
Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis

 , and
, and
Abstract
1. Introduction
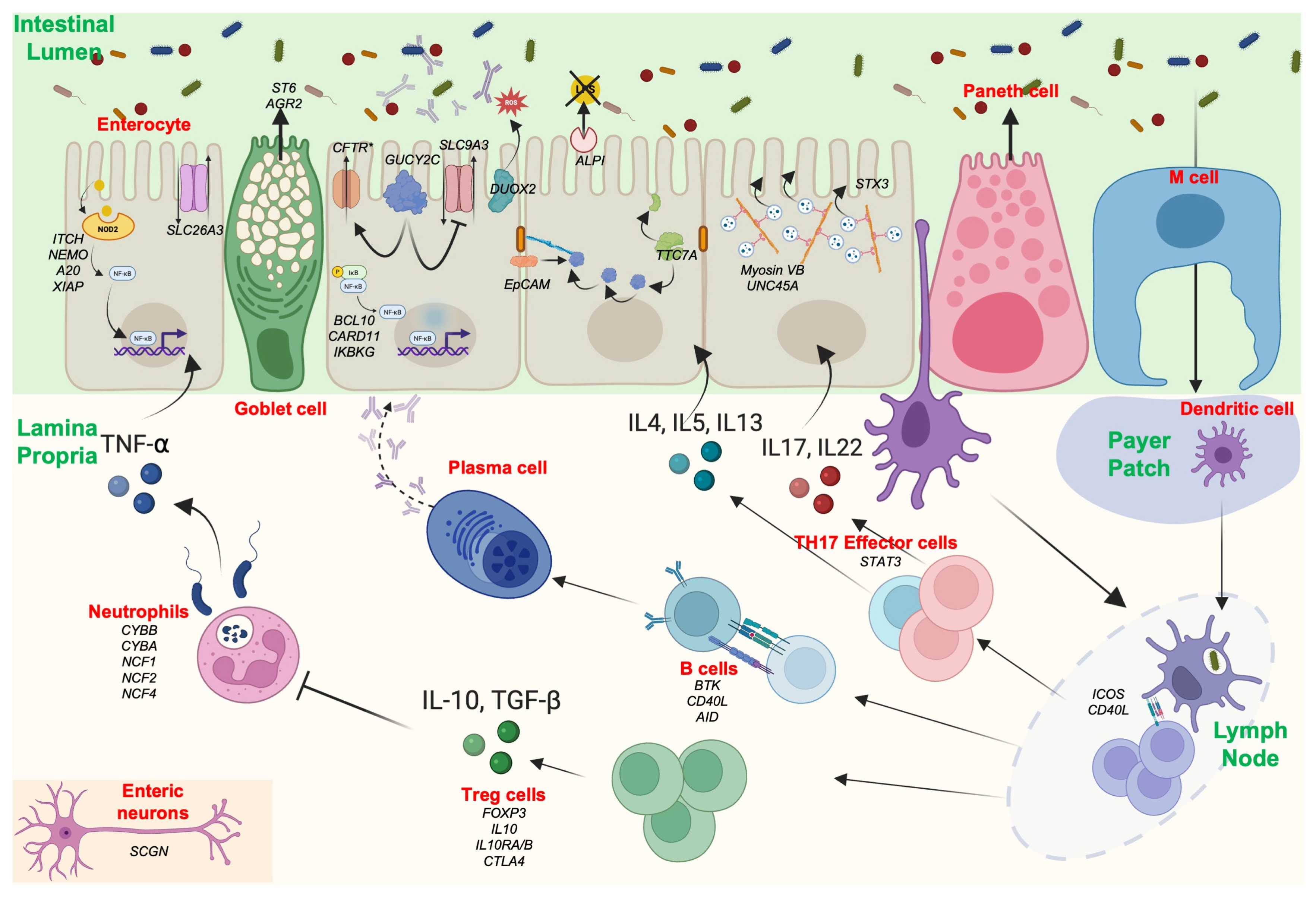
2. Altered Gastrointestinal Barrier in IBD and MID
2.1. Mucus and Goblet Cells
2.2. Enterocytes
- Junction Defect
- Transport Defect
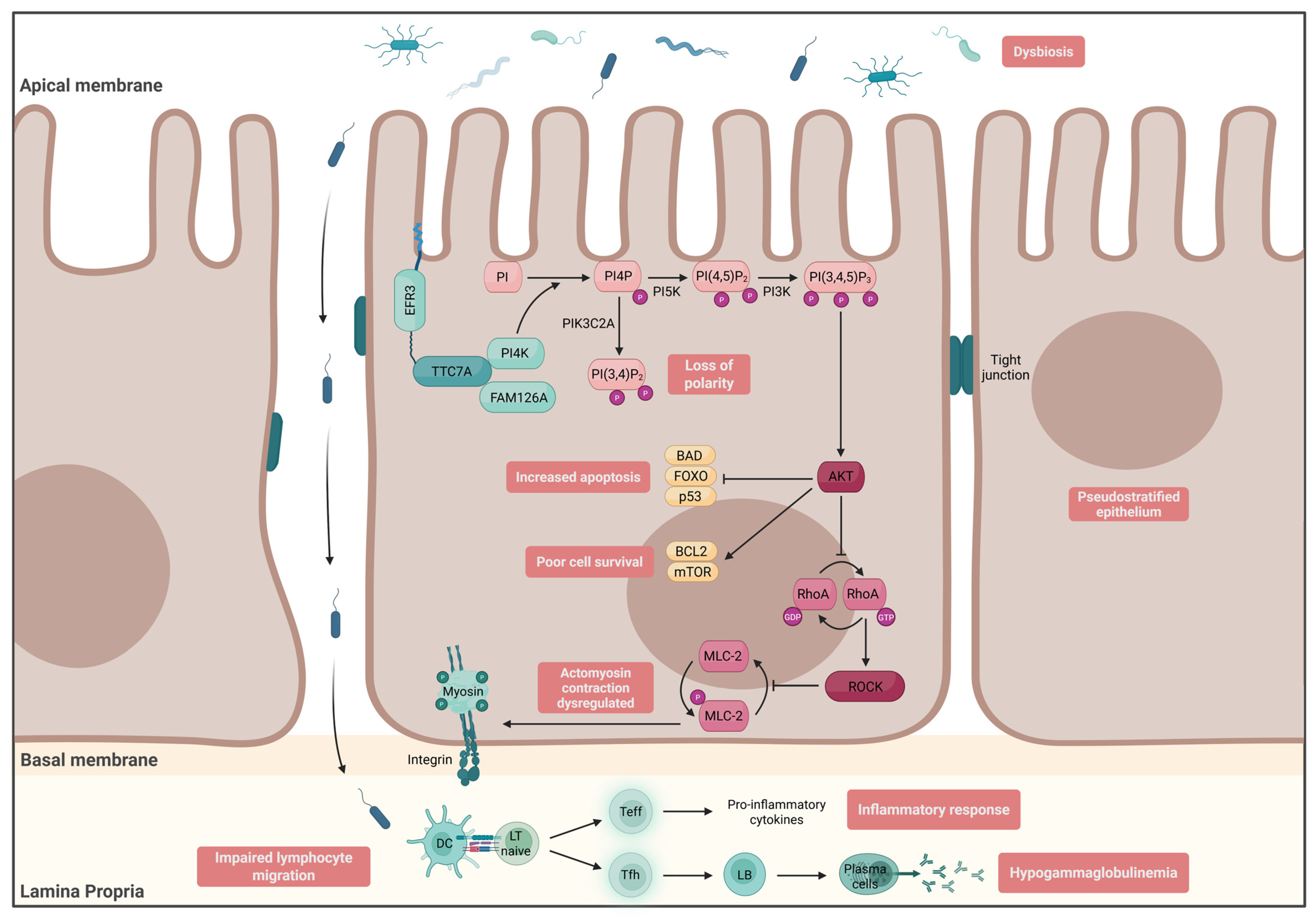
- Enterocyte Architecture
3. Altered GI Microbiota–Host Mucosal Interaction in IBD and MID
3.1. Innate Immunity in Epithelial Cells
- GUCY2C
- ALPI
- DUOX2/NOX1
3.2. Innate Immunity in Immune Cells
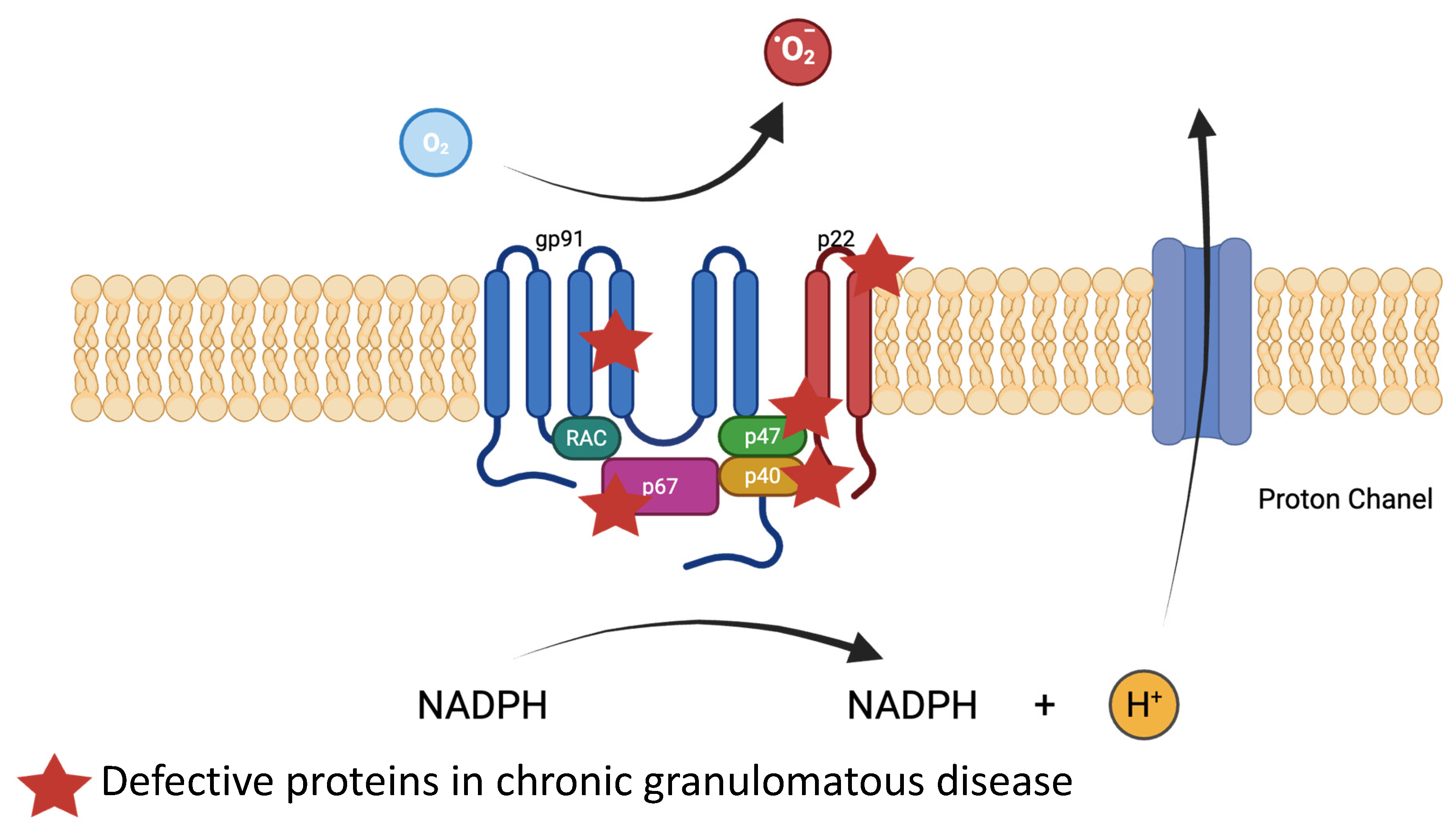
- Neutrophils’ and Macrophages’ Phagocytosis: NADPH Oxidase
3.3. Innate Immunity in Epithelial Cells and Immune Cells
- NOD2/CARD15
- Inflammasomes
- Autophagy
3.4. Enteroencrine and Neuroendocrine Cells
3.5. Microbiota in IBD and MID
4. Altered Adaptive Immunity in IBD and MID
4.1. T-Effectors: TH1/TH2/TH17
4.2. B Cells
5. Altered Regulatory Immunity in IBD and MID
6. Common Therapeutic Approach in IBD and MID
7. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ADA | Adenosine deaminase |
| AGR2 | Anterior gradient 2 |
| AIEC | Adherent-invasive E. coli |
| AIM2 | Absent in melanoma 2 |
| AJs | Adherens junctions |
| ALPI | Intestinal alkaline phosphatase |
| AMPs | Antimicrobial peptides |
| AR | Autosomal recessive |
| BTK | Bruton’s tyrosine kinase |
| CARD15 | Caspase recruitment domain-containing protein 15 |
| CCD | Congenital chronic diarrhea |
| CD | Crohn’s disease |
| CFTR | Cystic fibrosis transmembrane conductance regulator |
| CGD | Chronic granulomatous disease |
| CNS | Central nervous system |
| CTE | Congenital tufting enteropathy |
| CTLA4 | T-lymphocyte antigen 4 |
| cGMP | Guanosine 3′, 5′-cyclic monophosphate |
| DAMPs | Danger-associated molecular patterns |
| DCs | Dendritic cells |
| DOCK8 | Dedicator of cytokinesis 8 |
| DUOX2 | ROS production through dual oxidase 2 |
| EECs | Enteroendocrine cells |
| EpCAM | Epithelial cell adhesion molecule |
| EBV | Epstein–Barr virus |
| FMF | Familial Mediterranean fever |
| FOXP3 | Forkhead box P3 |
| GC-C | Guanylate cyclase C |
| GI | Gastrointestinal |
| GLILD | Granulomatous lymphocytic interstitial lung disease |
| GM-CSF | Granulocyte-macrophage colony-stimulating factor |
| GOF | Gain-of-function |
| GUCY2C | Guanylate cyclase 2C |
| HEIS | Hyper-IgE syndrome |
| HLH | Hemophagocytic lymphohistiocytosis |
| hnRNP A1 | Heterogeneous nuclear ribonucleoprotein A1 |
| HSCT | Hematopoietic stem cell transplantation |
| IBD | Inflammatory bowel disease |
| ICOS | Inducible T cell co-stimulator |
| IECs | Intestinal epithelial cells |
| IKKγ | Inhibitor of kappaB kinase |
| JAK | Janus kinase |
| LOF | loss-of-function |
| LPS | Lipopolysaccharide |
| MID | Monogenic intestinal disease |
| MHC | Major histocompatibility complex |
| MLCK | Myosin light chain kinase |
| MPO | Myeloperoxidase |
| MVID | Microvillus inclusion disease |
| MYO5B | Myosin 5b |
| MDP | Muramyl dipeptide |
| NEMO | NF-kB essential modulator |
| NETs | Neutrophil extracellular traps |
| NfkB | Nuclear factor-kappa B |
| NHE3 | Na+/H+ antiporter 3 |
| NLRP3 | NOD-like receptor family, pyrin domain containing 3 |
| NOD2 | Nucleotide-binding oligomerization domain 2 |
| NOX1 | NADPH oxidase 1 |
| PAD4 | Peptidylarginine Deiminase 4 |
| PAMPs | Pathogen-associated molecular patterns |
| PERCC1 | PERCC1 |
| PI4P | PI4-phosphate |
| RFX6 | RFX6 |
| RIPK2 | Receptor-interacting protein kinase 2 |
| ROS | Reactive oxygen species |
| SCFA | Short-chain fatty acid |
| SCGN | Secretagogin |
| SLC26A3 | Solute carrier family 26 member 3 |
| SNVs | Single-nucleotide variants |
| ST6 | ST6GALNAC1 |
| TJs | Tight junctions |
| TLRs | Toll-like receptors |
| TNFAIP3 | TNFα-induced protein 3 |
| Tregs | Regulatory T cells |
| TTC7A | Tetratricopeptide repeat domain 7A |
| UC | Ulcerative colitis |
| VEOIBD | Very early-onset IBD |
| WES | Whole exome sequencing |
| XIAP | X-linked inhibitor of apoptosis protein |
References
- Uhlig, H.H.; Schwerd, T.; Koletzko, S.; Shah, N.; Kammermeier, J.; Elkadri, A.; Ouahed, J.; Wilson, D.C.; Travis, S.P.; Turner, D.; et al. The diagnostic approach to monogenic very early onset inflammatory bowel disease. Gastroenterology 2014, 147, 990–1007.e3. [Google Scholar] [CrossRef] [PubMed]
- Nambu, R.; Warner, N.; Mulder, D.J.; Kotlarz, D.; McGovern, D.P.; Cho, J.; Klein, C.; Snapper, S.B.; Griffiths, A.M.; Iwama, I.; et al. A Systematic Review of Monogenic Inflammatory Bowel Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2022, 20, e653–e663. [Google Scholar] [CrossRef] [PubMed]
- Charbit-Henrion, F.; Parlato, M.; Malamut, G.; Ruemmele, F.; Cerf-Bensussan, N. Intestinal immunoregulation: Lessons from human mendelian diseases. Mucosal Immunol. 2021, 14, 1017–1037. [Google Scholar] [CrossRef] [PubMed]
- Bildstein, T.; Charbit-Henrion, F.; Azabdaftari, A.; Cerf-Bensussan, N.; Uhlig, H.H. Cellular and molecular basis of proximal small intestine disorders. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 687–709. [Google Scholar] [CrossRef]
- Charbit-Henrion, F.; Parlato, M.; Hanein, S.; Duclaux-Loras, R.; Nowak, J.; Begue, B.; Rakotobe, S.; Bruneau, J.; Fourrage, C.; Alibeu, O.; et al. Diagnostic Yield of Next-Generation Sequencing in Very Early-Onset Inflammatory Bowel Diseases: A Multicenter Study. J. Crohns Colitis 2018, 12, 1104–1112. [Google Scholar] [CrossRef]
- Norsa, L.; Berni Canani, R.; Duclaux-Loras, R.; Bequet, E.; Köglmeier, J.; Russell, R.K.; Uhlig, H.H.; Travis, S.; Hollis, J.; Koletzko, S.; et al. Inflammatory Bowel Disease in Patients with Congenital Chloride Diarrhoea. J. Crohns Colitis 2021, 15, 1679–1685. [Google Scholar] [CrossRef]
- Fiskerstrand, T.; Arshad, N.; Haukanes, B.I.; Tronstad, R.R.; Pham, K.D.-C.; Johansson, S.; Håvik, B.; Tønder, S.L.; Levy, S.E.; Brackman, D.; et al. Familial diarrhea syndrome caused by an activating GUCY2C mutation. N. Engl. J. Med. 2012, 366, 1586–1595. [Google Scholar] [CrossRef]
- McGuckin, M.A.; Eri, R.; Simms, L.A.; Florin, T.H.J.; Radford-Smith, G. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm. Bowel Dis. 2009, 15, 100–113. [Google Scholar] [CrossRef]
- Müller, T.; Rasool, I.; Heinz-Erian, P.; Mildenberger, E.; Hülstrunk, C.; Müller, A.; Michaud, L.; Koot, B.G.P.; Ballauff, A.; Vodopiutz, J.; et al. Congenital secretory diarrhoea caused by activating germline mutations in GUCY2C. Gut 2016, 65, 1306–1313. [Google Scholar] [CrossRef]
- Müller, T.; Hess, M.W.; Schiefermeier, N.; Pfaller, K.; Ebner, H.L.; Heinz-Erian, P.; Ponstingl, H.; Partsch, J.; Röllinghoff, B.; Köhler, H.; et al. MYO5B mutations cause microvillus inclusion disease and disrupt epithelial cell polarity. Nat. Genet. 2008, 40, 1163–1165. [Google Scholar] [CrossRef]
- Segain, J.-P.; Raingeard de la Blétière, D.; Sauzeau, V.; Bourreille, A.; Hilaret, G.; Cario-Toumaniantz, C.; Pacaud, P.; Galmiche, J.-P.; Loirand, G. Rho kinase blockade prevents inflammation via nuclear factor kappa B inhibition: Evidence in Crohn’s disease and experimental colitis. Gastroenterology 2003, 124, 1180–1187. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Kim, G.; Shafer, S.; Chen, Z.; Kubo, S.; Ji, Y.; Luo, J.; Yang, W.; Perner, S.P.; Kanellopoulou, C.; et al. Mucus sialylation determines intestinal host-commensal homeostasis. Cell 2022, 185, 1172–1188.e28. [Google Scholar] [CrossRef] [PubMed]
- Al-Shaibi, A.A.; Abdel-Motal, U.M.; Hubrack, S.Z.; Bullock, A.N.; Al-Marri, A.A.; Agrebi, N.; Al-Subaiey, A.A.; Ibrahim, N.A.; Charles, A.K.; Elawad, M.; et al. Human AGR2 Deficiency Causes Mucus Barrier Dysfunction and Infantile Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 1809–1830. [Google Scholar] [CrossRef] [PubMed]
- Parlato, M.; Charbit-Henrion, F.; Pan, J.; Romano, C.; Duclaux-Loras, R.; Le Du, M.-H.; Warner, N.; Francalanci, P.; Bruneau, J.; Bras, M.; et al. Human ALPI deficiency causes inflammatory bowel disease and highlights a key mechanism of gut homeostasis. EMBO Mol. Med. 2018, 10, e8483. [Google Scholar] [CrossRef]
- Hayes, P.; Dhillon, S.; O’Neill, K.; Thoeni, C.; Hui, K.Y.; Elkadri, A.; Guo, C.H.; Kovacic, L.; Aviello, G.; Alvarez, L.A.; et al. Defects in NADPH Oxidase Genes NOX1 and DUOX2 in Very Early Onset Inflammatory Bowel Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 489–502. [Google Scholar] [CrossRef]
- Ament, M.E.; Ochs, H.D. Gastrointestinal manifestations of chronic granulomatous disease. N. Engl. J. Med. 1973, 288, 382–387. [Google Scholar] [CrossRef]
- Lohr, N.J.; Molleston, J.P.; Strauss, K.A.; Torres-Martinez, W.; Sherman, E.A.; Squires, R.H.; Rider, N.L.; Chikwava, K.R.; Cummings, O.W.; Morton, D.H.; et al. Human ITCH E3 ubiquitin ligase deficiency causes syndromic multisystem autoimmune disease. Am. J. Hum. Genet. 2010, 86, 447–453. [Google Scholar] [CrossRef]
- Hall, C.H.T.; de Zoeten, E.F. Understanding very early onset inflammatory bowel disease (VEOIBD) in relation to inborn errors of immunity. Immunol. Rev. 2024, 322, 329–338. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef]
- Abolhassani, H.; El-Sherbiny, Y.M.; Arumugakani, G.; Carter, C.; Richards, S.; Lawless, D.; Wood, P.; Buckland, M.; Heydarzadeh, M.; Aghamohammadi, A.; et al. Expanding Clinical Phenotype and Novel Insights into the Pathogenesis of ICOS Deficiency. J. Clin. Immunol. 2020, 40, 277–288. [Google Scholar] [CrossRef]
- Pillay, B.A.; Fusaro, M.; Gray, P.E.; Statham, A.L.; Burnett, L.; Bezrodnik, L.; Kane, A.; Tong, W.; Abdo, C.; Winter, S.; et al. Somatic reversion of pathogenic DOCK8 variants alters lymphocyte differentiation and function to effectively cure DOCK8 deficiency. J. Clin. Investig. 2021, 131, e142434. [Google Scholar] [CrossRef] [PubMed]
- Loeys, B.L.; Schwarze, U.; Holm, T.; Callewaert, B.L.; Thomas, G.H.; Pannu, H.; De Backer, J.F.; Oswald, G.L.; Symoens, S.; Manouvrier, S.; et al. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N. Engl. J. Med. 2006, 355, 788–798. [Google Scholar] [CrossRef] [PubMed]
- Renner, E.D.; Torgerson, T.R.; Rylaarsdam, S.; Añover-Sombke, S.; Golob, K.; LaFlam, T.; Zhu, Q.; Ochs, H.D. STAT3 mutation in the original patient with Job’s syndrome. N. Engl. J. Med. 2007, 357, 1667–1668. [Google Scholar] [CrossRef]
- Flanagan, S.E.; Haapaniemi, E.; Russell, M.A.; Caswell, R.; Allen, H.L.; De Franco, E.; McDonald, T.J.; Rajala, H.; Ramelius, A.; Barton, J.; et al. Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nat. Genet. 2014, 46, 812–814. [Google Scholar] [CrossRef]
- Uhlig, H.H. Monogenic diseases associated with intestinal inflammation: Implications for the understanding of inflammatory bowel disease. Gut 2013, 62, 1795–1805. [Google Scholar] [CrossRef]
- Duclaux-Loras, R.; Charbit-Henrion, F.; Neven, B.; Nowak, J.; Collardeau-Frachon, S.; Malcus, C.; Ray, P.F.; Moshous, D.; Beltrand, J.; Goulet, O.; et al. Clinical Heterogeneity of Immune Dysregulation, Polyendocrinopathy, Enteropathy, X-Linked Syndrome: A French Multicenter Retrospective Study. Clin. Transl. Gastroenterol. 2018, 9, e201. [Google Scholar] [CrossRef]
- Begue, B.; Verdier, J.; Rieux-Laucat, F.; Goulet, O.; Morali, A.; Canioni, D.; Hugot, J.-P.; Daussy, C.; Verkarre, V.; Pigneur, B.; et al. Defective IL10 signaling defining a subgroup of patients with inflammatory bowel disease. Am. J. Gastroenterol. 2011, 106, 1544–1555. [Google Scholar] [CrossRef]
- Kuehn, H.S.; Ouyang, W.; Lo, B.; Deenick, E.K.; Niemela, J.E.; Avery, D.T.; Schickel, J.-N.; Tran, D.Q.; Stoddard, J.; Zhang, Y.; et al. Immune dysregulation in human subjects with heterozygous germline mutations in CTLA4. Science 2014, 345, 1623–1627. [Google Scholar] [CrossRef]
- Godl, K.; Johansson, M.E.V.; Lidell, M.E.; Mörgelin, M.; Karlsson, H.; Olson, F.J.; Gum, J.R.; Kim, Y.S.; Hansson, G.C. The N terminus of the MUC2 mucin forms trimers that are held together within a trypsin-resistant core fragment. J. Biol. Chem. 2002, 277, 47248–47256. [Google Scholar] [CrossRef]
- Grootjans, J.; Hundscheid, I.H.R.; Lenaerts, K.; Boonen, B.; Renes, I.B.; Verheyen, F.K.; Dejong, C.H.; von Meyenfeldt, M.F.; Beets, G.L.; Buurman, W.A. Ischaemia-induced mucus barrier loss and bacterial penetration are rapidly counteracted by increased goblet cell secretory activity in human and rat colon. Gut 2013, 62, 250–258. [Google Scholar] [CrossRef]
- Chang, M.; Alsaigh, T.; Kistler, E.B.; Schmid-Schönbein, G.W. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS ONE 2012, 7, e40087. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wei, B.; Wen, T.; Johansson, M.E.V.; Liu, X.; Bradford, E.; Thomsson, K.A.; McGee, S.; Mansour, L.; Tong, M.; et al. Loss of intestinal core 1-derived O-glycans causes spontaneous colitis in mice. J. Clin. Investig. 2011, 121, 1657–1666. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.M.H.; Karlsson, H.; Crespo, J.G.; Johansson, M.E.V.; Eklund, L.; Sjövall, H.; Hansson, G.C. Altered O-glycosylation profile of MUC2 mucin occurs in active ulcerative colitis and is associated with increased inflammation. Inflamm. Bowel Dis. 2011, 17, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- An, G.; Wei, B.; Xia, B.; McDaniel, J.M.; Ju, T.; Cummings, R.D.; Braun, J.; Xia, L. Increased susceptibility to colitis and colorectal tumors in mice lacking core 3-derived O-glycans. J. Exp. Med. 2007, 204, 1417–1429. [Google Scholar] [CrossRef]
- Buhner, S.; Buning, C.; Genschel, J.; Kling, K.; Herrmann, D.; Dignass, A.; Kuechler, I.; Krueger, S.; Schmidt, H.H.-J.; Lochs, H. Genetic basis for increased intestinal permeability in families with Crohn’s disease: Role of CARD15 3020insC mutation? Gut 2006, 55, 342–347. [Google Scholar] [CrossRef]
- Irvine, E.J.; Marshall, J.K. Increased intestinal permeability precedes the onset of Crohn’s disease in a subject with familial risk. Gastroenterology 2000, 119, 1740–1744. [Google Scholar] [CrossRef]
- Söderholm, J.D.; Olaison, G.; Peterson, K.H.; Franzén, L.E.; Lindmark, T.; Wirén, M.; Tagesson, C.; Sjödahl, R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of Crohn’s disease. Gut 2002, 50, 307–313. [Google Scholar] [CrossRef]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermüller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Kiesslich, R.; Duckworth, C.A.; Moussata, D.; Gloeckner, A.; Lim, L.G.; Goetz, M.; Pritchard, D.M.; Galle, P.R.; Neurath, M.F.; Watson, A.J.M. Local barrier dysfunction identified by confocal laser endomicroscopy predicts relapse in inflammatory bowel disease. Gut 2012, 61, 1146–1153. [Google Scholar] [CrossRef]
- Choi, W.; Yeruva, S.; Turner, J.R. Contributions of intestinal epithelial barriers to health and disease. Exp. Cell Res. 2017, 358, 71–77. [Google Scholar] [CrossRef]
- Salomon, J.; Goulet, O.; Canioni, D.; Brousse, N.; Lemale, J.; Tounian, P.; Coulomb, A.; Marinier, E.; Hugot, J.-P.; Ruemmele, F.; et al. Genetic characterization of congenital tufting enteropathy: Epcam associated phenotype and involvement of SPINT2 in the syndromic form. Hum. Genet. 2014, 133, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Delacour, D.; Salomon, J.; Robine, S.; Louvard, D. Plasticity of the brush border—The yin and yang of intestinal homeostasis. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Turner, J.R. Molecular basis of epithelial barrier regulation: From basic mechanisms to clinical application. Am. J. Pathol. 2006, 169, 1901–1909. [Google Scholar] [CrossRef]
- Prasad, S.; Mingrino, R.; Kaukinen, K.; Hayes, K.L.; Powell, R.M.; MacDonald, T.T.; Collins, J.E. Inflammatory processes have differential effects on claudins 2, 3 and 4 in colonic epithelial cells. Lab. Investig. J. Tech. Methods Pathol. 2005, 85, 1139–1162. [Google Scholar] [CrossRef]
- Landy, J.; Ronde, E.; English, N.; Clark, S.K.; Hart, A.L.; Knight, S.C.; Ciclitira, P.J.; Al-Hassi, H.O. Tight junctions in inflammatory bowel diseases and inflammatory bowel disease associated colorectal cancer. World, J. Gastroenterol. 2016, 22, 3117–3126. [Google Scholar] [CrossRef]
- Asano, K.; Matsushita, T.; Umeno, J.; Hosono, N.; Takahashi, A.; Kawaguchi, T.; Matsumoto, T.; Matsui, T.; Kakuta, Y.; Kinouchi, Y.; et al. A genome-wide association study identifies three new susceptibility loci for ulcerative colitis in the Japanese population. Nat. Genet. 2009, 41, 1325–1329. [Google Scholar] [CrossRef]
- Miranda, P.M.; De Palma, G.; Serkis, V.; Lu, J.; Louis-Auguste, M.P.; McCarville, J.L.; Verdu, E.F.; Collins, S.M.; Bercik, P. High salt diet exacerbates colitis in mice by decreasing Lactobacillus levels and butyrate production. Microbiome 2018, 6, 57. [Google Scholar] [CrossRef]
- Wilck, N.; Matus, M.G.; Kearney, S.M.; Olesen, S.W.; Forslund, K.; Bartolomaeus, H.; Haase, S.; Mähler, A.; Balogh, A.; Markó, L.; et al. Salt-responsive gut commensal modulates TH17 axis and disease. Nature 2017, 551, 585–589. [Google Scholar] [CrossRef]
- Janecke, A.R.; Heinz-Erian, P.; Yin, J.; Petersen, B.-S.; Franke, A.; Lechner, S.; Fuchs, I.; Melancon, S.; Uhlig, H.H.; Travis, S.; et al. Reduced sodium/proton exchanger NHE3 activity causes congenital sodium diarrhea. Hum. Mol. Genet. 2015, 24, 6614–6623. [Google Scholar] [CrossRef]
- Bernardazzi, C.; Saha, T.; Gurney, M.A.; Laubitz, D.; Dey, P.D.; Masannat, T.; Sheikh, I.A.; Midura-Kiela, M.T.; Ghishan, F.K.; Kiela, P.R. NHE3 Controls Proliferation and Migration of Colonic Epithelial Cells. Inflamm. Bowel Dis. 2025. [Google Scholar] [CrossRef]
- Engevik, A.C.; Kaji, I.; Engevik, M.A.; Meyer, A.R.; Weis, V.G.; Goldstein, A.; Hess, M.W.; Müller, T.; Koepsell, H.; Dudeja, P.K.; et al. Loss of MYO5B Leads to Reductions in Na+ Absorption With Maintenance of CFTR-Dependent Cl- Secretion in Enterocytes. Gastroenterology 2018, 155, 1883–1897.e10. [Google Scholar] [CrossRef] [PubMed]
- Bowman, D.M.; Meenderink, L.M.; Thomas, K.S.; Manning, E.H.; Tyska, M.J.; Goldenring, J.R. Microvillus inclusion disease-causing MYO5B point mutations exert differential effects on motor function. J. Biol. Chem. 2025, 301, 108328. [Google Scholar] [CrossRef] [PubMed]
- Lou, Y.; Lv, Y.; Yu, J.; Gu, W.; Jiang, M.; Chen, J. MYO5B gene mutations may promote the occurrence of very early onset inflammatory bowel disease: A case report. BMC Med. Genom. 2024, 17, 187. [Google Scholar] [CrossRef] [PubMed]
- Wiegerinck, C.L.; Janecke, A.R.; Schneeberger, K.; Vogel, G.F.; van Haaften-Visser, D.Y.; Escher, J.C.; Adam, R.; Thöni, C.E.; Pfaller, K.; Jordan, A.J.; et al. Loss of syntaxin 3 causes variant microvillus inclusion disease. Gastroenterology 2014, 147, 65–68.e10. [Google Scholar] [CrossRef]
- Duclaux-Loras, R.; Lebreton, C.; Berthelet, J.; Charbit-Henrion, F.; Nicolle, O.; Revenu des Courtils, C.; Waich, S.; Valovka, T.; Khiat, A.; Rabant, M.; et al. UNC45A deficiency causes microvillus inclusion disease-like phenotype by impairing myosin VB-dependent apical trafficking. J. Clin. Investig. 2022, 132, e154997. [Google Scholar] [CrossRef]
- Watson, A.; Harris, R.A.; Engevik, A.C.; Oezguen, N.; Nicholson, M.R.; Dooley, S.; Stubler, R.; Satter, L.F.; Karam, L.B.; Kellermayer, R. MYO5B and the Polygenic Landscape of Very Early-Onset Inflammatory Bowel Disease in an Ethnically Diverse Population. Inflamm. Bowel Dis. 2024, 31, 189–199. [Google Scholar] [CrossRef]
- Núñez, C.; Oliver, J.; Mendoza, J.L.; Gómez-García, M.; Piñero, A.; Taxonera, C.; Díaz-Rubio, M.; López-Nevot, M.A.; de la Concha, E.G.; Nieto, A.; et al. MYO9B polymorphisms in patients with inflammatory bowel disease. Gut 2007, 56, 1321–1322. [Google Scholar] [CrossRef]
- Marchiando, A.M.; Shen, L.; Graham, W.V.; Edelblum, K.L.; Duckworth, C.A.; Guan, Y.; Montrose, M.H.; Turner, J.R.; Watson, A.J.M. The epithelial barrier is maintained by in vivo tight junction expansion during pathologic intestinal epithelial shedding. Gastroenterology 2011, 140, 1208–1218.e2. [Google Scholar] [CrossRef]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.R.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Investig. J. Tech. 2006, 86, 191–201. [Google Scholar] [CrossRef]
- López-Posadas, R.; Becker, C.; Günther, C.; Tenzer, S.; Amann, K.; Billmeier, U.; Atreya, R.; Fiorino, G.; Vetrano, S.; Danese, S.; et al. Rho-A prenylation and signaling link epithelial homeostasis to intestinal inflammation. J. Clin. Investig. 2016, 126, 611–626. [Google Scholar] [CrossRef]
- Avitzur, Y.; Guo, C.; Mastropaolo, L.A.; Bahrami, E.; Chen, H.; Zhao, Z.; Elkadri, A.; Dhillon, S.; Murchie, R.; Fattouh, R.; et al. Mutations in tetratricopeptide repeat domain 7A result in a severe form of very early onset inflammatory bowel disease. Gastroenterology 2014, 146, 1028–1039. [Google Scholar] [CrossRef] [PubMed]
- Bigorgne, A.E.; Farin, H.F.; Lemoine, R.; Mahlaoui, N.; Lambert, N.; Gil, M.; Schulz, A.; Philippet, P.; Schlesser, P.; Abrahamsen, T.G.; et al. TTC7A mutations disrupt intestinal epithelial apicobasal polarity. J. Clin. Investig. 2014, 124, 328–337. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Banerjee, S.; Visweswariah, S.S. Mutational landscape of receptor guanylyl cyclase C: Functional analysis and disease-related mutations. IUBMB Life 2020, 72, 1145–1159. [Google Scholar] [CrossRef]
- Zlatkina, A.R.; Belousova, E.A.; Vinitsky, L.I.; Avtandilov, G.G.; Chervonnaya, L.V. Cyclic nucleotide concentrations of rectal mucosa in ulcerative colitis. Scand. J. Gastroenterol. 1990, 25, 341–344. [Google Scholar] [CrossRef]
- Brenna, Ø.; Bruland, T.; Furnes, M.W.; Granlund, A.v.B.; Drozdov, I.; Emgård, J.; Brønstad, G.; Kidd, M.; Sandvik, A.K.; Gustafsson, B.I. The guanylate cyclase-C signaling pathway is down-regulated in inflammatory bowel disease. Scand. J. Gastroenterol. 2015, 50, 1241–1252. [Google Scholar] [CrossRef]
- Prasad, H.; Visweswariah, S.S. Impaired Intestinal Sodium Transport in Inflammatory Bowel Disease: From the Passenger to the Driver’s Seat. Cell. Mol. Gastroenterol. Hepatol. 2021, 12, 277–292. [Google Scholar] [CrossRef]
- Rakoff-Nahoum, S.; Paglino, J.; Eslami-Varzaneh, F.; Edberg, S.; Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 2004, 118, 229–241. [Google Scholar] [CrossRef]
- Lu, Y.-C.; Yeh, W.-C.; Ohashi, P.S. LPS/TLR4 signal transduction pathway. Cytokine 2008, 42, 145–151. [Google Scholar] [CrossRef]
- Xavier, R.J.; Podolsky, D.K. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007, 448, 427–434. [Google Scholar] [CrossRef]
- Molnár, K.; Vannay, A.; Szebeni, B.; Bánki, N.F.; Sziksz, E.; Cseh, A.; Győrffy, H.; Lakatos, P.L.; Papp, M.; Arató, A.; et al. Intestinal alkaline phosphatase in the colonic mucosa of children with inflammatory bowel disease. World J. Gastroenterol. 2012, 18, 3254–3259. [Google Scholar] [CrossRef]
- Grasberger, H.; Magis, A.T.; Sheng, E.; Conomos, M.P.; Zhang, M.; Garzotto, L.S.; Hou, G.; Bishu, S.; Nagao-Kitamoto, H.; El-Zaatari, M.; et al. DUOX2 variants associate with preclinical disturbances in microbiota-immune homeostasis and increased inflammatory bowel disease risk. J. Clin. Investig. 2021, 131, e141676. [Google Scholar] [CrossRef] [PubMed]
- Hazime, H.; Ducasa, G.M.; Santander, A.M.; Brito, N.; González, E.E.; Ban, Y.; Kaunitz, J.; Akiba, Y.; Fernández, I.; Burgueño, J.F.; et al. Intestinal Epithelial Inactivity of Dual Oxidase 2 Results in Microbiome-Mediated Metabolic Syndrome. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.C.; Bikker, H.; Kempers, M.J.E.; van Trotsenburg, A.S.P.; Baas, F.; de Vijlder, J.J.M.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Csillag, C.; Borup, R.; Olsen, J.; Nielsen, F.C.; Nielsen, O.H. Treatment response and colonic gene expression in patients with Crohn’s disease. Scand. J. Gastroenterol. 2007, 42, 834–840. [Google Scholar] [CrossRef]
- Makhezer, N.; Ben Khemis, M.; Liu, D.; Khichane, Y.; Marzaioli, V.; Tlili, A.; Mojallali, M.; Pintard, C.; Letteron, P.; Hurtado-Nedelec, M.; et al. NOX1-derived ROS drive the expression of Lipocalin-2 in colonic epithelial cells in inflammatory conditions. Mucosal Immunol. 2019, 12, 117–131. [Google Scholar] [CrossRef]
- Danne, C.; Skerniskyte, J.; Marteyn, B.; Sokol, H. Neutrophils: From IBD to the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 184–197. [Google Scholar] [CrossRef]
- Buell, M.G.; Berin, M.C. Neutrophil-independence of the initiation of colonic injury. Comparison of results from three models of experimental colitis in the rat. Dig. Dis. Sci. 1994, 39, 2575–2588. [Google Scholar] [CrossRef]
- Natsui, M.; Kawasaki, K.; Takizawa, H.; Hayashi, S.I.; Matsuda, Y.; Sugimura, K.; Seki, K.; Narisawa, R.; Sendo, F.; Asakura, H. Selective depletion of neutrophils by a monoclonal antibody, RP-3, suppresses dextran sulphate sodium-induced colitis in rats. J. Gastroenterol. Hepatol. 1997, 12, 801–808. [Google Scholar] [CrossRef]
- Kühl, A.A.; Kakirman, H.; Janotta, M.; Dreher, S.; Cremer, P.; Pawlowski, N.N.; Loddenkemper, C.; Heimesaat, M.M.; Grollich, K.; Zeitz, M.; et al. Aggravation of different types of experimental colitis by depletion or adhesion blockade of neutrophils. Gastroenterology 2007, 133, 1882–1892. [Google Scholar] [CrossRef]
- Li, T.; Wang, C.; Liu, Y.; Li, B.; Zhang, W.; Wang, L.; Yu, M.; Zhao, X.; Du, J.; Zhang, J.; et al. Neutrophil Extracellular Traps Induce Intestinal Damage and Thrombotic Tendency in Inflammatory Bowel Disease. J. Crohns Colitis. 2019, 14, 240–253. [Google Scholar] [CrossRef]
- Chen, F.; Yang, W.; Huang, X.; Cao, A.T.; Bilotta, A.J.; Xiao, Y.; Sun, M.; Chen, L.; Ma, C.; Liu, X.; et al. Neutrophils Promote Amphiregulin Production in Intestinal Epithelial Cells through TGF-β and Contribute to Intestinal Homeostasis. J. Immunol. 2018, 201, 2492–2501. [Google Scholar] [CrossRef] [PubMed]
- Zindl, C.L.; Lai, J.-F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 12768–12773. [Google Scholar] [CrossRef] [PubMed]
- Mortha, A.; Remark, R.; Del Valle, D.M.; Chuang, L.-S.; Chai, Z.; Alves, I.; Azevedo, C.; Gaifem, J.; Martin, J.; Petralia, F.; et al. Neutralizing Anti-Granulocyte Macrophage-Colony Stimulating Factor Autoantibodies Recognize Post-Translational Glycosylations on Granulocyte Macrophage-Colony Stimulating Factor Years Before Diagnosis and Predict Complicated Crohn’s Disease. Gastroenterology 2022, 163, 659–670. [Google Scholar] [CrossRef] [PubMed]
- Reeves, E.P.; Lu, H.; Jacobs, H.L.; Messina, C.G.M.; Bolsover, S.; Gabella, G.; Potma, E.O.; Warley, A.; Roes, J.; Segal, A.W. Killing activity of neutrophils is mediated through activation of proteases by K+ flux. Nature 2002, 416, 291–297. [Google Scholar] [CrossRef]
- Rieber, N.; Hector, A.; Kuijpers, T.; Roos, D.; Hartl, D. Current concepts of hyperinflammation in chronic granulomatous disease. Clin. Dev. Immunol. 2012, 2012, 252460. [Google Scholar] [CrossRef]
- Chiriaco, M.; Salfa, I.; Di Matteo, G.; Rossi, P.; Finocchi, A. Chronic granulomatous disease: Clinical, molecular, and therapeutic aspects. Pediatr. Allergy Immunol. Off. Publ. Eur. Soc. Pediatr. Allergy Immunol. 2016, 27, 242–253. [Google Scholar] [CrossRef]
- Limbergen, J.V.; Wilson, D.C.; Satsangi, J. The genetics of Crohn’s disease. Annu. Rev Genom. Hum Genet. 2009, 10, 89–116. [Google Scholar] [CrossRef]
- Hoffmann, P.; Lamerz, D.; Hill, P.; Kirchner, M.; Gauss, A. Gene Polymorphisms of NOD2, IL23R, PTPN2 and ATG16L1 in Patients with Crohn’s Disease: On the Way to Personalized Medicine? Genes. 2021, 12, 866. [Google Scholar] [CrossRef]
- Strober, W.; Watanabe, T. NOD2, an intracellular innate immune sensor involved in host defense and Crohn’s disease. Mucosal Immunol. 2011, 4, 484–495. [Google Scholar] [CrossRef]
- Rigaud, S.; Fondanèche, M.-C.; Lambert, N.; Pasquier, B.; Mateo, V.; Soulas, P.; Galicier, L.; Le Deist, F.; Rieux-Laucat, F.; Revy, P.; et al. XIAP deficiency in humans causes an X-linked lymphoproliferative syndrome. Nature 2006, 444, 110–114. [Google Scholar] [CrossRef]
- Mudde, A.C.A.; Booth, C.; Marsh, R.A. Evolution of Our Understanding of XIAP Deficiency. Front. Pediatr. 2021, 9, 660520. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, M.J.M.; Doiron, K.; Labbé, K.; Korneluk, R.G.; Barker, P.A.; Saleh, M. Cellular inhibitors of apoptosis cIAP1 and cIAP2 are required for innate immunity signaling by the pattern recognition receptors NOD1 and NOD2. Immunity 2009, 30, 789–801. [Google Scholar] [CrossRef]
- Brittain, H.K.; Feary, J.; Rosenthal, M.; Spoudeas, H.; Deciphering Developmental Disorders (DDD) Study; Wilson, L.C. Biallelic human ITCH variants causing a multisystem disease with dysmorphic features: A second report. Am. J. Med. Genet. A. 2019, 179, 1346–1350. [Google Scholar] [CrossRef]
- Hasegawa, M.; Fujimoto, Y.; Lucas, P.C.; Nakano, H.; Fukase, K.; Núñez, G.; Inohara, N. A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J. 2008, 27, 373–383. [Google Scholar] [CrossRef]
- Romberg, N.; Al Moussawi, K.; Nelson-Williams, C.; Stiegler, A.L.; Loring, E.; Choi, M.; Overton, J.; Meffre, E.; Khokha, M.K.; Huttner, A.J.; et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat. Genet. 2014, 46, 1135–1139. [Google Scholar] [CrossRef]
- Lapaquette, P.; Glasser, A.-L.; Huett, A.; Xavier, R.J.; Darfeuille-Michaud, A. Crohn’s disease-associated adherent-invasive E. coli are selectively favoured by impaired autophagy to replicate intracellularly. Cell. Microbiol. 2010, 12, 99–113. [Google Scholar] [CrossRef]
- Cadwell, K.; Liu, J.Y.; Brown, S.L.; Miyoshi, H.; Loh, J.; Lennerz, J.K.; Kishi, C.; Kc, W.; Carrero, J.A.; Hunt, S.; et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008, 456, 259–263. [Google Scholar] [CrossRef]
- Cadwell, K.; Patel, K.K.; Maloney, N.S.; Liu, T.-C.; Ng, A.C.Y.; Storer, C.E.; Head, R.D.; Xavier, R.; Stappenbeck, T.S.; Virgin, H.W. Virus-plus-susceptibility gene interaction determines Crohn’s disease gene Atg16L1 phenotypes in intestine. Cell 2010, 141, 1135–1145. [Google Scholar] [CrossRef]
- Quiniou, G.; Andromaque, L.; Duclaux-Loras, R.; Dinet, O.; Cervantes, O.; Verdet, M.; Meunier, C.; Boschetti, G.; Viret, C.; Nancey, S.; et al. Impaired reprogramming of the autophagy flux in maturing dendritic cells from crohn disease patients with core autophagy gene-related polymorphisms. Autophagy 2024, 20, 1837–1853. [Google Scholar] [CrossRef]
- Henckaerts, L.; Cleynen, I.; Brinar, M.; John, J.M.; Van Steen, K.; Rutgeerts, P.; Vermeire, S. Genetic variation in the autophagy gene ULK1 and risk of Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 1392–1397. [Google Scholar] [CrossRef]
- Ellinghaus, D.; Zhang, H.; Zeissig, S.; Lipinski, S.; Till, A.; Jiang, T.; Stade, B.; Bromberg, Y.; Ellinghaus, E.; Keller, A.; et al. Association between variants of PRDM1 and NDP52 and Crohn’s disease, based on exome sequencing and functional studies. Gastroenterology 2013, 145, 339–347. [Google Scholar] [CrossRef] [PubMed]
- Sazonovs, A.; Stevens, C.R.; Venkataraman, G.R.; Yuan, K.; Avila, B.; Abreu, M.T.; Ahmad, T.; Allez, M.; Ananthakrishnan, A.N.; Atzmon, G.; et al. Large-scale sequencing identifies multiple genes and rare variants associated with Crohn’s disease susceptibility. Nat. Genet. 2022, 54, 1275–1283. [Google Scholar] [CrossRef]
- Travassos, L.H.; Carneiro, L.A.M.; Ramjeet, M.; Hussey, S.; Kim, Y.-G.; Magalhães, J.G.; Yuan, L.; Soares, F.; Chea, E.; Le Bourhis, L.; et al. Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat. Immunol. 2010, 11, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Matsuzawa-Ishimoto, Y.; Hwang, S.; Cadwell, K. Autophagy and Inflammation. Annu. Rev. Immunol. 2018, 36, 73–101. [Google Scholar] [CrossRef] [PubMed]
- Parkes, M.; Barrett, J.C.; Prescott, N.J.; Tremelling, M.; Anderson, C.A.; Fisher, S.A.; Roberts, R.G.; Nimmo, E.R.; Cummings, F.R.; Soars, D.; et al. Sequence variants in the autophagy gene IRGM and multiple other replicating loci contribute to Crohn’s disease susceptibility. Nat. Genet. 2007, 39, 830–832. [Google Scholar] [CrossRef]
- Mehto, S.; Jena, K.K.; Nath, P.; Chauhan, S.; Kolapalli, S.P.; Das, S.K.; Sahoo, P.K.; Jain, A.; Taylor, G.A.; Chauhan, S. The Crohn’s Disease Risk Factor IRGM Limits NLRP3 Inflammasome Activation by Impeding Its Assembly and by Mediating Its Selective Autophagy. Mol. Cell 2019, 73, 429–445.e7. [Google Scholar] [CrossRef]
- Sifuentes-Dominguez, L.F.; Li, H.; Llano, E.; Liu, Z.; Singla, A.; Patel, A.S.; Kathania, M.; Khoury, A.; Norris, N.; Rios, J.J.; et al. SCGN deficiency results in colitis susceptibility. eLife 2019, 8, e49910. [Google Scholar] [CrossRef]
- Wagner, L.; Oliyarnyk, O.; Gartner, W.; Nowotny, P.; Groeger, M.; Kaserer, K.; Waldhäusl, W.; Pasternack, M.S. Cloning and expression of secretagogin, a novel neuroendocrine- and pancreatic islet of Langerhans-specific Ca2+-binding protein. J. Biol. Chem. 2000, 275, 24740–24751. [Google Scholar] [CrossRef]
- Rogstam, A.; Linse, S.; Lindqvist, A.; James, P.; Wagner, L.; Berggård, T. Binding of calcium ions and SNAP-25 to the hexa EF-hand protein secretagogin. Biochem. J. 2007, 401, 353–363. [Google Scholar] [CrossRef]
- Iliev, I.D.; Ananthakrishnan, A.N.; Guo, C.-J. Microbiota in inflammatory bowel disease: Mechanisms of disease and therapeutic opportunities. Nat. Rev. Microbiol. 2025, 1–16. [Google Scholar] [CrossRef]
- Sokol, H.; Mahlaoui, N.; Aguilar, C.; Bach, P.; Join-Lambert, O.; Garraffo, A.; Seksik, P.; Danion, F.; Jegou, S.; Straube, M.; et al. Intestinal dysbiosis in inflammatory bowel disease associated with primary immunodeficiency. J. Allergy Clin. Immunol. 2019, 143, 775–778.e6. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Freeborn, J.; Armbrister, S.A.; Tran, D.Q.; Rhoads, J.M. Treg-associated monogenic autoimmune disorders and gut microbial dysbiosis. Pediatr. Res. 2022, 91, 35–43. [Google Scholar] [CrossRef] [PubMed]
- Basso, L.; Boué, J.; Augé, C.; Deraison, C.; Blanpied, C.; Cenac, N.; Lluel, P.; Vergnolle, N.; Dietrich, G. Mobilization of CD4+ T lymphocytes in inflamed mucosa reduces pain in colitis mice: Toward a vaccinal strategy to alleviate inflammatory visceral pain. Pain. 2018, 159, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Saez, A.; Herrero-Fernandez, B.; Gomez-Bris, R.; Sánchez-Martinez, H.; Gonzalez-Granado, J.M. Pathophysiology of Inflammatory Bowel Disease: Innate Immune System. Int. J. Mol. Sci. 2023, 24, 1526. [Google Scholar] [CrossRef]
- Fujino, S.; Andoh, A.; Bamba, S.; Ogawa, A.; Hata, K.; Araki, Y.; Bamba, T.; Fujiyama, Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut 2003, 52, 65–70. [Google Scholar] [CrossRef]
- Fuss, I.J.; Heller, F.; Boirivant, M.; Leon, F.; Yoshida, M.; Fichtner-Feigl, S.; Yang, Z.; Exley, M.; Kitani, A.; Blumberg, R.S.; et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J. Clin. Investig. 2004, 113, 1490–1497. [Google Scholar] [CrossRef]
- Heller, F.; Florian, P.; Bojarski, C.; Richter, J.; Christ, M.; Hillenbrand, B.; Mankertz, J.; Gitter, A.H.; Bürgel, N.; Fromm, M.; et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005, 129, 550–564. [Google Scholar] [CrossRef]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar] [CrossRef]
- Maloy, K.J.; Kullberg, M.C. IL-23 and Th17 cytokines in intestinal homeostasis. Mucosal Immunol. 2008, 1, 339–349. [Google Scholar] [CrossRef]
- Feagan, B.G.; Sandborn, W.J.; Gasink, C.; Jacobstein, D.; Lang, Y.; Friedman, J.R.; Blank, M.A.; Johanns, J.; Gao, L.-L.; Miao, Y.; et al. Ustekinumab as Induction and Maintenance Therapy for Crohn’s Disease. N. Engl. J. Med. 2016, 375, 1946–1960. [Google Scholar] [CrossRef]
- Sands, B.E.; Sandborn, W.J.; Panaccione, R.; O’Brien, C.D.; Zhang, H.; Johanns, J.; Adedokun, O.J.; Li, K.; Peyrin-Biroulet, L.; Van Assche, G.; et al. Ustekinumab as Induction and Maintenance Therapy for Ulcerative Colitis. N. Engl. J. Med. 2019, 381, 1201–1214. [Google Scholar] [CrossRef] [PubMed]
- Peyrin-Biroulet, L.; Chapman, J.C.; Colombel, J.-F.; Caprioli, F.; D’Haens, G.; Ferrante, M.; Schreiber, S.; Atreya, R.; Danese, S.; Lindsay, J.O.; et al. Risankizumab versus Ustekinumab for Moderate-to-Severe Crohn’s Disease. N. Engl. J. Med. 2024, 391, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Fabre, A.; Marchal, S.; Barlogis, V.; Mari, B.; Barbry, P.; Rohrlich, P.-S.; Forbes, L.R.; Vogel, T.P.; Giovannini-Chami, L. Clinical Aspects of STAT3 Gain-of-Function Germline Mutations: A Systematic Review. J. Allergy Clin. Immunol. Pract. 2019, 7, 1958–1969.e9. [Google Scholar] [CrossRef]
- Rodari, M.M.; Cerf-Bensussan, N.; Parlato, M. Dysregulation of the immune response in TGF-β signalopathies. Front. Immunol. 2022, 13, 1066375. [Google Scholar] [CrossRef]
- Brandtzaeg, P.; Baklien, K.; Fausa, O.; Hoel, P.S. Immunohistochemical characterization of local immunoglobulin formation in ulcerative colitis. Gastroenterology 1974, 66, 1123–1136. [Google Scholar] [CrossRef]
- Castro-Dopico, T.; Dennison, T.W.; Ferdinand, J.R.; Mathews, R.J.; Fleming, A.; Clift, D.; Stewart, B.J.; Jing, C.; Strongili, K.; Labzin, L.I.; et al. Anti-commensal IgG Drives Intestinal Inflammation and Type 17 Immunity in Ulcerative Colitis. Immunity 2019, 50, 1099–1114.e10. [Google Scholar] [CrossRef]
- Lin, Z.; Hegarty, J.P.; Yu, W.; Cappel, J.A.; Chen, X.; Faber, P.W.; Wang, Y.; Poritz, L.S.; Fan, J.-B.; Koltun, W.A. Identification of disease-associated DNA methylation in B cells from Crohn’s disease and ulcerative colitis patients. Dig. Dis. Sci. 2012, 57, 3145–3153. [Google Scholar] [CrossRef]
- Lopez-Herrera, G.; Tampella, G.; Pan-Hammarström, Q.; Herholz, P.; Trujillo-Vargas, C.M.; Phadwal, K.; Simon, A.K.; Moutschen, M.; Etzioni, A.; Mory, A.; et al. Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. Am. J. Hum. Genet. 2012, 90, 986–1001. [Google Scholar] [CrossRef]
- Ohnmacht, C.; Park, J.-H.; Cording, S.; Wing, J.B.; Atarashi, K.; Obata, Y.; Gaboriau-Routhiau, V.; Marques, R.; Dulauroy, S.; Fedoseeva, M.; et al. The microbiota regulates type 2 immunity through RORγt+ T cells. Science 2015, 349, 989–993. [Google Scholar] [CrossRef]
- Mottet, C.; Uhlig, H.H.; Powrie, F. Cutting edge: Cure of colitis by CD4+CD25+ regulatory T cells. J. Immunol. 2003, 170, 3939–3943. [Google Scholar] [CrossRef]
- Saruta, M.; Yu, Q.T.; Fleshner, P.R.; Mantel, P.-Y.; Schmidt-Weber, C.B.; Banham, A.H.; Papadakis, K.A. Characterization of FOXP3+CD4+ regulatory T cells in Crohn’s disease. Clin. Immunol. 2007, 125, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Maul, J.; Loddenkemper, C.; Mundt, P.; Berg, E.; Giese, T.; Stallmach, A.; Zeitz, M.; Duchmann, R. Peripheral and intestinal regulatory CD4+ CD25(high) T cells in inflammatory bowel disease. Gastroenterology 2005, 128, 1868–1878. [Google Scholar] [CrossRef] [PubMed]
- Uhlig, H.H.; Coombes, J.; Mottet, C.; Izcue, A.; Thompson, C.; Fanger, A.; Tannapfel, A.; Fontenot, J.D.; Ramsdell, F.; Powrie, F. Characterization of Foxp3+CD4+CD25+ and IL-10-secreting CD4+CD25+ T cells during cure of colitis. J. Immunol. 2006, 177, 5852–5860. [Google Scholar] [CrossRef]
- Kotlarz, D.; Beier, R.; Murugan, D.; Diestelhorst, J.; Jensen, O.; Boztug, K.; Pfeifer, D.; Kreipe, H.; Pfister, E.-D.; Baumann, U.; et al. Loss of interleukin-10 signaling and infantile inflammatory bowel disease: Implications for diagnosis and therapy. Gastroenterology 2012, 143, 347–355. [Google Scholar] [CrossRef]
- Kotlarz, D.; Marquardt, B.; Barøy, T.; Lee, W.S.; Konnikova, L.; Hollizeck, S.; Magg, T.; Lehle, A.S.; Walz, C.; Borggraefe, I.; et al. Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat. Genet. 2018, 50, 344–348. [Google Scholar] [CrossRef]
- Grotra, R.; Karri, P.S.; Gupta, A.; Malik, R.; Gupta, A.K.; Meena, J.P.; Seth, R. Matched Unrelated Donor Hematopoietic Stem Cell Transplant as Successful Curative Therapy for IL10RB Mutation-Associated Very Early Onset IBD. Pediatr. Transplant. 2024, 28, e14891. [Google Scholar] [CrossRef]
- Parlato, M.; Charbit-Henrion, F.; Abi Nader, E.; Begue, B.; Guegan, N.; Bruneau, J.; Khater, S.; Macintyre, E.; Picard, C.; Frédéric, R.-L.; et al. Efficacy of Ruxolitinib Therapy in a Patient With Severe Enterocolitis Associated With a STAT3 Gain-of-Function Mutation. Gastroenterology 2019, 156, 1206–1210.e1. [Google Scholar] [CrossRef]
- Leiding, J.W.; Vogel, T.P.; Santarlas, V.G.J.; Mhaskar, R.; Smith, M.R.; Carisey, A.; Vargas-Hernández, A.; Silva-Carmona, M.; Heeg, M.; Rensing-Ehl, A.; et al. Monogenic early-onset lymphoproliferation and autoimmunity: Natural history of STAT3 gain-of-function syndrome. J. Allergy Clin. Immunol. 2023, 151, 1081–1095. [Google Scholar] [CrossRef]
- Bolton, C.; Smillie, C.S.; Pandey, S.; Elmentaite, R.; Wei, G.; Argmann, C.; Aschenbrenner, D.; James, K.R.; McGovern, D.P.B.; Macchi, M.; et al. An Integrated Taxonomy for Monogenic Inflammatory Bowel Disease. Gastroenterology 2022, 162, 859–876. [Google Scholar] [CrossRef]
- Kaji, I.; Roland, J.T.; Watanabe, M.; Engevik, A.C.; Goldstein, A.E.; Hodges, C.A.; Goldenring, J.R. Lysophosphatidic Acid Increases Maturation of Brush Borders and SGLT1 Activity in MYO5B-deficient Mice, a Model of Microvillus Inclusion Disease. Gastroenterology 2020, 159, 1390–1405.e20. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IBD Pathophysiology | Genes Related to Monogenic Disease |
|---|---|
| Gastrointestinal barrier | |
| |
| Tight junctions Transport defect Enterocyte architecture | EpCAM [5] SLC26A3 [6], GUCY2C [7,8,9] MYO5B [10], TTC7A [11] |
| ST6 [12], AGR2 [13] |
| Innate Immunity | |
| ALPI [14], DUOX2 [15] |
| CYBB, CYBA, NCF1, NCF2, NCF4 [16] |
| ITCH [17], NEMO [18], A20 [19] |
| Adaptive immunity | |
| ICOS [20], DOCK8 [21], TGFbR1/2/3, SMAD2/3, IPO8 [22], STAT3 [23,24] |
| BTK, CD40L, AID [25] |
| Immune regulation | |
| FOXP3 [26], IL10, IL10RA/B [27], CTLA4 [28] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petit, C.; Rozières, A.; Boschetti, G.; Viret, C.; Faure, M.; Nancey, S.; Duclaux-Loras, R. Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis. Int. J. Mol. Sci. 2025, 26, 6133. https://doi.org/10.3390/ijms26136133
Petit C, Rozières A, Boschetti G, Viret C, Faure M, Nancey S, Duclaux-Loras R. Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis. International Journal of Molecular Sciences. 2025; 26(13):6133. https://doi.org/10.3390/ijms26136133
Chicago/Turabian StylePetit, Céline, Aurore Rozières, Gilles Boschetti, Christophe Viret, Mathias Faure, Stéphane Nancey, and Rémi Duclaux-Loras. 2025. "Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis" International Journal of Molecular Sciences 26, no. 13: 6133. https://doi.org/10.3390/ijms26136133
APA StylePetit, C., Rozières, A., Boschetti, G., Viret, C., Faure, M., Nancey, S., & Duclaux-Loras, R. (2025). Advances in Understanding Intestinal Homeostasis: Lessons from Inflammatory Bowel Disease and Monogenic Intestinal Disorder Pathogenesis. International Journal of Molecular Sciences, 26(13), 6133. https://doi.org/10.3390/ijms26136133