of up to 60 markers in situ. | The approach is associated with high costs, primarily due to the use of antibodies and tagged DNA oligonucleotides. Additionally, the detection of low-abundance proteins often requires signals’ amplification, as the native signal is typically insufficient for reliable quantification. | Future advancements in this technology may involve the incorporation of nucleic acid labeling, enabling the simultaneous detection of nucleic acids. This could open new avenues for investigating causative genetic mutations and post-transcriptional modifications. |
| Single-cell RNA sequencing: TME gene expression and T-cell receptor sequencing | Powerful tool for thoroughly analyzing the tumor microenvironment (TME) to identify new and effective immunotherapies. | Single-cell RNA sequencing data is inherently noisy, making it difficult to establish clear correlations between genotype and phenotype due to technical limitations. These challenges are further amplified when analyzing cells derived from solid tumor tissues, where variability in tissue dissociation methods and cryopreservation conditions can significantly impact data’s quality and consistency. | Additional capabilities may include the inference of splice variants, chromosomal copy-number aberrations, and even the prediction of future cellular states. However, these advanced analyses require specialized expertise and careful interpretation to ensure accuracy and reliability. |
| Visium Spatial Gene Expression: barcoding transcriptomes across TMEs | It has facilitated the identification of different B-cell maturation states within tertiary lymphoid structures, utilizing Visium Spatial Gene Expression (SGE) in conjunction with pooled CRISPR screens. | Visium SGE is subject to several significant limitations, including suboptimal capture efficiency, restricted sequencing depth, and a high incidence of dropout events. These factors complicate the study of cell–cell interactions and the organization of higher-order tissue structures that influence immune responses. A key challenge is its lack of single-cell resolution, which restricts its ability to resolve fine-grained spatial details. | Integrating this approach with single-cell transcriptomics holds promise for overcoming current limitations in spatial resolution. |
| GeoMx: protein and transcriptome barcoding TMEs | It allows for a high-plex evaluation of transcripts (over 18,000 genes) and/or proteins (more than 100 proteins) within a single tissue sample. This technology is applicable to formalin-fixed, paraffin-embedded (FFPE), and fresh-frozen tissues. Interactive software facilitates collaboration, enabling the profiling of RNA transcripts and proteins according to the tissue’s spatial distribution. | The platform is relatively expensive, and whole-tissue analysis is more efficiently performed using alternatives like Visium or PhenoCycler-Fusion, which can profile the entire slide. Moreover, GeoMx lacks single-cell and subcellular resolution, limiting its utility for high-resolution spatial studies. | This approach has been employed to detect biomarkers associated with responses to bispecific antibody therapy in bone marrow biopsies. It has also been utilized to identify biomarkers linked to responses to cellular immunotherapies, including CAR T cell and transgenic T cell treatments. |
| Spatially resolved proteomics: mass spectrometry imaging and related technologies | This technique permits the in situ analysis of the spatial proteome, lipidome, glycome, and metabolome directly within tissue sections, without the need for specific staining or labeling, unlike many conventional visualization methods. It has been extensively applied in high-resolution studies of small molecular markers, including lipids, metabolites, and elemental species, as well as in the spatial characterization of drug compounds. | It lacks compatibility with the characterization of high-molecular-weight compounds. | It is anticipated that key advancements in MSImg will include the precise in situ analysis and visualization of novel biomarkers via single-cell spatial multi-omics, accelerated real-time examination of living tissues, and precision-guided surgery—all of which represent cutting-edge frontiers in the field. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roznovan, C.N.; Măruțescu, L.G.; Gradisteanu Pircalabioru, G. Immuno-Oncology at the Crossroads: Confronting Challenges in the Quest for Effective Cancer Therapies. Int. J. Mol. Sci. 2025, 26, 6177. https://doi.org/10.3390/ijms26136177
Roznovan CN, Măruțescu LG, Gradisteanu Pircalabioru G. Immuno-Oncology at the Crossroads: Confronting Challenges in the Quest for Effective Cancer Therapies. International Journal of Molecular Sciences. 2025; 26(13):6177. https://doi.org/10.3390/ijms26136177
Chicago/Turabian StyleRoznovan, Claudiu Natanael, Luminița Gabriela Măruțescu, and Gratiela Gradisteanu Pircalabioru. 2025. "Immuno-Oncology at the Crossroads: Confronting Challenges in the Quest for Effective Cancer Therapies" International Journal of Molecular Sciences 26, no. 13: 6177. https://doi.org/10.3390/ijms26136177
APA StyleRoznovan, C. N., Măruțescu, L. G., & Gradisteanu Pircalabioru, G. (2025). Immuno-Oncology at the Crossroads: Confronting Challenges in the Quest for Effective Cancer Therapies. International Journal of Molecular Sciences, 26(13), 6177. https://doi.org/10.3390/ijms26136177