
Multi-Omics Integration: Predicting Progression and Optimizing Clinical Treatment of Hepatocellular Carcinoma Through Malignant-Cell-Related Genes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
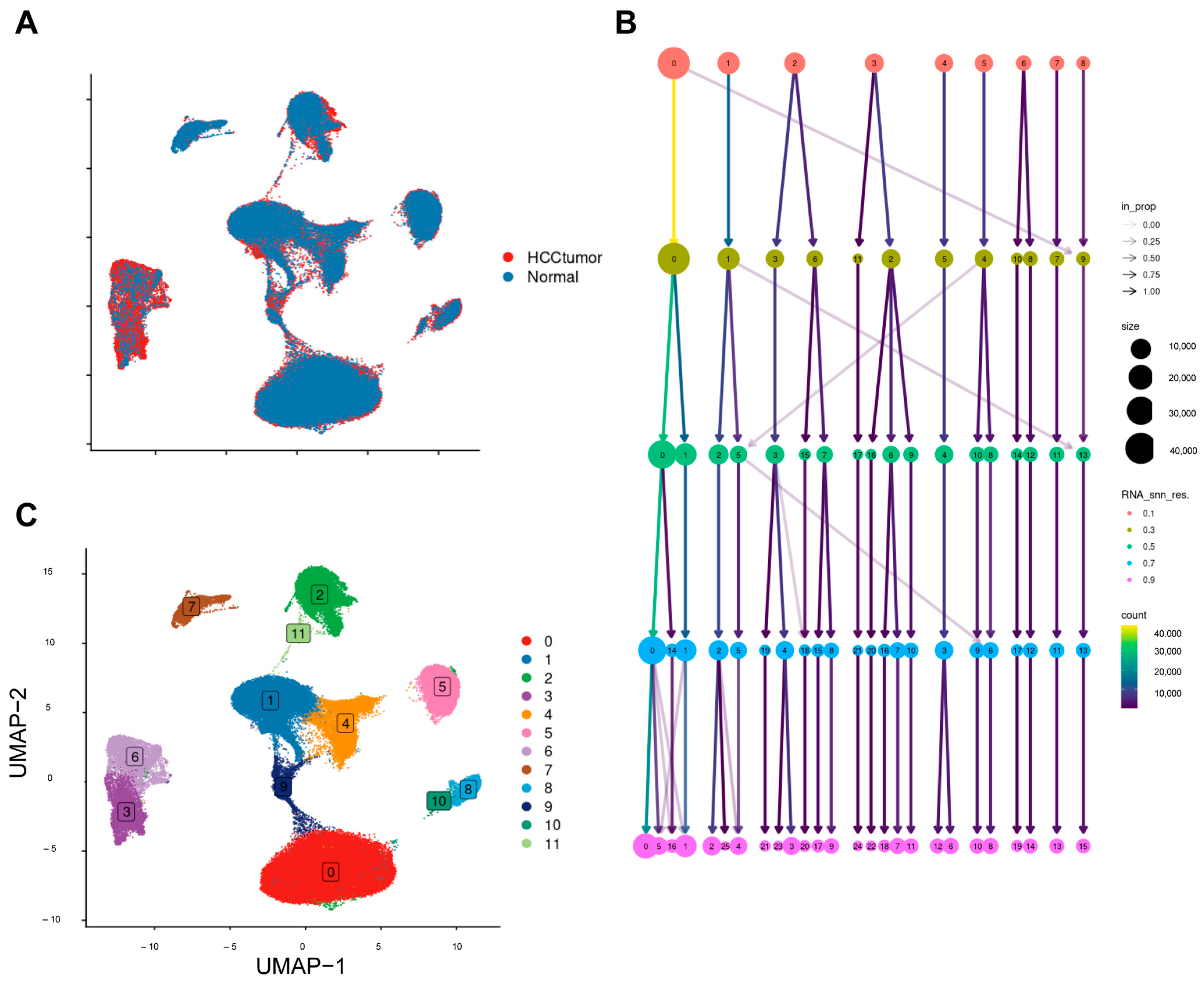
2.1. Processing Single-Cell Data
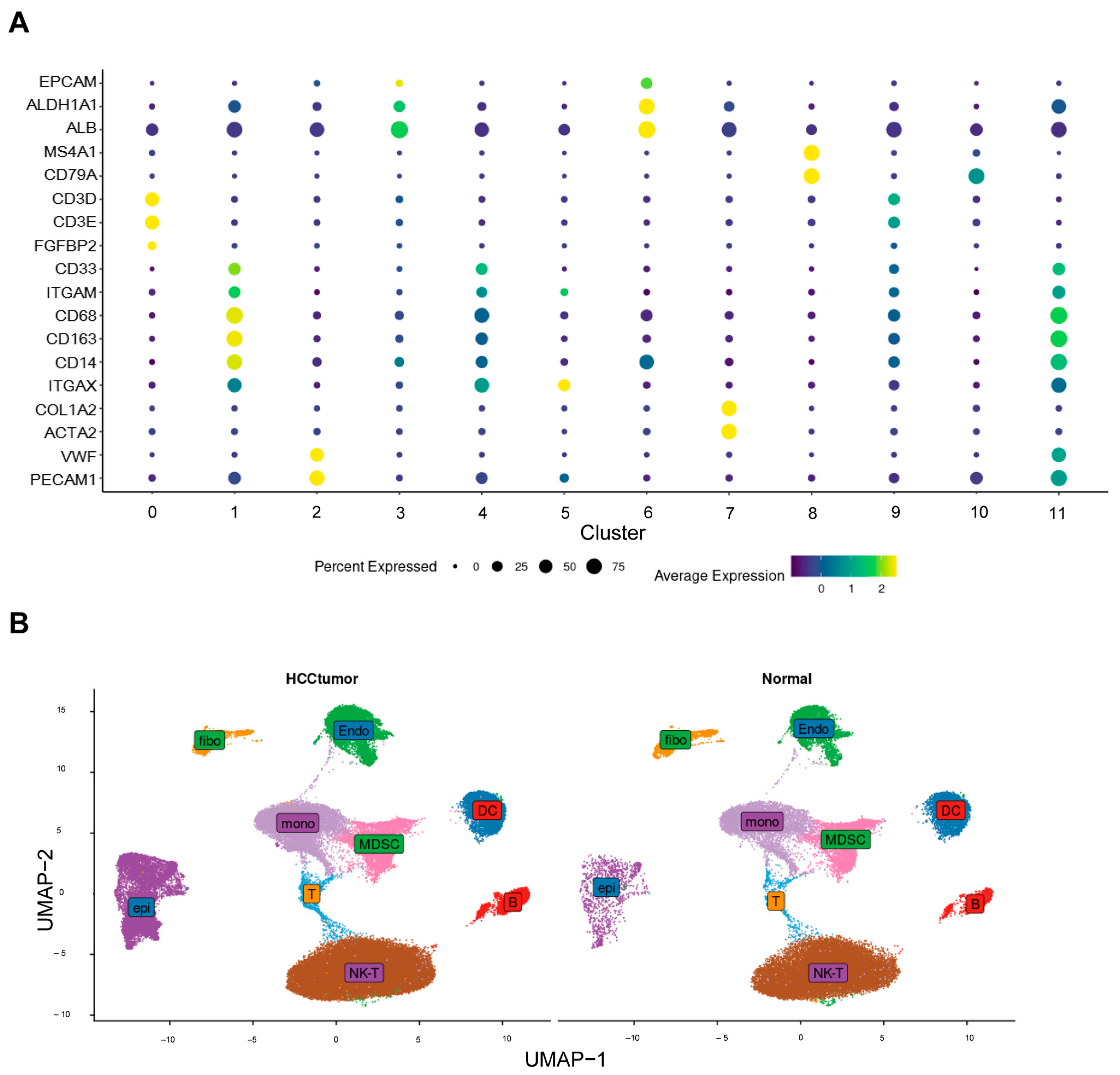
2.2. Annotations for Cell Types
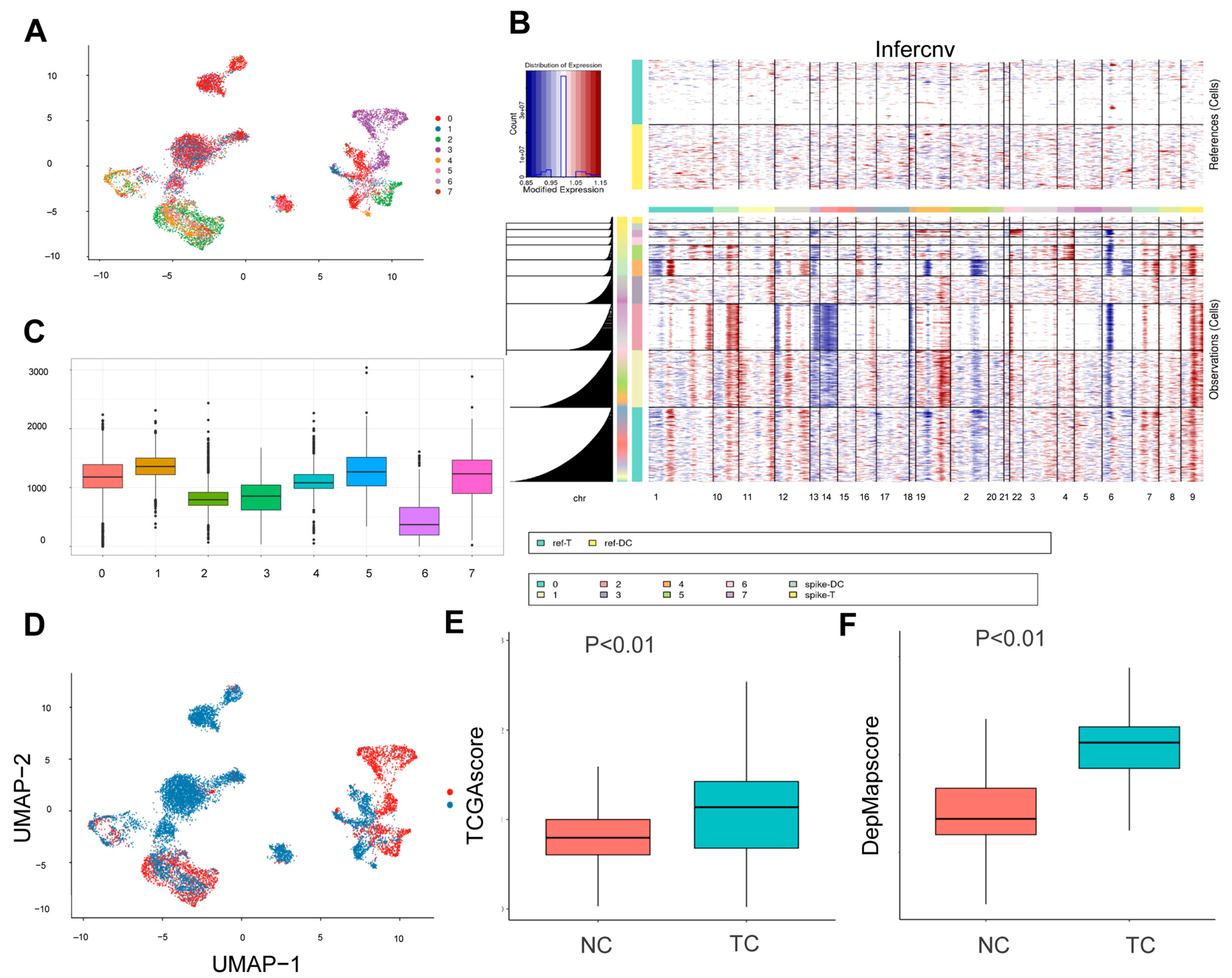
2.3. InferCNV
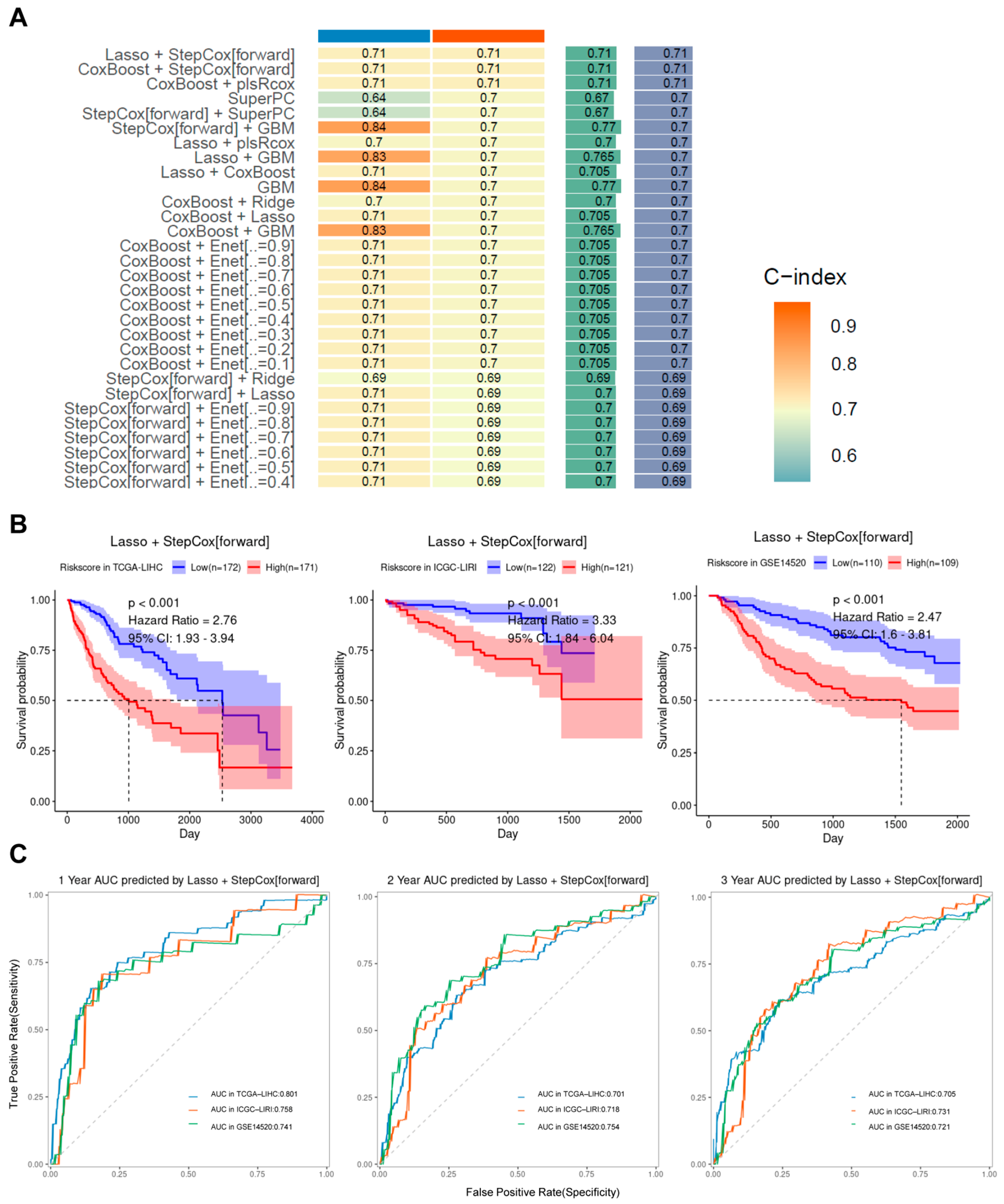
2.4. Machine Learning to Build Models
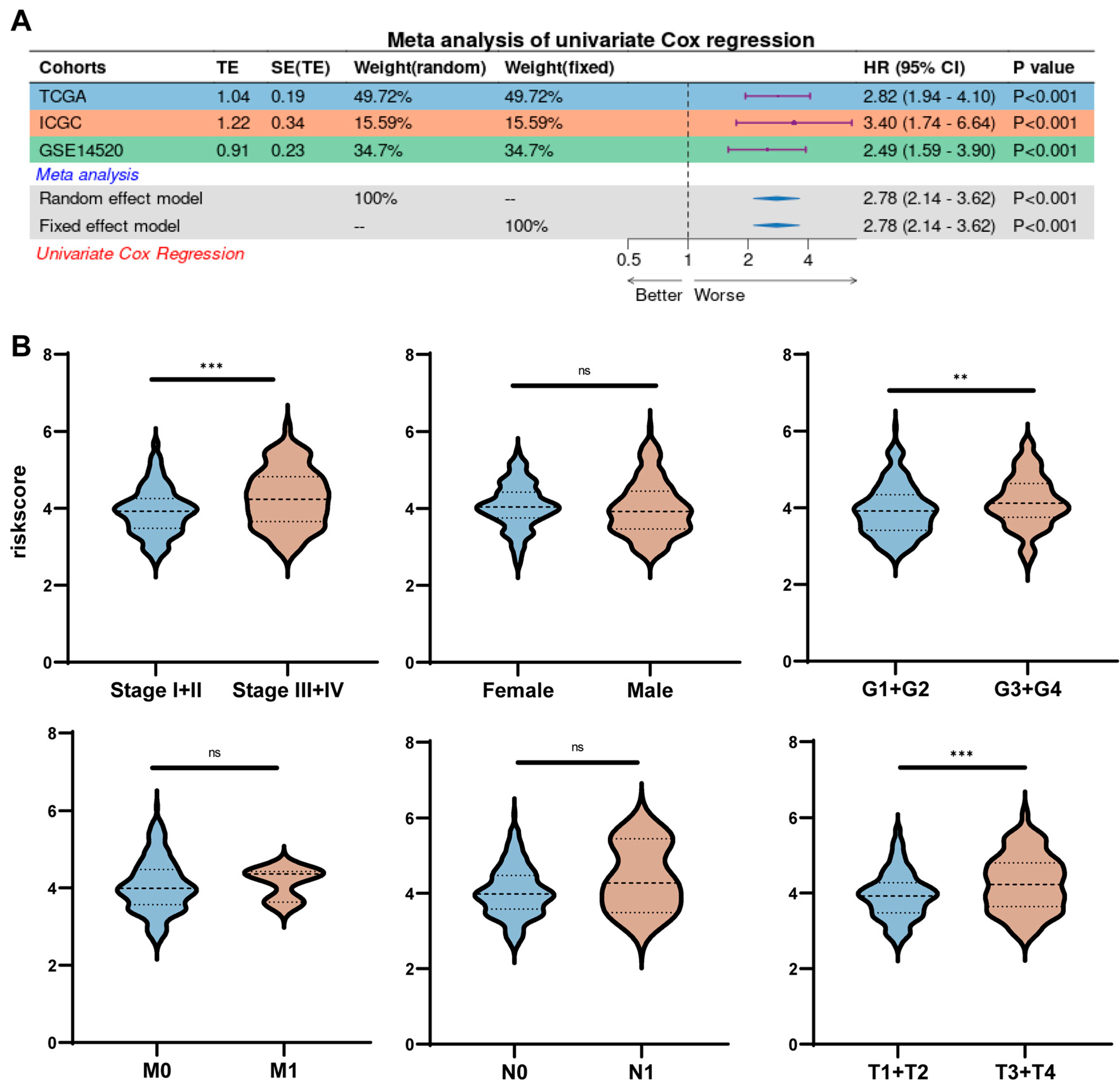
2.5. Risk Scores Correlate Strongly with Clinical Characteristics
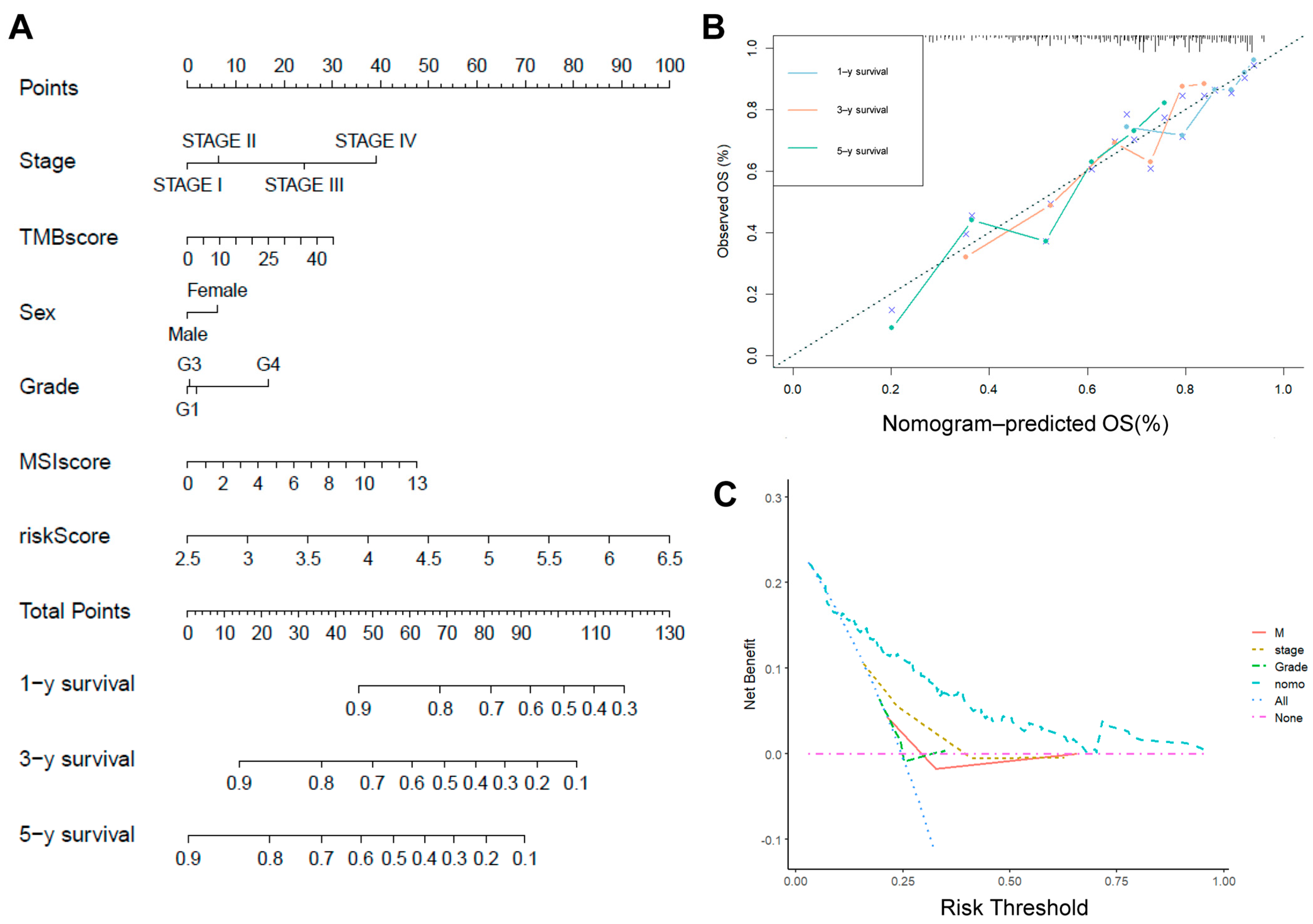
2.6. Risk Scores in Combination with Other Clinicopathological Characteristics
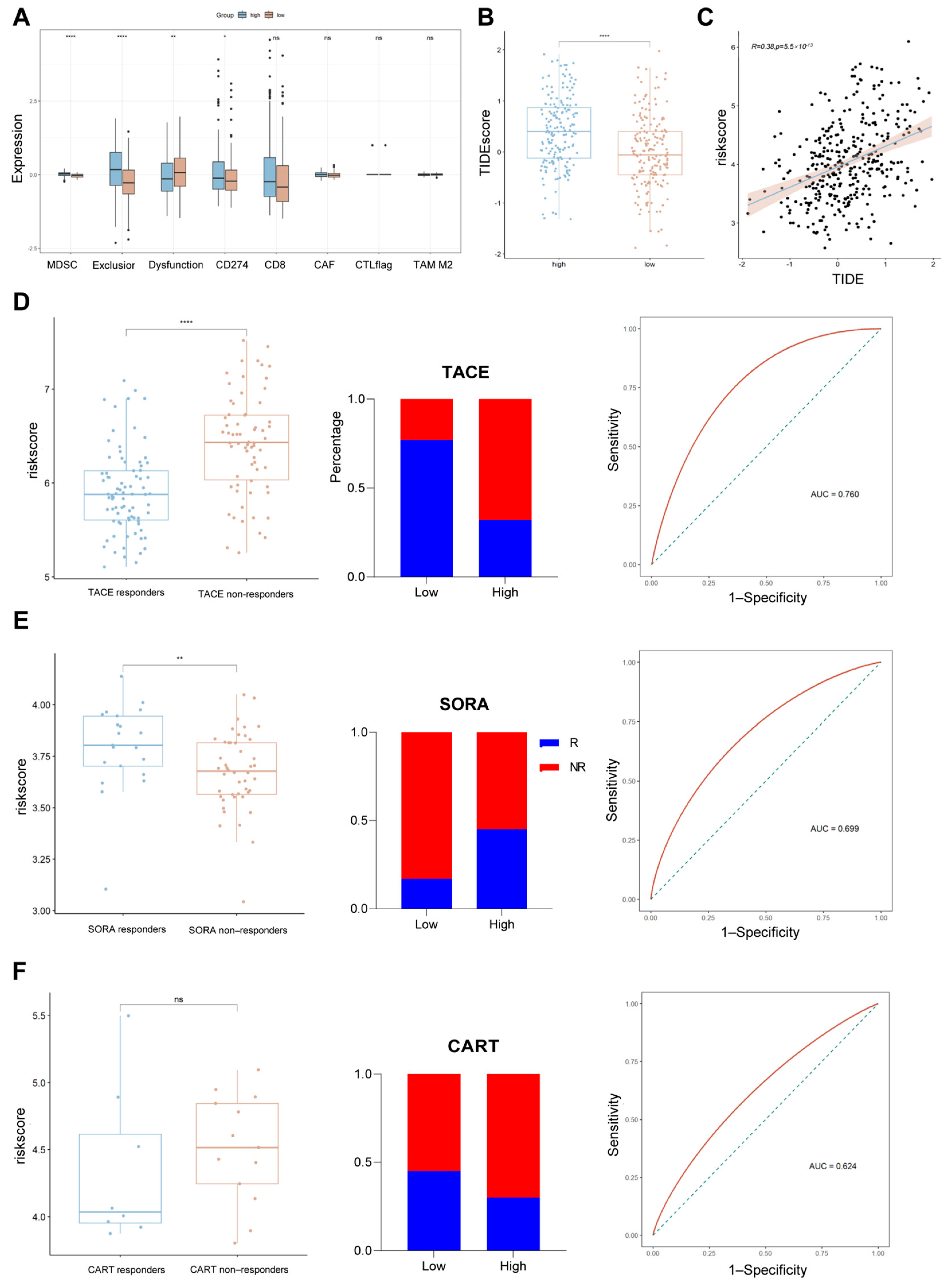
2.7. Assessing the Relationship Between Risk Scores and Common Treatments
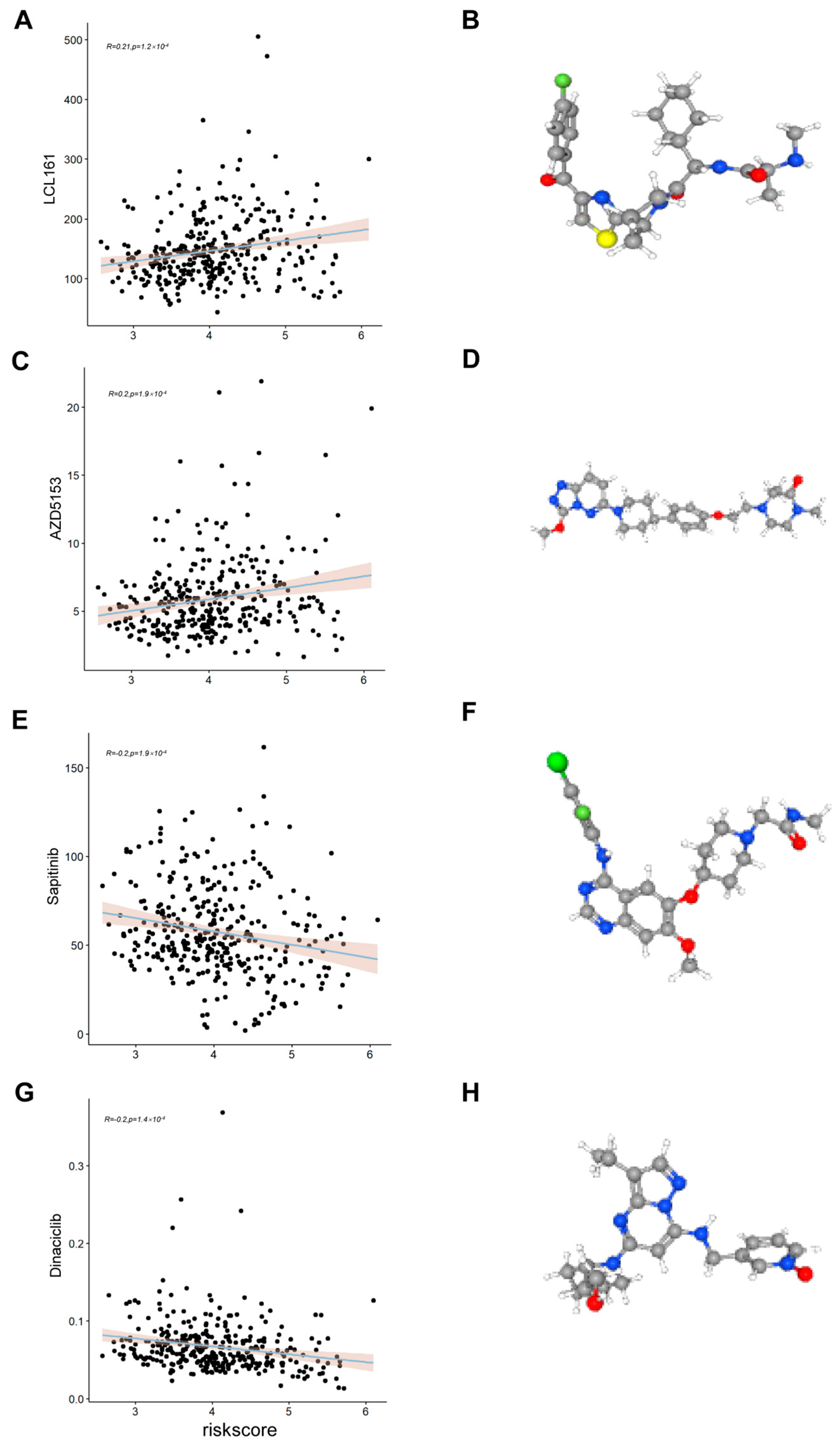
2.8. Assessing the Relationship Between Risk Scores and Drug Sensitivity
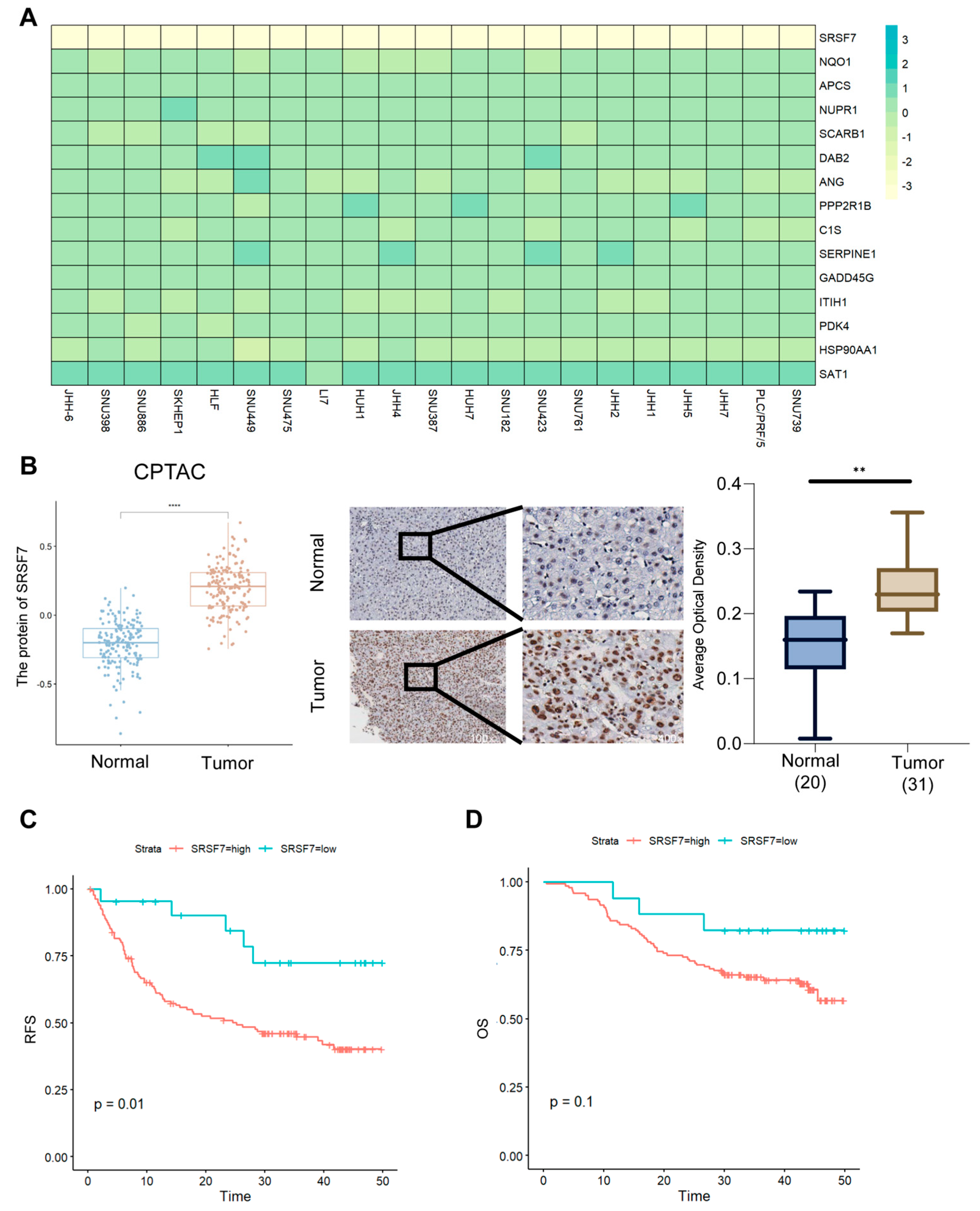
2.9. SRSF7 Is Highly Expressed in Hepatocellular Carcinoma Tissues and Is Associated with a Poor Prognosis
3. Discussion
4. Materials and Methods
4.1. Data Sources and Processing
4.2. Processing of Single-Cell Data
4.3. Identifying Tumor Cells
4.4. Calculation of Malignancy Scores
4.5. Machine Learning
4.6. Evaluating and Comparing Models
4.7. Immunohistochemistry
4.8. Cell Experiments
4.9. RT-PCR
4.10. Western Blot
4.11. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Laversanne, M.; Sung, H.; Ferlay, J.; Siegel, R.L.; Soerjomataram, I.; Jemal, A. Global Cancer Statistics 2022: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2024, 74, 229–263. [Google Scholar] [CrossRef]
- Qu, Z.; Wu, J.; Wu, J.; Luo, D.; Jiang, C.; Ding, Y. Exosomes Derived from HCC Cells Induce Sorafenib Resistance in Hepatocellular Carcinoma Both In Vivo and In Vitro. J. Exp. Clin. Cancer Res. 2016, 35, 159. [Google Scholar] [CrossRef] [PubMed]
- Grohmann, M.; Wiede, F.; Dodd, G.T.; Gurzov, E.N.; Ooi, G.J.; Butt, T.; Rasmiena, A.A.; Kaur, S.; Gulati, T.; Goh, P.K.; et al. Obesity Drives STAT-1-Dependent NASH and STAT-3-Dependent HCC. Cell 2018, 175, 1289–1306.e20. [Google Scholar] [CrossRef] [PubMed]
- Yeo, Y.H.; Lee, Y.-T.; Tseng, H.-R.; Zhu, Y.; You, S.; Agopian, V.G.; Yang, J.D. Alpha-Fetoprotein: Past, Present, and Future. Hepatol. Commun. 2024, 8, e0422. [Google Scholar] [CrossRef]
- Zeng, Z.; Cao, Z.; Tang, Y. Increased E2F2 Predicts Poor Prognosis in Patients with HCC Based on TCGA Data. BMC Cancer 2020, 20, 1037. [Google Scholar] [CrossRef]
- Peng, H.; Zou, Z.; Xiang, Z.; Lu, X.; Zhang, Y.; Peng, X. Cuproptosis-Related Prognostic Signatures Predict the Prognosis and Immunotherapy in HCC Patients. Medicine 2023, 102, e34741. [Google Scholar] [CrossRef] [PubMed]
- Du, B.; Wang, F.; Jarad, B.; Wang, Z.; Zhang, Y. A Novel Signature Based on Microvascular Invasion Predicts the Recurrence of HCC. J. Transl. Med. 2020, 18, 272. [Google Scholar] [CrossRef]
- Tanay, A.; Regev, A. Scaling Single-Cell Genomics from Phenomenology to Mechanism. Nature 2017, 541, 331–338. [Google Scholar] [CrossRef]
- Zheng, C.; Zheng, L.; Yoo, J.-K.; Guo, H.; Zhang, Y.; Guo, X.; Kang, B.; Hu, R.; Huang, J.Y.; Zhang, Q.; et al. Landscape of Infiltrating T Cells in Liver Cancer Revealed by Single-Cell Sequencing. Cell 2017, 169, 1342–1356.e16. [Google Scholar] [CrossRef]
- Ma, L.; Hernandez, M.O.; Zhao, Y.; Mehta, M.; Tran, B.; Kelly, M.; Rae, Z.; Hernandez, J.M.; Davis, J.L.; Martin, S.P.; et al. Tumor Cell Biodiversity Drives Microenvironmental Reprogramming in Liver Cancer. Cancer Cell 2019, 36, 418–430.e6. [Google Scholar] [CrossRef]
- Zhang, F.; Gu, W.; Hurles, M.E.; Lupski, J.R. Copy Number Variation in Human Health, Disease, and Evolution. Annu. Rev. Genomics Hum. Genet. 2009, 10, 451–481. [Google Scholar] [CrossRef]
- Mirzaei, G.; Petreaca, R.C. Distribution of Copy Number Variations and Rearrangement Endpoints in Human Cancers with a Review of Literature. Mutat. Res. 2022, 824, 111773. [Google Scholar] [CrossRef]
- Weir, B.; Zhao, X.; Meyerson, M. Somatic Alterations in the Human Cancer Genome. Cancer Cell 2004, 6, 433–438. [Google Scholar] [CrossRef] [PubMed]
- Shao, X.; Lv, N.; Liao, J.; Long, J.; Xue, R.; Ai, N.; Xu, D.; Fan, X. Copy Number Variation Is Highly Correlated with Differential Gene Expression: A Pan-Cancer Study. BMC Med. Genet. 2019, 20, 175. [Google Scholar] [CrossRef]
- Cai, X.; Liang, C.; Zhang, M.; Dong, Z.; Weng, Y.; Yu, W. Mitochondrial DNA Copy Number and Cancer Risks: A Comprehensive Mendelian Randomization Analysis. Int. J. Cancer 2024, 154, 1504–1513. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.-Q.; Tang, Z.; Huang, R.; Qu, W.-F.; Fang, Y.; Yang, R.; Tao, C.-Y.; Gao, J.; Wu, X.-L.; Sun, H.-X.; et al. CD36+ Cancer-Associated Fibroblasts Provide Immunosuppressive Microenvironment for Hepatocellular Carcinoma via Secretion of Macrophage Migration Inhibitory Factor. Cell Discov. 2023, 9, 25. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, T.; Xu, Y.; Zhang, X.; Li, F.; Bai, J.; Chen, J.; Jiang, W.; Yang, K.; Ou, Q.; et al. CellMarker 2.0: An Updated Database of Manually Curated Cell Markers in Human/Mouse and Web Tools Based on scRNA-Seq Data. Nucleic Acids Res. 2023, 51, D870–D876. [Google Scholar] [CrossRef]
- Wang, L.; Li, X.; Xu, C.; Wang, D.; Ma, C.; Wang, Z.; Li, Y.; Li, Z. Unveiling Novel Cell Clusters and Biomarkers in Glioblastoma and Its Peritumoral Microenvironment at the Single-Cell Perspective. J. Transl. Med. 2024, 22, 551. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.-O.; Lee, K.-M.; Lee, H.-B.; Kim, K.-T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-Cell RNA-Seq Enables Comprehensive Tumour and Immune Cell Profiling in Primary Breast Cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef]
- Tirosh, I.; Izar, B.; Prakadan, S.M.; Wadsworth, M.H.; Treacy, D.; Trombetta, J.J.; Rotem, A.; Rodman, C.; Lian, C.; Murphy, G.; et al. Dissecting the Multicellular Ecosystem of Metastatic Melanoma by Single-Cell RNA-Seq. Science 2016, 352, 189–196. [Google Scholar] [CrossRef]
- Fan, Y.; Xue, H.; Zheng, H. Systemic Therapy for Hepatocellular Carcinoma: Current Updates and Outlook. J. Hepatocell. Carcinoma 2022, 9, 233–263. [Google Scholar] [CrossRef]
- Chang, M.-H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lai, M.-W.; Wu, T.-C.; Wu, S.-F.; Lee, C.-M.; Yang, S.-S.; Chu, H.-C.; et al. Long-Term Effects of Hepatitis B Immunization of Infants in Preventing Liver Cancer. Gastroenterology 2016, 151, 472–480.e1. [Google Scholar] [CrossRef] [PubMed]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Gavriilidis, P.; Pawlik, T.M.; Azoulay, D. Comprehensive Review of Hepatocellular Carcinoma with Portal Vein Tumor Thrombus: State of Art and Future Perspectives. Hepatobiliary Pancreat. Dis. Int. 2024, 23, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Holczbauer, Á.; Wangensteen, K.J.; Shin, S. Cellular Origins of Regenerating Liver and Hepatocellular Carcinoma. JHEP Rep. 2022, 4, 100416. [Google Scholar] [CrossRef]
- Yao, C.; Wu, S.; Kong, J.; Sun, Y.; Bai, Y.; Zhu, R.; Li, Z.; Sun, W.; Zheng, L. Angiogenesis in Hepatocellular Carcinoma: Mechanisms and Anti-Angiogenic Therapies. Cancer Biol. Med. 2023, 20, 25–43. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhang, Q. Insights on Epithelial Cells at the Single-Cell Level in Hepatocellular Carcinoma Prognosis and Response to Chemotherapy. Front. Pharmacol. 2023, 14, 1292831. [Google Scholar] [CrossRef]
- Campana, L.; Esser, H.; Huch, M.; Forbes, S. Liver Regeneration and Inflammation: From Fundamental Science to Clinical Applications. Nat. Rev. Mol. Cell Biol. 2021, 22, 608–624. [Google Scholar] [CrossRef]
- Shen, W.; Yuan, L.; Cheng, F.; Wu, Z.; Li, X. SRSF7 Is a Promising Prognostic Biomarker in Hepatocellular Carcinoma and Is Associated with Immune Infiltration. Genes. Genom. 2024, 46, 49–64. [Google Scholar] [CrossRef]
- Shi, W.; Yao, X.; Cao, X.; Fu, Y.; Wang, Y. Serine/Arginine-Rich Splicing Factor 7 Knockdown Inhibits Aerobic Glycolysis and Growth in HepG2 Cells by Regulating PKM2 Expression. Curr. Issues Mol. Biol. 2024, 46, 5023–5036. [Google Scholar] [CrossRef]
- Lan, W.; Santofimia-Castaño, P.; Xia, Y.; Zhou, Z.; Huang, C.; Fraunhoffer, N.; Barea, D.; Cervello, M.; Giannitrapani, L.; Montalto, G.; et al. Targeting NUPR1 with the Small Compound ZZW-115 Is an Efficient Strategy to Treat Hepatocellular Carcinoma. Cancer Lett. 2020, 486, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Li, D.; Liang, Y.; Long, Y.; Liu, Y. SEC14L2 Regulates the Transport of Cholesterol in Non-Small Cell Lung Cancer through SCARB1. Lipids Health Dis. 2024, 23, 407. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yao, X.; Jiang, Q.; Sun, X. miR-106b Targets DAB2 to Promote Hepatocellular Carcinoma Cell Proliferation and Metastasis. Oncol. Lett. 2018, 16, 3063–3069. [Google Scholar] [CrossRef] [PubMed]
- Ji, Y.; Chen, H.; Gow, W.; Ma, L.; Jin, Y.; Hui, B.; Yang, Z.; Wang, Z. Potential Biomarkers Ang II/AT1R and S1P/S1PR1 Predict the Prognosis of Hepatocellular Carcinoma. Oncol. Lett. 2020, 20, 208. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, Z.; Ma, A.; Qu, Y.; Xia, S.; Xu, D.; Ge, C.; Qiu, B.; Xia, Q.; Li, J.; et al. Growth Arrest and DNA Damage 45G Down-Regulation Contributes to Janus Kinase/Signal Transducer and Activator of Transcription 3 Activation and Cellular Senescence Evasion in Hepatocellular Carcinoma. Hepatology 2014, 59, 178–189. [Google Scholar] [CrossRef]
- Qin, Y.-J.; Lin, T.-Y.; Lin, X.-L.; Liu, Y.; Zhao, W.-T.; Li, X.-Y.; Lian, M.; Chen, H.-W.; Li, Y.-L.; Zhang, X.-L.; et al. Loss of PDK4 Expression Promotes Proliferation, Tumorigenicity, Motility and Invasion of Hepatocellular Carcinoma Cells. J. Cancer 2020, 11, 4397–4405. [Google Scholar] [CrossRef]
- Wang, Z.; Fan, L.; Xu, H.; Qin, Z.; Zhu, Z.; Wu, D.; Zhang, Y.; Liu, R.; Wei, J.; Qian, Z.; et al. HSP90AA1 Is an Unfavorable Prognostic Factor for Hepatocellular Carcinoma and Contributes to Tumorigenesis and Chemotherapy Resistance. Transl. Oncol. 2024, 50, 102148. [Google Scholar] [CrossRef]
- Li, D.; Yu, T.; Hu, J.; Wu, J.; Feng, S.; Xu, Q.; Zhu, H.; Zhang, X.; Zhang, Y.; Zhou, B.; et al. Downregulation of CYP39A1 Serves as a Novel Biomarker in Hepatocellular Carcinoma with Worse Clinical Outcome. Oxid. Med. Cell. Longev. 2021, 2021, 5175581. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, X.; Li, D.; Li, J.; Yuan, J.; Gu, L.; Xiong, X. Expression, Location, Clinical Implication, and Bioinformatics Analysis of RNASET2 in Gastric Adenocarcinoma. Front. Oncol. 2020, 10, 836. [Google Scholar] [CrossRef]
- Hart, T.; Brown, K.R.; Sircoulomb, F.; Rottapel, R.; Moffat, J. Measuring Error Rates in Genomic Perturbation Screens: Gold Standards for Human Functional Genomics. Mol. Syst. Biol. 2014, 10, 733. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, W.; Zhang, Y.; Adegboro, A.A.; Fasoranti, D.O.; Dai, L.; Pan, Z.; Liu, H.; Xiong, Y.; Li, W.; et al. Mime: A Flexible Machine-Learning Framework to Construct and Visualize Models for Clinical Characteristics Prediction and Feature Selection. Comput. Struct. Biotechnol. J. 2024, 23, 2798–2810. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Cheng, L.; Yan, H.; Yuan, J. Multi-Omics Integration: Predicting Progression and Optimizing Clinical Treatment of Hepatocellular Carcinoma Through Malignant-Cell-Related Genes. Int. J. Mol. Sci. 2025, 26, 6135. https://doi.org/10.3390/ijms26136135
Wang Q, Cheng L, Yan H, Yuan J. Multi-Omics Integration: Predicting Progression and Optimizing Clinical Treatment of Hepatocellular Carcinoma Through Malignant-Cell-Related Genes. International Journal of Molecular Sciences. 2025; 26(13):6135. https://doi.org/10.3390/ijms26136135
Chicago/Turabian StyleWang, Qianwen, Lingli Cheng, Honglin Yan, and Jingping Yuan. 2025. "Multi-Omics Integration: Predicting Progression and Optimizing Clinical Treatment of Hepatocellular Carcinoma Through Malignant-Cell-Related Genes" International Journal of Molecular Sciences 26, no. 13: 6135. https://doi.org/10.3390/ijms26136135
APA StyleWang, Q., Cheng, L., Yan, H., & Yuan, J. (2025). Multi-Omics Integration: Predicting Progression and Optimizing Clinical Treatment of Hepatocellular Carcinoma Through Malignant-Cell-Related Genes. International Journal of Molecular Sciences, 26(13), 6135. https://doi.org/10.3390/ijms26136135