

Amprenavir Mitigates Pepsin-Induced Transcriptomic Changes in Normal and Precancerous Esophageal Cells

 , , ,
, , ,  and
and 
Abstract
1. Introduction
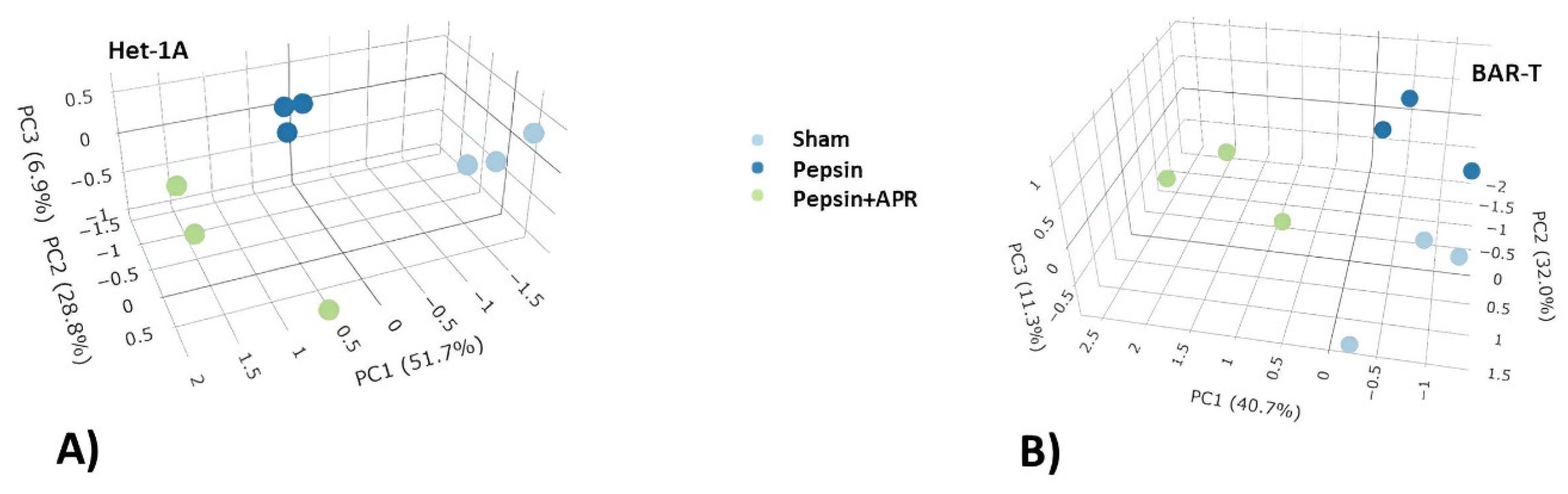
2. Results
3. Discussion
3.1. Pepsin Causes Cell Injury and Disrupts Cytoskeletal Organization
3.2. Amprenavir Enhances Repair Pathways and Supports Epithelial Integrity Against Peptic Damage
3.3. Summary and Limitations
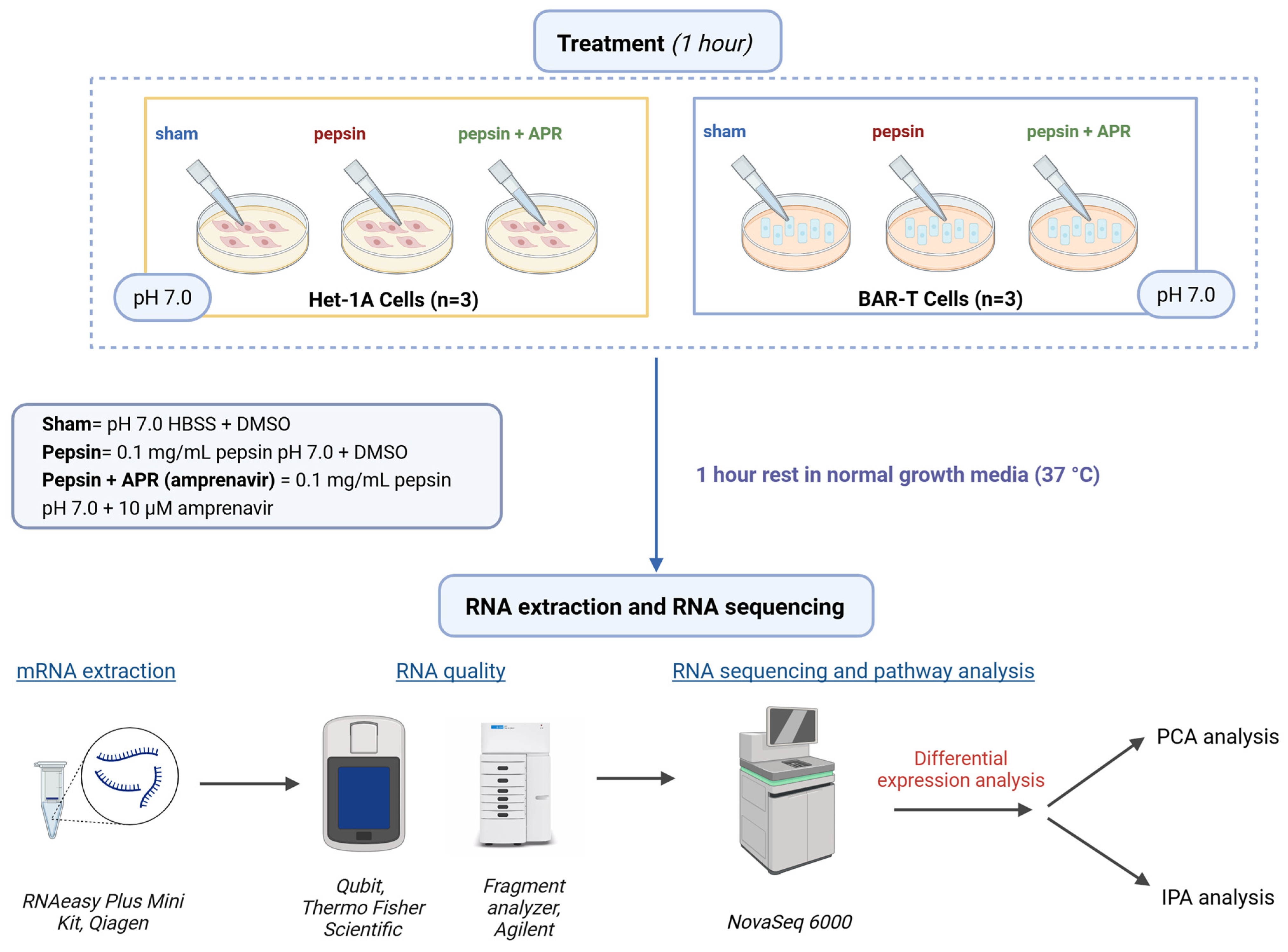
4. Materials and Methods
4.1. Cell Culture and Treatment
4.2. RNA-seq and Ingenuity Pathway Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Gorgulu, V.; Ergun, P.; Kipcak, S.; Doganavsargil, B.; Sifrim, D.; Bor, S. Revisiting the Role of Esophageal Mucosal Dilated Intercellular Spaces in the Diagnosis and Pathophysiology of Heartburn. J. Neurogastroenterol. Motil. 2023, 29, 436–445. [Google Scholar] [CrossRef] [PubMed]
- Zerbib, F.; Bredenoord, A.J.; Fass, R.; Kahrilas, P.J.; Roman, S.; Savarino, E.; Sifrim, D.; Vaezi, M.; Yadlapati, R.; Gyawali, C.P. ESNM/ANMS consensus paper: Diagnosis and management of refractory gastro-esophageal reflux disease. Neurogastroenterol. Motil. 2021, 33, e14075. [Google Scholar] [CrossRef] [PubMed]
- Nirwan, J.S.; Hasan, S.S.; Babar, Z.U.; Conway, B.R.; Ghori, M.U. Global Prevalence and Risk Factors of Gastro-oesophageal Reflux Disease (GORD): Systematic Review with Meta-analysis. Sci. Rep. 2020, 10, 5814. [Google Scholar] [CrossRef] [PubMed]
- Yadlapati, R.; Gyawali, C.P.; Pandolfino, J.E.; Participants, C.G.C.C. AGA Clinical Practice Update on the Personalized Approach to the Evaluation and Management of GERD: Expert Review. Clin. Gastroenterol. Hepatol. 2022, 20, 984–994.e1. [Google Scholar] [CrossRef]
- Fass, R.; Boeckxstaens, G.E.; El-Serag, H.; Rosen, R.; Sifrim, D.; Vaezi, M.F. Gastro-oesophageal reflux disease. Nat. Rev. Dis. Primers 2021, 7, 55. [Google Scholar] [CrossRef]
- Ergun, P.; Kipcak, S.; Bor, S. Epigenetic Alterations from Barrett’s Esophagus to Esophageal Adenocarcinoma. Int. J. Mol. Sci. 2023, 24, 7817. [Google Scholar] [CrossRef]
- Fossmark, R.; Martinsen, T.C.; Waldum, H.L. Adverse Effects of Proton Pump Inhibitors-Evidence and Plausibility. Int. J. Mol. Sci. 2019, 20, 5203. [Google Scholar] [CrossRef]
- Davis, T.A.; Gyawali, C.P. Refractory Gastroesophageal Reflux Disease: Diagnosis and Management. J. Neurogastroenterol. Motil. 2024, 30, 17–28. [Google Scholar] [CrossRef]
- Ergun, P.; Kipcak, S.; Gorgulu, V.; Doganavsargil, B.; Bor, S. Molecular and Functional Recovery of Esophageal Barrier Integrity After Laparoscopic Anti-reflux Surgery. Dig. Dis. Sci. 2025. [Google Scholar] [CrossRef]
- Patel, D.; Fass, R.; Vaezi, M. Untangling Nonerosive Reflux Disease From Functional Heartburn. Clin. Gastroenterol. Hepatol. 2021, 19, 1314–1326. [Google Scholar] [CrossRef]
- Ergun, P.; Kipcak, S.; Dettmar, P.W.; Fisher, J.; Woodcock, A.D.; Bor, S. Pepsin and pH of Gastric Juice in Patients With Gastrointestinal Reflux Disease and Subgroups. J. Clin. Gastroenterol. 2022, 56, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Ergun, P.; Kipcak, S.; Gunel, N.S.; Bor, S.; Sozmen, E.Y. Roles of Cytokines in Pathological and Physiological Gastroesophageal Reflux Exposure. J. Neurogastroenterol. Motil. 2024, 30, 290–302. [Google Scholar] [CrossRef] [PubMed]
- Ergun, P.; Kipcak, S.; Selvi Gunel, N.; Yildirim Sozmen, E.; Bor, S. Inflammatory responses in esophageal mucosa before and after laparoscopic antireflux surgery. World J. Gastrointest. Surg. 2024, 16, 871–881. [Google Scholar] [CrossRef]
- Samuels, T.L.; Johnston, N. Pepsin in gastroesophageal and extraesophageal reflux: Molecular pathophysiology and diagnostic utility. Curr. Opin. Otolaryngol. Head Neck Surg. 2020, 28, 401–409. [Google Scholar] [CrossRef]
- Samuels, T.L.; Altman, K.W.; Gould, J.C.; Kindel, T.; Bosler, M.; MacKinnon, A.; Hagen, C.E.; Johnston, N. Esophageal pepsin and proton pump synthesis in barrett’s esophagus and esophageal adenocarcinoma. Laryngoscope 2019, 129, 2687–2695. [Google Scholar] [CrossRef]
- Han, D.; Zhang, C. The Oxidative Damage and Inflammation Mechanisms in GERD-Induced Barrett’s Esophagus. Front. Cell Dev. Biol. 2022, 10, 885537. [Google Scholar] [CrossRef]
- Yibirin, M.; De Oliveira, D.; Valera, R.; Plitt, A.E.; Lutgen, S. Adverse Effects Associated with Proton Pump Inhibitor Use. Cureus 2021, 13, e12759. [Google Scholar] [CrossRef]
- Pauwels, A.; Boecxstaens, V.; Andrews, C.N.; Attwood, S.E.; Berrisford, R.; Bisschops, R.; Boeckxstaens, G.E.; Bor, S.; Bredenoord, A.J.; Cicala, M.; et al. How to select patients for antireflux surgery? The ICARUS guidelines (international consensus regarding preoperative examinations and clinical characteristics assessment to select adult patients for antireflux surgery). Gut 2019, 68, 1928–1941. [Google Scholar] [CrossRef]
- Johnston, N.; Samuels, T.L.; Goetz, C.J.; Arnold, L.A.; Smith, B.C.; Seabloom, D.; Wuertz, B.; Ondrey, F.; Wiedmann, T.S.; Vuksanovic, N.; et al. Oral and Inhaled Fosamprenavir Reverses Pepsin-Induced Damage in a Laryngopharyngeal Reflux Mouse Model. Laryngoscope 2023, 133 (Suppl. 1), S1–S11. [Google Scholar] [CrossRef]
- Sayer, J.M.; Louis, J.M. Interactions of different inhibitors with active-site aspartyl residues of HIV-1 protease and possible relevance to pepsin. Proteins 2009, 75, 556–568. [Google Scholar] [CrossRef]
- Johnston, N.; Dettmar, P.W.; Ondrey, F.G.; Nanchal, R.; Lee, S.H.; Bock, J.M. Pepsin: Biomarker, mediator, and therapeutic target for reflux and aspiration. Ann. N. Y. Acad. Sci. 2018, 1434, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Wells, C.W.; Blumin, J.H.; Toohill, R.J.; Merati, A.L. Receptor-mediated uptake of pepsin by laryngeal epithelial cells. Ann. Otol. Rhinol. Laryngol. 2007, 116, 934–938. [Google Scholar] [CrossRef] [PubMed]
- Johnston, N.; Wells, C.W.; Samuels, T.L.; Blumin, J.H. Pepsin in nonacidic refluxate can damage hypopharyngeal epithelial cells. Ann. Otol. Rhinol. Laryngol. 2009, 118, 677–685. [Google Scholar] [CrossRef]
- Blaine-Sauer, S.; Samuels, T.L.; Yan, K.; Johnston, N. The Protease Inhibitor Amprenavir Protects against Pepsin-Induced Esophageal Epithelial Barrier Disruption and Cancer-Associated Changes. Int. J. Mol. Sci. 2023, 24, 6765. [Google Scholar] [CrossRef]
- Samuels, T.L.; Blaine-Sauer, S.; Yan, K.; Johnston, N. Amprenavir inhibits pepsin-mediated laryngeal epithelial disruption and E-cadherin cleavage in vitro. Laryngoscope Investig. Otolaryngol. 2023, 8, 953–962. [Google Scholar] [CrossRef]
- Ergun, P.; Samuels, T.L.; Mathison, A.J.; Plehhova, K.; Coyle, C.; Horvath, L.; Johnston, N. Global Transcriptomic Analysis of Topical Sodium Alginate Protection against Peptic Damage in an In Vitro Model of Treatment-Resistant Gastroesophageal Reflux Disease. Int. J. Mol. Sci. 2024, 25, 10714. [Google Scholar] [CrossRef]
- Quilty, F.; Freeley, M.; Gargan, S.; Gilmer, J.; Long, A. Deoxycholic acid induces proinflammatory cytokine production by model oesophageal cells via lipid rafts. J. Steroid Biochem. Mol. Biol. 2021, 214, 105987. [Google Scholar] [CrossRef]
- Rafiee, P.; Nelson, V.M.; Manley, S.; Wellner, M.; Floer, M.; Binion, D.G.; Shaker, R. Effect of curcumin on acidic pH-induced expression of IL-6 and IL-8 in human esophageal epithelial cells (HET-1A): Role of PKC, MAPKs, and NF-kappaB. Am. J. Physiol.-Gastrointest. Liver Physiol. 2009, 296, G388–G398. [Google Scholar] [CrossRef]
- Jaiswal, K.R.; Morales, C.P.; Feagins, L.A.; Gandia, K.G.; Zhang, X.; Zhang, H.Y.; Hormi-Carver, K.; Shen, Y.; Elder, F.; Ramirez, R.D.; et al. Characterization of telomerase-immortalized, non-neoplastic, human Barrett’s cell line (BAR-T). Dis. Esophagus 2007, 20, 256–264. [Google Scholar] [CrossRef]
- Samuels, T.; Hoekzema, C.; Gould, J.; Goldblatt, M.; Frelich, M.; Bosler, M.; Lee, S.H.; Johnston, N. Local Synthesis of Pepsin in Barrett’s Esophagus and the Role of Pepsin in Esophageal Adenocarcinoma. Ann. Otol. Rhinol. Laryngol. 2015, 124, 893–902. [Google Scholar] [CrossRef]
- Bus, P.; Siersema, P.D.; van Baal, J.W. Cell culture models for studying the development of Barrett’s esophagus: A systematic review. Cell Oncol. 2012, 35, 149–161. [Google Scholar] [CrossRef] [PubMed]
- Park, D.H.; Seo, S.I.; Lee, K.J.; Kim, J.; Kim, Y.; Seo, W.W.; Lee, H.S.; Shin, W.G.; Yoo, J.J. Long-term proton pump inhibitor use and risk of osteoporosis and hip fractures: A nationwide population-based and multicenter cohort study using a common data model. J. Gastroenterol. Hepatol. 2022, 37, 1534–1543. [Google Scholar] [CrossRef] [PubMed]
- Lespessailles, E.; Toumi, H. Proton Pump Inhibitors and Bone Health: An Update Narrative Review. Int. J. Mol. Sci. 2022, 23, 10733. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ramos-Alvarez, I.; Jensen, R.T. Long-Term Proton Pump Inhibitor-Acid Suppressive Treatment Can Cause Vitamin B(12) Deficiency in Zollinger-Ellison Syndrome (ZES) Patients. Int. J. Mol. Sci. 2024, 25, 7286. [Google Scholar] [CrossRef]
- Brusnic, O.; Onisor, D.; Boicean, A.; Hasegan, A.; Ichim, C.; Guzun, A.; Chicea, R.; Todor, S.B.; Vintila, B.I.; Anderco, P.; et al. Fecal Microbiota Transplantation: Insights into Colon Carcinogenesis and Immune Regulation. J. Clin. Med. 2024, 13, 6578. [Google Scholar] [CrossRef]
- Spechler, S.J.; Hunter, J.G.; Jones, K.M.; Lee, R.; Smith, B.R.; Mashimo, H.; Sanchez, V.M.; Dunbar, K.B.; Pham, T.H.; Murthy, U.K.; et al. Randomized Trial of Medical versus Surgical Treatment for Refractory Heartburn. N. Engl. J. Med. 2019, 381, 1513–1523. [Google Scholar] [CrossRef]
- Yadlapati, R.; DeLay, K. Proton Pump Inhibitor-Refractory Gastroesophageal Reflux Disease. Med. Clin. North. Am. 2019, 103, 15–27. [Google Scholar] [CrossRef]
- Zhuang, Q.; Liao, A.; He, Q.; Liu, C.; Zheng, C.; Li, X.; Liu, Y.; Wang, B.; Liu, S.; Zhang, Y.; et al. The efficacy and safety of fexuprazan in treating erosive esophagitis: A phase III, randomized, double-blind, multicenter study. J. Gastroenterol. Hepatol. 2024, 39, 658–666. [Google Scholar] [CrossRef]
- Laine, L.; DeVault, K.; Katz, P.; Mitev, S.; Lowe, J.; Hunt, B.; Spechler, S. Vonoprazan Versus Lansoprazole for Healing and Maintenance of Healing of Erosive Esophagitis: A Randomized Trial. Gastroenterology 2023, 164, 61–71. [Google Scholar] [CrossRef]
- Hvid-Jensen, F.; Pedersen, L.; Funch-Jensen, P.; Drewes, A.M. Proton pump inhibitor use may not prevent high-grade dysplasia and oesophageal adenocarcinoma in Barrett’s oesophagus: A nationwide study of 9883 patients. Aliment. Pharmacol. Ther. 2014, 39, 984–991. [Google Scholar] [CrossRef]
- Li, L.; Cao, Z.; Zhang, C.; Pan, W. Risk of esophageal adenocarcinoma in patients with Barrett’s esophagus using proton pump inhibitors: A systematic review with meta-analysis and sequential trial analysis. Transl. Cancer Res. 2021, 10, 1620–1627. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Sun, T.T.; Hong, J.; Fang, J.Y.; Xiong, H.; Meltzer, S.J. Proton Pump Inhibitors Do Not Reduce the Risk of Esophageal Adenocarcinoma in Patients with Barrett’s Esophagus: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169691. [Google Scholar] [CrossRef] [PubMed]
- Que, J.; Garman, K.S.; Souza, R.F.; Spechler, S.J. Pathogenesis and Cells of Origin of Barrett’s Esophagus. Gastroenterology 2019, 157, 349–364.e1. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.H. From reflux esophagitis to Barrett’s esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 2015, 21, 5210–5219. [Google Scholar] [CrossRef] [PubMed]
- Ergün, P.; Capanoglu, D.; Kipcak, S.; Bor, S. Response of Esophageal Epithelium to Acute and Chronic Stress in Rabbits. Bull. Exp. Biol. Med. 2021, 171, 582–587. [Google Scholar] [CrossRef]
- Johnston, N.; Wells, C.W.; Samuels, T.L.; Blumin, J.H. Rationale for targeting pepsin in the treatment of reflux disease. Ann. Otol. Rhinol. Laryngol. 2010, 119, 547–558. [Google Scholar] [CrossRef]
- Dai, Y.F.; Tan, J.J.; Deng, C.Q.; Liu, X.; Lv, Z.H.; Li, X.P. Association of pepsin and DNA damage in laryngopharyngeal reflux-related vocal fold polyps. Am. J. Otolaryngol. 2020, 41, 102681. [Google Scholar] [CrossRef]
- Sales, T.; Sidou, F.; da Costa Filho, H.B.; de Melo Nogueira, K.; Dias Júnior, G.J.; de Sousa Lima, M.A.; da Silva, L.M.G.; Nicolau, L.A.D.; Soares, P.M.G.; Nobre, E.S.M.; et al. Pepsin Inhibitors Prevent Inflammation and Loss of Laryngeal Barrier Function in Mice with Gastroesophageal Reflux. Laryngoscope 2024, 134, 3080–3085. [Google Scholar] [CrossRef]
- Samuels, T.L.; Zimmermann, M.T.; Zeighami, A.; Demos, W.; Southwood, J.E.; Blumin, J.H.; Bock, J.M.; Johnston, N. RNA Sequencing Reveals Cancer-Associated Changes in Laryngeal Cells Exposed to Non-Acid Pepsin. Laryngoscope 2021, 131, 121–129. [Google Scholar] [CrossRef]
- Hou, C.; Zhou, L.; Zheng, Y.; Chen, T.; Hu, R.; Zheng, J.; Liu, C.; Liu, Y. Weak acid and pepsin reflux induce laryngopharyngeal mucosal barrier injury: A rabbit-model-based study. PLoS ONE 2025, 20, e0315083. [Google Scholar] [CrossRef]
- Choi, Y.S.; Na, H.G.; Bae, C.H.; Song, S.Y.; Kim, Y.D. Pepsin exposure in a non-acidic environment upregulates mucin 5AC (MUC5AC) expression via matrix metalloproteinase 9 (MMP9)/nuclear factor κB (NF-κB) in human airway epithelial cells. Int. Forum Allergy Rhinol. 2021, 11, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Doukas, P.G.; Vageli, D.P.; Sasaki, C.T.; Judson, B.L. Pepsin Promotes Activation of Epidermal Growth Factor Receptor and Downstream Oncogenic Pathways, at Slightly Acidic and Neutral pH, in Exposed Hypopharyngeal Cells. Int. J. Mol. Sci. 2021, 22, 4275. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, C.T.; Toman, J.; Vageli, D. The In Vitro Effect of Acidic-Pepsin on Nuclear Factor KappaB Activation and Its Related Oncogenic Effect on Normal Human Hypopharyngeal Cells. PLoS ONE 2016, 11, e0168269. [Google Scholar] [CrossRef] [PubMed]
- Bulmer, D.M.; Ali, M.S.; Brownlee, I.A.; Dettmar, P.W.; Pearson, J.P. Laryngeal mucosa: Its susceptibility to damage by acid and pepsin. Laryngoscope 2010, 120, 777–782. [Google Scholar] [CrossRef]
- Yu, O.M.; Brown, J.H. G Protein-Coupled Receptor and RhoA-Stimulated Transcriptional Responses: Links to Inflammation, Differentiation, and Cell Proliferation. Mol. Pharmacol. 2015, 88, 171–180. [Google Scholar] [CrossRef]
- Wu, S.Y.; Fu, T.; Jiang, Y.Z.; Shao, Z.M. Natural killer cells in cancer biology and therapy. Mol. Cancer 2020, 19, 120. [Google Scholar] [CrossRef]
- Zhang, Y.; Fai, T.G. Influence of the vessel wall geometry on the wall-induced migration of red blood cells. PLoS Comput. Biol. 2023, 19, e1011241. [Google Scholar] [CrossRef]
- Sun, L.; Ye, R.D. Role of G protein-coupled receptors in inflammation. Acta Pharmacol. Sin. 2012, 33, 342–350. [Google Scholar] [CrossRef]
- Gruden, E.; Kienzl, M.; Ristic, D.; Kindler, O.; Kaspret, D.M.; Schmid, S.T.; Kargl, J.; Sturm, E.; Doyle, A.D.; Wright, B.L.; et al. Mononuclear cell composition and activation in blood and mucosal tissue of eosinophilic esophagitis. Front. Immunol. 2024, 15, 1347259. [Google Scholar] [CrossRef]
- Liu, D.; Qian, T.; Sun, S.; Jiang, J.J. Laryngopharyngeal Reflux and Inflammatory Responses in Mucosal Barrier Dysfunction of the Upper Aerodigestive Tract. J. Inflamm. Res. 2020, 13, 1291–1304. [Google Scholar] [CrossRef]
- Tang, B.; Guo, M.; Zhai, Y.; Zhang, K.; Ni, K.; Zhang, Y.; Huang, L. Human esophageal cancer stem-like cells escape the cytotoxicity of natural killer cells via down-regulation of ULBP-1. J. Transl. Med. 2024, 22, 737. [Google Scholar] [CrossRef] [PubMed]
- Oparin, A.; Vnukova, A. The Role of Endothelial Dysfunction in the Mechanism of Gastroesophageal Reflux Disease Development in Patients with Ischemic Heart Disease. Acta Clin. Croat. 2017, 56, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ren, H.; Wang, J.; Xiao, X.; Zhu, L.; Wang, Y.; Yang, L. CHAC1: A master regulator of oxidative stress and ferroptosis in human diseases and cancers. Front. Cell Dev. Biol. 2024, 12, 1458716. [Google Scholar] [CrossRef] [PubMed]
- Ro, S.H.; Xue, X.; Ramakrishnan, S.K.; Cho, C.S.; Namkoong, S.; Jang, I.; Semple, I.A.; Ho, A.; Park, H.W.; Shah, Y.M.; et al. Tumor suppressive role of sestrin2 during colitis and colon carcinogenesis. eLife 2016, 5, e12204. [Google Scholar] [CrossRef]
- Lin, W.; Wang, C.; Liu, G.; Bi, C.; Wang, X.; Zhou, Q.; Jin, H. SLC7A11/xCT in cancer: Biological functions and therapeutic implications. Am. J. Cancer Res. 2020, 10, 3106–3126. [Google Scholar]
- Tian, G.; Hu, C.; Yun, Y.; Yang, W.; Dubiel, W.; Cheng, Y.; Wolf, D.A. Dual roles of HSP70 chaperone HSPA1 in quality control of nascent and newly synthesized proteins. EMBO J. 2021, 40, e106183. [Google Scholar] [CrossRef]
- Scieglinska, D.; Piglowski, W.; Chekan, M.; Mazurek, A.; Krawczyk, Z. Differential expression of HSPA1 and HSPA2 proteins in human tissues; tissue microarray-based immunohistochemical study. Histochem. Cell Biol. 2011, 135, 337–350. [Google Scholar] [CrossRef]
- Samuels, T.L.; Johnston, N. Pepsin as a causal agent of inflammation during nonacidic reflux. Otolaryngol. Head Neck Surg. 2009, 141, 559–563. [Google Scholar] [CrossRef]
- Du, L.; Xu, C.; Shi, J.; Tang, L.; Xiao, L.; Lei, C.; Liu, H.; Liang, Y.; Guo, Y.; Tang, K. Elevated CXCL14 in Induced Sputum Was Associated with Eosinophilic Inflammation and Airway Obstruction in Patients with Asthma. Int. Arch. Allergy Immunol. 2022, 183, 1216–1225. [Google Scholar] [CrossRef]
- Nestler, E.J. ∆FosB: A transcriptional regulator of stress and antidepressant responses. Eur. J. Pharmacol. 2015, 753, 66–72. [Google Scholar] [CrossRef]
- Wang, X.; Xu, G.; Zhang, F.; Wei, Y.; Deng, J.; Mu, L.; He, J.; He, D.; Yin, M.; Dal Pra, I.; et al. eIF6 modulates skin wound healing by upregulating keratin 6B. Stem Cells Transl. Med. 2024, 13, 1101–1112. [Google Scholar] [CrossRef] [PubMed]
- Souza, R.F.; Spechler, S.J. Mechanisms and pathophysiology of Barrett oesophagus. Nat. Rev. Gastroenterol. Hepatol. 2022, 19, 605–620. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Shen, Y.; Wang, J.; Zhou, M.; Chen, Y. Identification of key pathways and genes in Barrett’s esophagus using integrated bioinformatics methods. Mol. Med. Rep. 2018, 17, 3069–3077. [Google Scholar] [CrossRef]
- Korbut, E.; Janmaat, V.T.; Wierdak, M.; Hankus, J.; Wójcik, D.; Surmiak, M.; Magierowska, K.; Brzozowski, T.; Peppelenbosch, M.P.; Magierowski, M. Molecular Profile of Barrett’s Esophagus and Gastroesophageal Reflux Disease in the Development of Translational Physiological and Pharmacological Studies. Int. J. Mol. Sci. 2020, 21, 6436. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Wang, D.; Li, J.; Hou, Y.; Qiao, C. Screening and Identification of Prognostic Tumor-Infiltrating Immune Cells and Genes of Endometrioid Endometrial Adenocarcinoma: Based on The Cancer Genome Atlas Database and Bioinformatics. Front. Oncol. 2020, 10, 554214. [Google Scholar] [CrossRef]
- Wang, J.; Xiao, Y.; Yu, Q.; Zhang, C. KRT72 might serves as a prognostic biomarker for patients with prostate cancer. Asian J. Surg. 2023, 46, 5382–5384. [Google Scholar] [CrossRef]
- Oakley, R.H.; Cidlowski, J.A. The biology of the glucocorticoid receptor: New signaling mechanisms in health and disease. J. Allergy Clin. Immunol. 2013, 132, 1033–1044. [Google Scholar] [CrossRef]
- Ueki, S.; Fujishima, F.; Kumagai, T.; Ishida, H.; Okamoto, H.; Takaya, K.; Sato, C.; Taniyma, Y.; Kamei, T.; Sasano, H. GR, Sgk1, and NDRG1 in esophageal squamous cell carcinoma: Their correlation with therapeutic outcome of neoadjuvant chemotherapy. BMC Cancer 2020, 20, 161. [Google Scholar] [CrossRef]
- Zhang, T.; Li, M.; Zhao, S.; Zhou, M.; Liao, H.; Wu, H.; Mo, X.; Wang, H.; Guo, C.; Zhang, H.; et al. CaMK4 Promotes Acute Lung Injury Through NLRP3 Inflammasome Activation in Type II Alveolar Epithelial Cell. Front. Immunol. 2022, 13, 890710. [Google Scholar] [CrossRef]
- O’Farrell, N.J.; Feighery, R.; Picardo, S.L.; Lynam-Lennon, N.; Biniecka, M.; McGarrigle, S.A.; Phelan, J.J.; MacCarthy, F.; O’Toole, D.; Fox, E.J.; et al. Changes in mitochondrial stability during the progression of the Barrett’s esophagus disease sequence. BMC Cancer 2016, 16, 497. [Google Scholar] [CrossRef]
- Nadatani, Y.; Huo, X.; Zhang, X.; Yu, C.; Cheng, E.; Zhang, Q.; Dunbar, K.B.; Theiss, A.; Pham, T.H.; Wang, D.H.; et al. NOD-Like Receptor Protein 3 Inflammasome Priming and Activation in Barrett’s Epithelial Cells. Cell Mol. Gastroenterol. Hepatol. 2016, 2, 439–453. [Google Scholar] [CrossRef] [PubMed]
- Phelan, J.J.; MacCarthy, F.; Feighery, R.; O’Farrell, N.J.; Lynam-Lennon, N.; Doyle, B.; O’Toole, D.; Ravi, N.; Reynolds, J.V.; O’Sullivan, J. Differential expression of mitochondrial energy metabolism profiles across the metaplasia-dysplasia-adenocarcinoma disease sequence in Barrett’s oesophagus. Cancer Lett. 2014, 354, 122–131. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.C.; Su, J.; Zhou, J.J.; Yuan, Q.; Han, J.S. Roles of MT-ND1 in Cancer. Curr. Med. Sci. 2023, 43, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, J.; Yuan, Q.; Su, J.; Li, Q.; Lu, X.; Zhang, L.; Cai, Z.; Han, J. Quantitative detection of circulating MT-ND1 as a potential biomarker for colorectal cancer. Bosn. J. Basic. Med. Sci. 2021, 21, 577–586. [Google Scholar] [CrossRef]
- Cavalcante, G.C.; Ribeiro-Dos-Santos, Â.; de Araújo, G.S. Mitochondria in tumour progression: A network of mtDNA variants in different types of cancer. BMC Genom. Data 2022, 23, 16. [Google Scholar] [CrossRef]
- Tan, J.J.; Dai, Y.F.; Wang, F.; Lv, Z.H.; Huang, L.J.; Peng, L.Y.; Li, X.P. Pepsin-mediated inflammation in laryngopharyngeal reflux via the ROS/NLRP3/IL-1β signaling pathway. Cytokine 2024, 178, 156568. [Google Scholar] [CrossRef]
- Feagins, L.A.; Zhang, H.Y.; Zhang, X.; Hormi-Carver, K.; Thomas, T.; Terada, L.S.; Spechler, S.J.; Souza, R.F. Mechanisms of oxidant production in esophageal squamous cell and Barrett’s cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G411–G417. [Google Scholar] [CrossRef]
- Cordier, F.; Creytens, D. New kids on the block: FOS and FOSB gene. J. Clin. Pathol. 2023, 76, 721–726. [Google Scholar] [CrossRef]
- Sack, G.H., Jr. Serum Amyloid A (SAA) Proteins. Subcell. Biochem. 2020, 94, 421–436. [Google Scholar] [CrossRef]
- Huang, G.; Li, H.; Zhang, H. Abnormal Expression of Mitochondrial Ribosomal Proteins and Their Encoding Genes with Cell Apoptosis and Diseases. Int. J. Mol. Sci. 2020, 21, 8879. [Google Scholar] [CrossRef]
- Haag, S.; Sloan, K.E.; Ranjan, N.; Warda, A.S.; Kretschmer, J.; Blessing, C.; Hübner, B.; Seikowski, J.; Dennerlein, S.; Rehling, P.; et al. NSUN3 and ABH1 modify the wobble position of mt-tRNAMet to expand codon recognition in mitochondrial translation. EMBO J. 2016, 35, 2104–2119. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, A.; Dienemann, C.; Cramer, P.; Hillen, H.S. Structural basis of RNA processing by human mitochondrial RNase P. Nat. Struct. Mol. Biol. 2021, 28, 713–723. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Chen, Z.; Yu, F.; Liu, H.; Ma, C.; Xie, D.; Hu, X.; Leak, R.K.; Chou, S.H.Y.; Stetler, R.A.; et al. IL-4/STAT6 signaling facilitates innate hematoma resolution and neurological recovery after hemorrhagic stroke in mice. Proc. Natl. Acad. Sci. USA 2020, 117, 32679–32690. [Google Scholar] [CrossRef]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef]
- Gargus, M.; Niu, C.; Vallone, J.G.; Binkley, J.; Rubin, D.C.; Shaker, A. Human esophageal myofibroblasts secrete proinflammatory cytokines in response to acid and Toll-like receptor 4 ligands. Am. J. Physiol. Gastrointest. Liver Physiol. 2015, 308, G904–G923. [Google Scholar] [CrossRef]
- Cui, G.; Wang, M.; Li, X.; Wang, C.; Shon, K.; Liu, Z.; Ren, L.; Yang, X.; Li, X.; Wu, Y.; et al. Berberine in combination with evodiamine ameliorates gastroesophageal reflux disease through TAS2R38/TRPV1-mediated regulation of MAPK/NF-κB signaling pathways and macrophage polarization. Phytomedicine 2024, 135, 156251. [Google Scholar] [CrossRef]
- Basbous, S.; Azzarelli, R.; Pacary, E.; Moreau, V. Pathophysiological functions of Rnd proteins. Small GTPases 2021, 12, 336–357. [Google Scholar] [CrossRef]
- DiDonato, J.A.; Mercurio, F.; Karin, M. NF-κB and the link between inflammation and cancer. Immunol. Rev. 2012, 246, 379–400. [Google Scholar] [CrossRef]
- Luzina, I.G.; Rus, V.; Lockatell, V.; Courneya, J.P.; Hampton, B.S.; Fishelevich, R.; Misharin, A.V.; Todd, N.W.; Badea, T.C.; Rus, H.; et al. Regulator of Cell Cycle Protein (RGCC/RGC-32) Protects against Pulmonary Fibrosis. Am. J. Respir. Cell Mol. Biol. 2022, 66, 146–157. [Google Scholar] [CrossRef]
- Karantza, V. Keratins in health and cancer: More than mere epithelial cell markers. Oncogene 2011, 30, 127–138. [Google Scholar] [CrossRef]
- Crisponi, L.; Buers, I.; Rutsch, F. CRLF1 and CLCF1 in Development, Health and Disease. Int. J. Mol. Sci. 2022, 23, 992. [Google Scholar] [CrossRef] [PubMed]
- Coelho-Rato, L.S.; Parvanian, S.; Modi, M.K.; Eriksson, J.E. Vimentin at the core of wound healing. Trends Cell Biol. 2024, 34, 239–254. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, N.; Uchiyama, K.; Kuroda, M.; Sakuma, K.; Kokura, S.; Ichikawa, H.; Naito, Y.; Takemura, T.; Yoshikawa, T.; Okanoue, T. Interleukin-8 expression in the esophageal mucosa of patients with gastroesophageal reflux disease. Scand. J. Gastroenterol. 2004, 39, 816–822. [Google Scholar] [CrossRef]
- Łukaszewicz-Zając, M.; Pączek, S.; Mroczko, B. The significance of chemokine CXCL-8 in esophageal carcinoma. Arch. Med. Sci. 2020, 16, 475–480. [Google Scholar] [CrossRef]
- Johnston, N.; Yan, J.C.; Hoekzema, C.R.; Samuels, T.L.; Stoner, G.D.; Blumin, J.H.; Bock, J.M. Pepsin promotes proliferation of laryngeal and pharyngeal epithelial cells. Laryngoscope 2012, 122, 1317–1325. [Google Scholar] [CrossRef]
- Tan, J.J.; Wang, L.; Mo, T.T.; Wang, J.; Wang, M.G.; Li, X.P. Pepsin promotes IL-8 signaling-induced epithelial-mesenchymal transition in laryngeal carcinoma. Cancer Cell Int. 2019, 19, 64. [Google Scholar] [CrossRef]
- Mertens, V.; Blondeau, K.; Vanaudenaerde, B.; Vos, R.; Farre, R.; Pauwels, A.; Verleden, G.; Van Raemdonck, D.; Dupont, L.; Sifrim, D. Gastric juice from patients “on” acid suppressive therapy can still provoke a significant inflammatory reaction by human bronchial epithelial cells. J. Clin. Gastroenterol. 2010, 44, e230–e235. [Google Scholar] [CrossRef]
- Ten Kate, R.W.; Tuynman, H.A.; Festen, H.P.; Pals, G.; Meuwissen, S.G. Effect of high dose omeprazole on gastric pepsin secretion and serum pepsinogen levels in man. Eur. J. Clin. Pharmacol. 1988, 35, 173–176. [Google Scholar] [CrossRef]
- Wang, Y.J.; Lang, X.Q.; Wu, D.; He, Y.Q.; Lan, C.H.; Xiao, X.; Wang, B.; Zou, D.W.; Wu, J.M.; Zhao, Y.B.; et al. Salivary Pepsin as an Intrinsic Marker for Diagnosis of Sub-types of Gastroesophageal Reflux Disease and Gastroesophageal Reflux Disease-related Disorders. J. Neurogastroenterol. Motil. 2020, 26, 74–84. [Google Scholar] [CrossRef]
- Olp, M.D.; Kalous, K.S.; Smith, B.C. ICEKAT: An interactive online tool for calculating initial rates from continuous enzyme kinetic traces. BMC Bioinform. 2020, 21, 186. [Google Scholar] [CrossRef] [PubMed]
- Wire, M.B.; Shelton, M.J.; Studenberg, S. Fosamprenavir: Clinical pharmacokinetics and drug interactions of the amprenavir prodrug. Clin. Pharmacokinet. 2006, 45, 137–168. [Google Scholar] [CrossRef]
- De Felice, F.; Malerba, S.; Nardone, V.; Salvestrini, V.; Calomino, N.; Testini, M.; Boccardi, V.; Desideri, I.; Gentili, C.; De Luca, R.; et al. Progress and Challenges in Integrating Nutritional Care into Oncology Practice: Results from a National Survey on Behalf of the NutriOnc Research Group. Nutrients 2025, 17, 188. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. Fastqc A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 March 2024).
- Wang, L.; Wang, S.; Li, W. RSeQC: Quality control of RNA-seq experiments. Bioinformatics 2012, 28, 2184–2185. [Google Scholar] [CrossRef]
- Kalari, K.R.; Nair, A.A.; Bhavsar, J.D.; O’Brien, D.R.; Davila, J.I.; Bockol, M.A.; Nie, J.; Tang, X.; Baheti, S.; Doughty, J.B.; et al. MAP-RSeq: Mayo Analysis Pipeline for RNA sequencing. BMC Bioinform. 2014, 15, 224. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Krämer, A.; Green, J.; Pollard, J., Jr.; Tugendreich, S. Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 2014, 30, 523–530. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Pepsin vs. Sham | ||
| Top Canonical Pathways | p-Value | Overlap |
| Class A/1 (Rhodopsin-like receptors) | 3.53 × 10−3 | 4/317 (1.3%) |
| Natural Killer Cell Signaling | 7.06 × 10−3 | 3/198 (1.5%) |
| Cell surface interactions at the vascular wall | 8.40 × 10−3 | 2/64 (3.1%) |
| Top Diseases | p-value range | #Genes |
| Metabolic Disease | 1.71 × 10−2–1.01 × 10−6 | 22 |
| Organismal Injury and Abnormalities | 1.71 × 10−2–1.01 × 10−6 | 48 |
| Gastrointestinal Disease | 1.71 × 10−2–2.60 × 10−6 | 45 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cellular Development | 1.71 × 10−2–6.38 × 10−6 | 27 |
| Cellular Growth and Proliferation | 1.71 × 10−2–6.38 × 10−6 | 27 |
| Cell Death and Survival | 1.68 × 10−2–3.44 × 10−5 | 23 |
| Top Upstream Regulators | p-value range | |
| nelfinavir | 9.27 × 10−9 | |
| EIF2S1 | 6.11 × 10−8 | |
| tosedostat | 2.89 × 10−7 | |
| Pepsin + Amprenavir vs. Sham | ||
| Top Canonical Pathways | p-value | Overlap |
| Hematoma Resolution Signaling Pathway | 2.34 × 10−8 | 7/258 (2.7%) |
| rRNA processing | 6.65× 10−8 | 4/32 (12.5%) |
| NGF-stimulated transcription | 1.51 × 10−7 | 4/39 (10.3%) |
| Top Diseases | p-value range | #Genes |
| Organismal Injury and Abnormalities | 3.70 × 10−3–8.37 × 10−9 | 30 |
| Cancer | 3.70 × 10−3–2.66 × 10−8 | 30 |
| Metabolic Disease | 3.58 × 10−3–7.57× 10−8 | 21 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cellular Development | 3.70 × 10−3–2.24 × 10−9 | 18 |
| Cellular Growth and Proliferation | 3.70 × 10−3–2.24 × 10−9 | 18 |
| Cell Death and Survival | 3.70 × 10−3–9.54 × 10−8 | 17 |
| Top Upstream Regulators | p-value range | |
| HGF | 3.14 × 10−15 | |
| GLI1 | 3.07 × 10−13 | |
| PRL | 1.86 × 10−12 | |
| Pepsin + Amprenavir vs. Pepsin | ||
| Top Canonical Pathways | p-value | Overlap |
| rRNA processing | 1.16 × 10−24 | 10/32 (31.2%) |
| Oxidative Phosphorylation | 1.92 × 10−16 | 9/112 (8.0%) |
| Mitochondrial Dysfunction | 5.26 × 10−12 | 9/344 (2.6%) |
| Top Diseases | p-value range | #Genes |
| Developmental Disorder | 1.46 × 10−2–8.74 × 10−22 | 14 |
| Metabolic Disease | 1.97 × 10−2–8.74 × 10−22 | 13 |
| Neurological Disease | 2.06 × 10−2–8.74 × 10−22 | 15 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cell-To-Cell Signaling and Interaction | 1.97 × 10−2–8.81 × 10−17 | 10 |
| Cell Signaling | 1.33 × 10−2–1.70 × 10−7 | 6 |
| Post-Translational Modification | 1.33 × 10−2–1.70 × 10−7 | 7 |
| Top Upstream Regulators | p-value range | |
| DAP3 | 2.48 × 10−30 | |
| NSUN3 | 3.80 × 10−26 | |
| mtRNase P | 1.13 × 10−25 | |
| Pepsin vs. Sham | logFC | Pepsin + Amprenavir vs. Sham | logFC | Pepsin + Amprenavir vs. Pepsin | logFC | |||
|---|---|---|---|---|---|---|---|---|
| CHAC1 | −1.74 | ↓ | CHAC1 | −1.37 | ↓ | SAA2 | −0.42 | ↓ |
| STC2 | −0.46 | ↓ | POTEE/POTEF | −8.51 | ↓ | FOS | −0.53 | ↓ |
| SESN2 | −0.57 | ↓ | COL1A2 | −6.29 | ↓ | MT−ND1 | −0.59 | ↓ |
| SLC7A11 | −0.60 | ↓ | FOS | −0.57 | ↓ | SGPP2 | −0.39 | ↓ |
| COL1A2 | −3.99 | ↓ | CAV1 | −5.52 | ↓ | MT−CO2 | −0.47 | ↓ |
| POTEE/POTEF | −4.25 | ↓ | HSPA1A−B | 0.71 | ↑ | MT−ND2 | −0.53 | ↓ |
| CAV1 | −4.42 | ↓ | HES1 | 0.51 | ↑ | SAA2−SAA4 | −0.47 | ↓ |
| ADM2 | −0.68 | ↓ | MIDN | 0.47 | ↑ | MT−ND4 | −0.59 | ↓ |
| HSPA1A-B | 0.59 | ↑ | ID1 | 0.54 | ↑ | MT−CYB | −0.50 | ↓ |
| CXCL14 | 0.41 | ↑ | IL11 | 0.68 | ↑ | MT−CO1 | −0.41 | ↓ |
| Pepsin vs. Sham | ||
|---|---|---|
| Top Canonical Pathways | p-value | Overlap |
| Keratinization | 2.75 × 10−6 | 4/214 (1.9%) |
| Wound Healing Signaling Pathway | 2.27 × 10−4 | 3/252 (1.2%) |
| Glucocorticoid Receptor Signaling | 2.57 × 10−3 | 3/582 (0.5%) |
| Top Diseases | p-value range | #Genes |
| Dermatological Diseases and Conditions | 2.35 × 10−3–4.94 × 10−4 | 2 |
| Developmental Disorder | 2.96 × 10−3–4.94 × 10−4 | 2 |
| Organismal Injury and Abnormalities | 4.72 × 10−2–4.94 × 10−4 | 12 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cell Morphology | 4.79 × 10−2–1.23 × 10−5 | 3 |
| Cellular Assembly and Organization | 3.22 × 10−2–1.48 × 10−4 | 4 |
| Cellular Function and Maintenance | 4.65 × 10−2–1.48 × 10−4 | 4 |
| Top Upstream Regulators | p-value range | |
| CAMK4 | 2.44 × 10−4 | |
| miR-7002-5p (miRNAs w/seed UGGCUUC) | 4.98 × 10−4 | |
| cytisine | 4.98 × 10−4 | |
| Pepsin + Amprenavir vs. Sham | ||
| Top Canonical Pathways | p-value | Overlap |
| Keratinization | 7.15 × 10−7 | 4/214 (1.9%) |
| Glucocorticoid Receptor Signaling | 1.03 × 10−3 | 3/582 (0.5%) |
| Wound Healing Signaling Pathway | 3.68 × 10−3 | 2/252 (0.8%) |
| Top Diseases | p-value range | #Genes |
| Cancer | 3.61 × 10−2–3.71 × 10−4 | 8 |
| Dermatological Diseases and Conditions | 3.54 × 10−2–3.71 × 10−4 | 3 |
| Gastrointestinal Disease | 4.07 × 10−2–3.71 × 10−4 | 3 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cell Morphology | 2.82 × 10−2–6.71 × 10−6 | 3 |
| Cellular Assembly and Organization | 2.78 × 10−2–8.09 × 10−5 | 3 |
| Cellular Function and Maintenance | 1.58 × 10−2–8.09 × 10−5 | 3 |
| Top Upstream Regulators | p-value range | |
| GSTP1 | 2.37 × 10−5 | |
| CREB:CRTC1:PER1 gene | 3.45 × 10−4 | |
| BMAL1:CLOCK,NPAS2:PER1 gene | 3.45 × 10−4 | |
| Pepsin + Amprenavir vs. Pepsin | ||
| Top Canonical Pathways | p-value | Overlap |
| Wound Healing Signaling Pathway | 9.44 × 10−8 | 7/252 (2.8%) |
| HMGB1 Signaling | 5.30 × 10−6 | 5/167 (3.0%) |
| Macrophage Classical Activation Signaling Pathway | 9.67 × 10−6 | 5/189 (2.6%) |
| Top Diseases | p-value range | #Genes |
| Cancer | 1.00 × 10−2–7.95 × 10−8 | 37 |
| Immunological Disease | 9.79 × 10−3–7.95 × 10−8 | 25 |
| Organismal Injury and Abnormalities | 1.00 × 10−2–7.95× 10−8 | 37 |
| Molecular and Cellular Functions | p-value range | #Genes |
| Cell-To-Cell Signaling and Interaction | 9.11 × 10−3–1.08 × 10−7 | 19 |
| Lipid Metabolism | 9.11 × 10−3–3.68 × 10−7 | 15 |
| Cellular Development | 9.53 × 10−3–6.86 × 10−7 | 22 |
| Top Upstream Regulators | p-value range | |
| TBK1 | 1.40 × 10−8 | |
| SB203580 | 2.85 × 10−8 | |
| VEGF | 4.44 × 10−8 | |
| Pepsin vs. Sham | logFC | Pepsin + Amprenavir vs. Sham | logFC | Pepsin + Amprenavir vs. Pepsin | LogFC | |||
|---|---|---|---|---|---|---|---|---|
| FOSB | −0.45 | ↓ | PER1 | −0.46 | ↓ | RND1 | −0.69 | ↓ |
| AVIL | −0.84 | ↓ | RND1 | −0.56 | ↓ | KRT84 | −5.16 | ↓ |
| KRT6B | −1.03 | ↓ | KRT6B | −1.02 | ↓ | CHD7 | −0.46 | ↓ |
| GOLGA8A-B | −0.52 | ↓ | RASGEF1B | −0.62 | ↓ | RNF152 | −0.49 | ↓ |
| ATG9B | −0.59 | ↓ | KRTAP2−4 | 0.87 | ↑ | REL | −0.50 | ↓ |
| CPT1B | −0.51 | ↓ | CEACAM5 | 0.51 | ↑ | CLCF1 | 0.50 | ↑ |
| AHSA2P | −0.55 | ↓ | KRT71 | 6.24 | ↑ | KRTAP2−4 | 0.71 | ↑ |
| KRT71 | 4.61 | ↑ | KRT72 | 6.02 | ↑ | RGCC | 0.51 | ↑ |
| KRT72 | 4.37 | ↑ | PRR15L | 0.54 | ↑ | YJEFN3 | 0.83 | ↑ |
| DOK3 | 0.68 | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ergun, P.; Samuels, T.L.; Mathison, A.J.; Liu, T.; Jin, V.X.; Johnston, N. Amprenavir Mitigates Pepsin-Induced Transcriptomic Changes in Normal and Precancerous Esophageal Cells. Int. J. Mol. Sci. 2025, 26, 6182. https://doi.org/10.3390/ijms26136182
Ergun P, Samuels TL, Mathison AJ, Liu T, Jin VX, Johnston N. Amprenavir Mitigates Pepsin-Induced Transcriptomic Changes in Normal and Precancerous Esophageal Cells. International Journal of Molecular Sciences. 2025; 26(13):6182. https://doi.org/10.3390/ijms26136182
Chicago/Turabian StyleErgun, Pelin, Tina L. Samuels, Angela J. Mathison, Tianxiang Liu, Victor X. Jin, and Nikki Johnston. 2025. "Amprenavir Mitigates Pepsin-Induced Transcriptomic Changes in Normal and Precancerous Esophageal Cells" International Journal of Molecular Sciences 26, no. 13: 6182. https://doi.org/10.3390/ijms26136182
APA StyleErgun, P., Samuels, T. L., Mathison, A. J., Liu, T., Jin, V. X., & Johnston, N. (2025). Amprenavir Mitigates Pepsin-Induced Transcriptomic Changes in Normal and Precancerous Esophageal Cells. International Journal of Molecular Sciences, 26(13), 6182. https://doi.org/10.3390/ijms26136182