
On-Line Preconcentration of Selected Kynurenine Pathway Metabolites and Amino Acids in Urine via Pressure-Assisted Electrokinetic Injection in a Mixed Micelle System


Abstract
1. Introduction
2. Results and Discussion
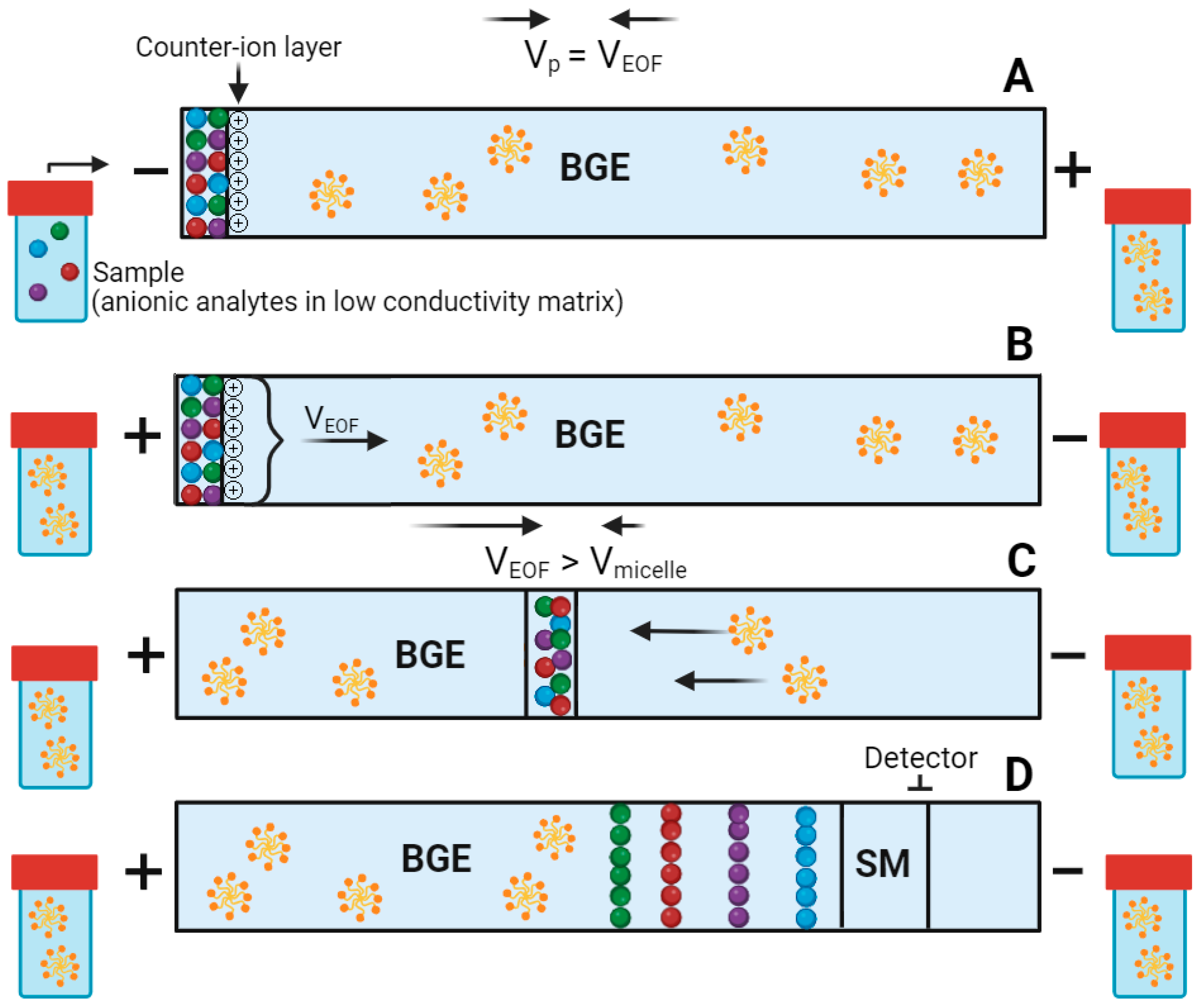
2.1. Mixed Micelle System
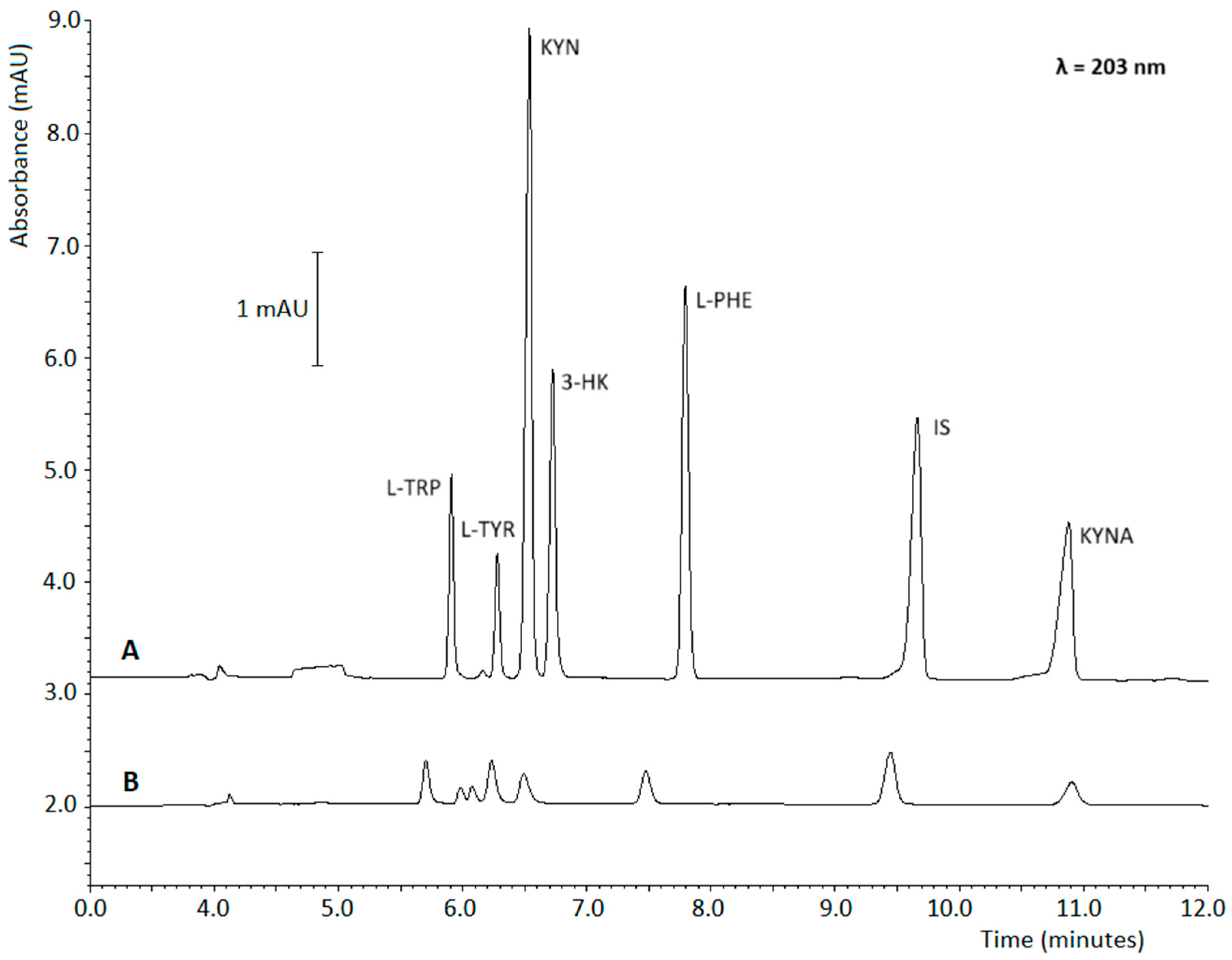
2.2. PAEKI Parameters Optimization
2.3. SPE Parameters Optimization
2.4. Validation Study
2.4.1. Linearity Range, LOD, and LOQ
2.4.2. Accuracy and Precision
2.4.3. Dilution Integrity and Carry-Over
2.4.4. Stability
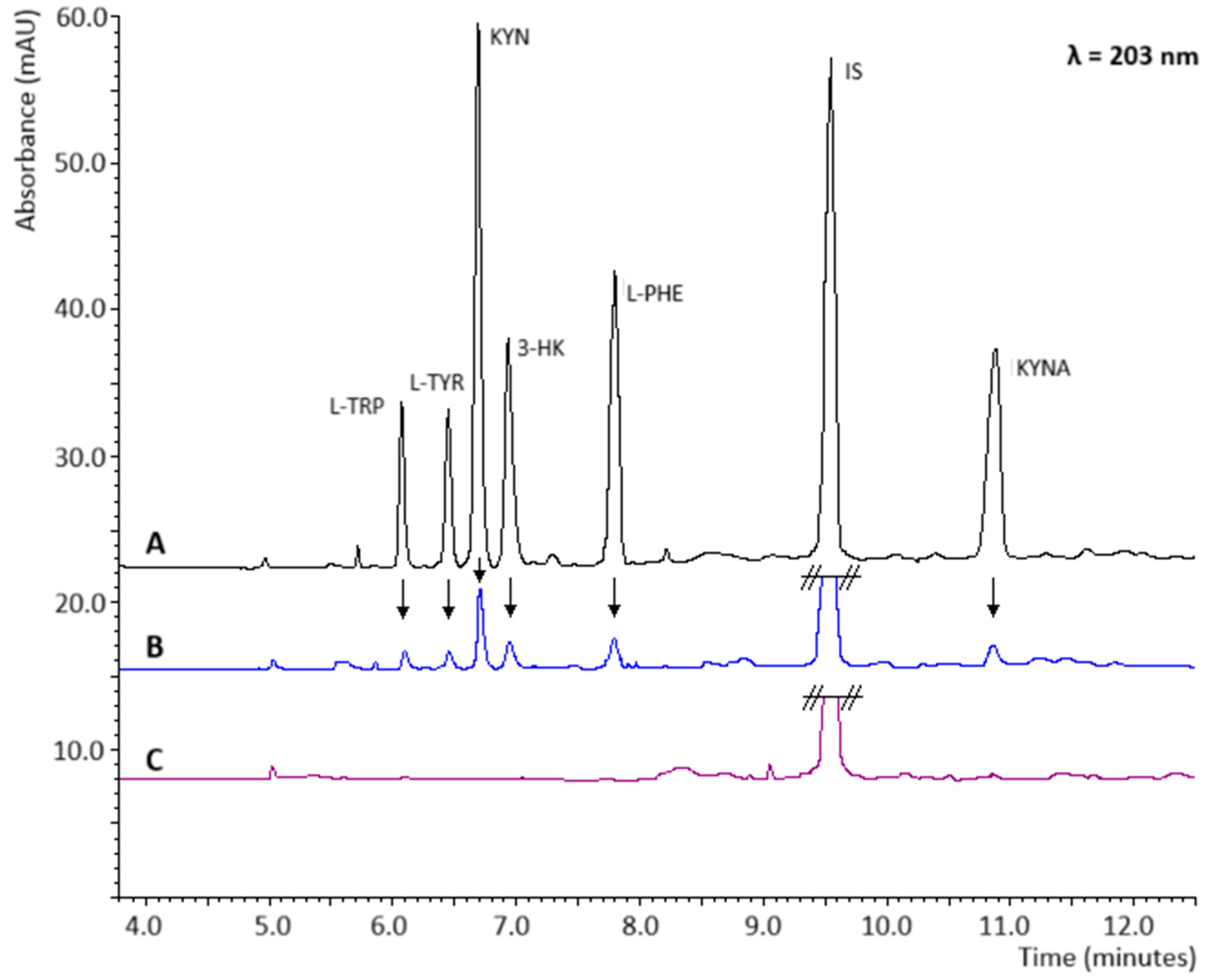
2.5. Signal Amplification
2.6. Recovery
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Urine Samples
3.3. Analyte Standard Solutions
3.4. Electrolyte and Surfactant Solutions
3.5. CE Instrumentation
3.6. Extraction Procedure
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Feng, Y.L.; Zhu, J. On-line enhancement technique for the analysis of nucleotides using capillary zone electrophoresis/mass spectrometry. Anal. Chem. 2006, 78, 6608–6613. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Gavina, J.; Feng, Y.L. Understanding mechanisms of pressure-assisted electrokinetic injection: Application to analysis of bromate, arsenic and selenium species in drinking water by capillary electrophoresis-mass spectrometry. J. Chromatogr. A 2011, 1218, 3095–3104. [Google Scholar] [CrossRef]
- Feng, Y.L.; Lian, H.; Zhu, J. Application of pressure assisted electrokinetic injection technique in the measurements of DNA oligonucleotides and their adducts using capillary electrophoresis-mass spectrometry. J. Chromatogr. A 2007, 1148, 244–249. [Google Scholar] [CrossRef]
- Feng, Y.L.; Zhu, J. Constant pressure-assisted electrokinetic injection for on-line enhanced detection of monophthalates in capillary electrophoresis-mass spectrometry with application to human urine. Electrophoresis 2008, 29, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhu, J.; Feng, Y.L. On-line enrichment and measurement of four halogenated phenols in water samples using pressure-assisted electrokinetic injection-tandem mass spectrometry. Anal. Sci. 2010, 26, 1157–1162. [Google Scholar] [CrossRef]
- Yang, S.; Ma, S.; Zhu, K.; Wang, M.; Li, J.; Arabi, M.; Liu, H.; Li, Y.; Chen, L. Simultaneous enrichment/determination of six sulfonamides in animal husbandry products and environmental waters by pressure-assisted electrokinetic injection coupled with capillary zone electrophoresis. J. Food Compos. Anal. 2020, 88, 103462. [Google Scholar] [CrossRef]
- Yang, X.; He, L.; Xu, Z. Pressure-assisted electrokinetic injection for the stacking of biogenic amines gives enhancement factor up to 1000 in CE with UV detection. Anal. Methods 2022, 14, 1782–1787. [Google Scholar] [CrossRef]
- Ma, S.; Gao, F.; Lu, W.; Zhou, N.; You, H.; Li, J.; Chen, L. Dispersive liquid-liquid microextraction coupled with pressure-assisted electrokinetic injection for simultaneous enrichment of seven phenolic compounds in water samples followed by determination using capillary electrophoresis. J. Sep. Sci. 2019, 42, 2263–2271. [Google Scholar] [CrossRef]
- Zhang, H.; Zhu, J.; Aranda-Rodriguez, R.; Feng, Y.L. Pressure-assisted electrokinetic injection for on-line enrichment in capillary electrophoresis-mass spectrometry: A sensitive method for measurement of ten haloacetic acids in drinking water. Anal. Chim. Acta 2011, 706, 176–183. [Google Scholar] [CrossRef]
- He, T.; Xu, Z.; Ren, J. Pressure-assisted electrokinetic injection stacking for seven typical antibiotics in waters to achieve μg/L level analysis by capillary electrophoresis with UV detection. Microchem. J. 2019, 146, 1295–1300. [Google Scholar] [CrossRef]
- Xu, Z.; Li, A.; Wang, Y.; Chen, Z.; Hirokawa, T. Pressure-assisted electrokinetic injection stacking for verteporfin drug to achieve highly sensitive enantioseparation and detection in artificial urine by capillary electrophoresis. J. Chromatogr. A 2014, 1355, 284–290. [Google Scholar] [CrossRef]
- Pieckowski, M.; Kowalski, P.; Olędzka, I.; Roszkowska, A.; Plenis, A.; Bączek, T. Nanoemulsion supported microemulsion electrokinetic chromatography coupled with selected preconcentration techniques as an approach for analysis of highly hydrophobic compounds. J. Chromatogr. A 2022, 1677, 463339. [Google Scholar] [CrossRef]
- Pieckowski, M.; Olędzka, I.; Bączek, T.; Kowalski, P. Strategies for Signal Amplification of Thyroid Hormones via Electromigration Techniques Coupled with UV Detection and Laser-Induced Fluorescence. Int. J. Mol. Sci. 2025, 26, 3708. [Google Scholar] [CrossRef] [PubMed]
- Badawy, A.A. The tryptophan utilization concept in pregnancy. Obstet. Gynecol. Sci. 2014, 57, 249–259. [Google Scholar] [CrossRef] [PubMed]
- Hényková, E.; Vránová, H.P.; Amakorová, P.; Pospíšil, T.; Žukauskaitė, A.; Vlčková, M.; Urbánek, L.; Novák, O.; Mareš, J.; Kaňovský, P.; et al. Stable isotope dilution ultra-high performance liquid chromatography-tandem mass spectrometry quantitative profiling of tryptophan-related neuroactive substances in human serum and cerebrospinal fluid. J. Chromatogr. A 2016, 1437, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gao, P.; Zhu, D. Optimization of Zn2+-containing mobile phase for simultaneous determination of kynurenine, kynurenic acid and tryptophan in human plasma by high performance liquid chromatography. J. Chromatogr. B Analyt Technol. Biomed. Life Sci. 2010, 878, 603–608. [Google Scholar] [CrossRef]
- Alves, L.F.; Moore, J.B.; Kell, D.B. The Biology and Biochemistry of Kynurenic Acid, a Potential Nutraceutical with Multiple Biological Effects. Int. J. Mol. Sci. 2024, 25, 9082. [Google Scholar] [CrossRef]
- Guidetti, P.; Hoffman, G.E.; Melendez-Ferro, M.; Albuquerque, E.X.; Schwarcz, R. Astrocytic localization of kynurenine aminotransferase II in the rat brain visualized by immunocytochemistry. Glia 2007, 55, 78–92. [Google Scholar] [CrossRef]
- Baran, H.; Jellinger, K.; Deecke, L. Kynurenine metabolism in Alzheimer’s disease. J. Neural Transm. 1999, 106, 165–181. [Google Scholar] [CrossRef]
- Parrott, J.M.; O’Connor, J.C. Kynurenine 3-Monooxygenase: An Influential Mediator of Neuropathology. Front. Psychiatry 2015, 6, 116. [Google Scholar] [CrossRef]
- Ligam, P.; Manuelpillai, U.; Wallace, E.M.; Walker, D. Localisation of indoleamine 2,3-dioxygenase and kynurenine hydroxylase in the human placenta and decidua: Implications for role of the kynurenine pathway in pregnancy. Placenta 2005, 26, 498–504. [Google Scholar] [CrossRef]
- Butler, M.I.; Long-Smith, C.; Moloney, G.M.; Morkl, S.; O'MAhony, S.M.; Cryan, J.F.; Clarke, G.; Dinan, T.G. The immune-kynurenine pathway in social anxiety disorder. Brain Behav. Immun. 2022, 99, 317–326. [Google Scholar] [CrossRef]
- Zinellu, A.; Sotgia, S.; Deiana, L.; Talanas, G.; Terrosu, P.; Carru, C. Simultaneous analysis of kynurenine and tryptophan in human plasma by capillary electrophoresis with UV detection. J. Sep. Sci. 2012, 35, 1146–1151. [Google Scholar] [CrossRef] [PubMed]
- Chawdhury, A.S.M.M.R.; Shamsi, S.A.; Miller, A.; Liu, A. Capillary electrochromatography-mass spectrometry of kynurenine pathway metabolites. J. Chromatogr. A 2021, 1651, 462294. [Google Scholar] [CrossRef] [PubMed]
- Sidorova, A.A.; Kartsova, L.A. Study of the kynurenine pathway of tryptophan metabolism by capillary electrophoresis and mass spectrometry. J. Anal. Chem. 2011, 66, 322–326. [Google Scholar] [CrossRef]
- Hansen, D.K.; Lunte, S.M. Determination of kynurenic acid by capillary electrophoresis with laser-induced fluorescence detection. J. Chromatogr. A 1997, 781, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Arvidsson, B.; Johannesson, N.; Citterio, A.; Righetti, P.G.; Bergquist, J. High throughput analysis of tryptophan metabolites in a complex matrix using capillary electrophoresis coupled to time-of-flight mass spectrometry. J. Chromatogr. A 2007, 1159, 154–158. [Google Scholar] [CrossRef]
- Rizvi, A.S.; Murtaza, G.; Irfan, M.; Xiao, Y.; Qu, F. Determination of Kynurenine Enantiomers by Alpha-Cyclodextrin, Cationic-β-Cyclodextrin and Their Synergy Complemented with Stacking Enrichment in Capillary Electrophoresis. J. Chromatogr. A 2020, 1622, 461128. [Google Scholar] [CrossRef]
- Rang, Y.; Zhang, W.; Chen, Z. Determination of kynurenine and tryptophan in human plasma by Stacking-Micellar electrokinetic chromatography. Anal. Lett. 2013, 46, 2503–2513. [Google Scholar] [CrossRef]
- Dehouck, P.; Van Schepdael, A.; Roets, E.; Hoogmartens, J. Analysis of clindamycin by micellar electrokinetic chromatography with a mixed micellar system. J. Chromatogr. A 2001, 932, 145–152. [Google Scholar] [CrossRef]
- Elarbi, F.M.; Ettarhouni, Z.O.; Abdussalam-Mohammed, W.; Mezoughi, A.B. Study on the Effects of Biologically Active Amino Acids on the Micellization of Anionic Surfactant Sodium Dodecyl Sulfate (SDS) at Different Temperatures. Chemistry 2022, 4, 146–155. [Google Scholar] [CrossRef]
- Palmer, J.; Munro, N.J.; Landers, J.P. A universal concept for stacking neutral analytes in micellar capillary electrophoresis. Anal. Chem. 1999, 71, 1679–1687. [Google Scholar] [CrossRef] [PubMed]
- EMA/CHMP/ICH/172948/2019; ICH Guideline M10 on Bioanalytical Method Validation and Study Sample Analysis. European Medicines Agency: Amsterdam, The Netherlands, 2022.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analytes | Electromigration Method | Detection | LOD | Separation Time [min] | Sample Matrix | Reference |
|---|---|---|---|---|---|---|
| L-TRP, KYN, 3-HK, KYNA, L-TYR, L-PHE | PAEKI-MEKC | UV | 1.2–3.0 ng/mL (5.7–15.8 pM) * | 13 | artificial urine, human urine | this study |
| L-TRP, KYN | CZE | UV | 0.4–0.15 µM | 9 | human plasma | [23] |
| L-TRP, KYN, 3-HK, KYNA, AA, XA | CZE | MS | 31 to 1000 nM | 26 | human plasma | [24] |
| L-TRP, KYN, 3-HK, KYNA | CD-CZE | UV | 0.3 µg/mL (1.3 nM) * | 8 | homogenized tissues of Drosophila melanogaster | [25] |
| KYNA | CZE | LIF | 1 nM | 1 | rat brain | [26] |
| L-TRP, KYN, KYNA | CZE | MS | 20–67 nM | 5 | human CSF | [27] |
| D, L-KYN | Acid-mediated stacking-CD-CZE | UV | 15 nM | 10 | human plasma and urine | [28] |
| L-TRP, KYN | LVSS-MEKC | UV | 0.15–0.30 μM | 17 | human plasma | [29] |
| Analyte | PAEKI-MEKC (Mixed Micelle System) | HDI-MEKC (5 s, 0.5 psi) | EF Enhancement Factor | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Slope | Intercept | R2 | LOD [ng/mL] | LOQ [ng/mL] | Precision (%RSD) | LOD [ng/mL] | |||
| Migration Time | Peak Hight | ||||||||
| L-TRP | 0.0023 | −0.0231 | 0.9995 | 2.6 | 8.6 | 2.3 | 5.2 | 140 | 53.8 |
| L-TYR | 0.0016 | −0.0039 | 0.9995 | 3 | 9.9 | 2.2 | 6.1 | 138 | 46.0 |
| KYN | 0.0042 | 0.0094 | 0.9996 | 1.2 | 4.0 | 1.9 | 7.9 | 172 | 143.3 |
| 3-HK | 0.0027 | −0.0398 | 0.9987 | 2.2 | 7.3 | 2.6 | 7.2 | 150 | 68.2 |
| L-PHE | 0.0030 | −0.0139 | 0.9993 | 1.5 | 5.0 | 2.8 | 6.7 | 153 | 102.0 |
| KYNA | 0.0014 | 0.0006 | 0.9998 | 2.9 | 9.6 | 2.9 | 6.4 | 180 | 62.1 |
| Analyte | Nominal Concentration [ng/mL] | Within-Run (n = 5) | Between-Run (n = 6) | ||||
|---|---|---|---|---|---|---|---|
| Measured Concentration [ng/mL] (Mean ± SD) | Accuracy [%] | Precision [CV%] | Measured Concentration [ng/mL] (mean ± SD) | Accuracy [%] | Precision [CV%] | ||
| L-TRP | LLOQ 10 | 10.8 ± 1.3 | 108.1 | 11.4 | 10.9 ± 1.6 | 109.2 | 12.4 |
| LQC 25 | 23.0 ± 2.4 | 92.0 | 9.8 | 22.9 ± 2.7 | 91.6 | 11.5 | |
| MQC 500 | 492.6 ± 23.5 | 98.4 | 7.0 | 485.6 ± 24.9 | 97.1 | 7.9 | |
| HQC 1000 | 994.3 ± 32.6 | 99.4 | 4.1 | 992.7 ± 34.7 | 99.3 | 5.3 | |
| L-TYR | LLOQ 10 | 11.0 ± 1.4 | 109.7 | 11.0 | 11.1 ± 1.7 | 110.6 | 12.1 |
| LQC 25 | 23.4 ± 2.4 | 93.6 | 8.0 | 22.9 ± 2.8 | 91.6 | 8.3 | |
| MQC 500 | 486.2 ± 24.2 | 97.2 | 3.9 | 484.8 ± 25.4 | 96.9 | 4.5 | |
| HQC 1000 | 1012.4 ± 30.9 | 101.2 | 3.0 | 1017.4 ± 32.7 | 101.7 | 3.7 | |
| KYN | LLOQ 4 | 4.30 ± 0.4 | 107.5 | 11.9 | 4.5 ± 0.7 | 112.5 | 12.2 |
| LQC 25 | 25.8 ± 2.1 | 103.2 | 9.1 | 25.9 ± 2.4 | 103.6 | 9.5 | |
| MQC 500 | 480.4 ± 17.6 | 96.1 | 5.9 | 477.4 ± 19.8 | 95.5 | 6.2 | |
| HQC 1000 | 993.7 ± 30.1 | 99.4 | 5.0 | 989.6 ± 33.8 | 99.0 | 5.7 | |
| 3-HK | LLOQ 10 | 11.0 ± 1.9 | 110.0 | 12.0 | 11.0 ± 2.1 | 110 | 12.2 |
| LQC 25 | 26.1 ± 2.4 | 104.4 | 9.9 | 26.2 ± 2.9 | 104.8 | 10.4 | |
| MQC 500 | 476.0 ± 24.8 | 95.2 | 6.8 | 476.0 ± 26.4 | 95.2 | 7.4 | |
| HQC 1000 | 1000.9 ± 34.9 | 100.1 | 3.0 | 1006.0 ± 36.9 | 100.6 | 3.3 | |
| L-PHE | LLOQ 10 | 10.8 ± 1.6 | 107.7 | 11.5 | 11.3 ± 1.9 | 112.7 | 12.8 |
| LQC 25 | 24.2 ± 2.5 | 96.8 | 10.0 | 24.0 ± 2.8 | 96.0 | 10.4 | |
| MQC 500 | 484.5 ± 22.7 | 96.2 | 5.7 | 481.7 ± 24.4 | 96.4 | 6.1 | |
| HQC 1000 | 1010.4 ± 35.7 | 101.0 | 4.8 | 1013.3 ± 37.7 | 101.3 | 5.1 | |
| KYNA | LLOQ 10 | 11.0 ± 1.7 | 109.9 | 10.1 | 11.2 ± 1.9 | 112.2 | 10.3 |
| LQC 25 | 26.4 ± 3.1 | 105.6 | 9.1 | 26.3 ± 3.3 | 105.2 | 9.7 | |
| MQC 500 | 495.1 ± 22.8 | 99.0 | 6.0 | 490.3 ± 24.2 | 98.1 | 6.8 | |
| HQC 1000 | 1011.1 ± 35.4 | 101.1 | 5.1 | 1023.0 ± 37.4 | 102.3 | 5.4 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pieckowski, M.; Olędzka, I.; Bączek, T.; Kowalski, P. On-Line Preconcentration of Selected Kynurenine Pathway Metabolites and Amino Acids in Urine via Pressure-Assisted Electrokinetic Injection in a Mixed Micelle System. Int. J. Mol. Sci. 2025, 26, 6125. https://doi.org/10.3390/ijms26136125
Pieckowski M, Olędzka I, Bączek T, Kowalski P. On-Line Preconcentration of Selected Kynurenine Pathway Metabolites and Amino Acids in Urine via Pressure-Assisted Electrokinetic Injection in a Mixed Micelle System. International Journal of Molecular Sciences. 2025; 26(13):6125. https://doi.org/10.3390/ijms26136125
Chicago/Turabian StylePieckowski, Michał, Ilona Olędzka, Tomasz Bączek, and Piotr Kowalski. 2025. "On-Line Preconcentration of Selected Kynurenine Pathway Metabolites and Amino Acids in Urine via Pressure-Assisted Electrokinetic Injection in a Mixed Micelle System" International Journal of Molecular Sciences 26, no. 13: 6125. https://doi.org/10.3390/ijms26136125
APA StylePieckowski, M., Olędzka, I., Bączek, T., & Kowalski, P. (2025). On-Line Preconcentration of Selected Kynurenine Pathway Metabolites and Amino Acids in Urine via Pressure-Assisted Electrokinetic Injection in a Mixed Micelle System. International Journal of Molecular Sciences, 26(13), 6125. https://doi.org/10.3390/ijms26136125