

Rare Movement Disorders—An Approach for Clinicians


Abstract
1. Introduction
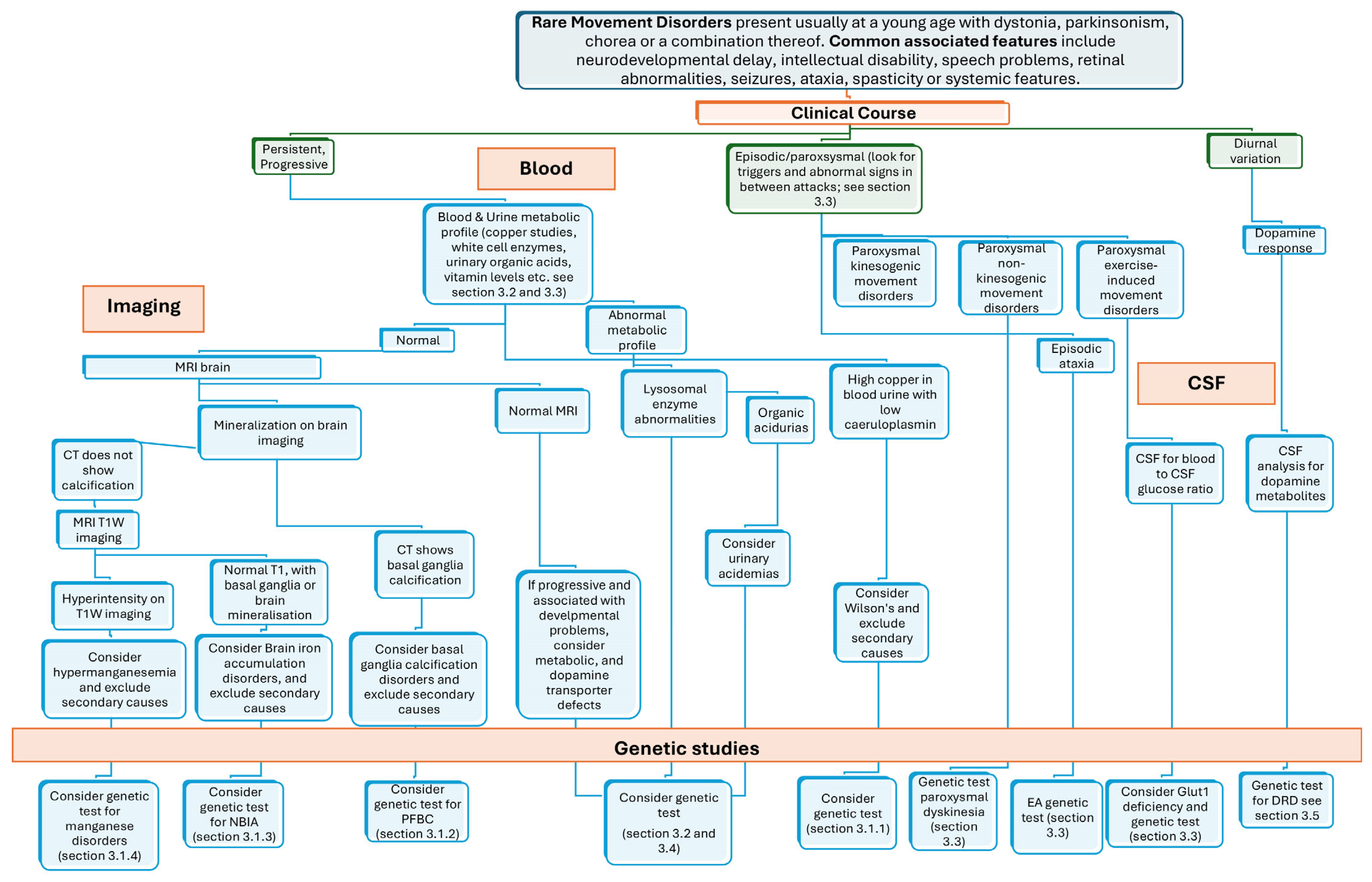
2. General Clinical Approach to RMDs
3. Overview and Phenotypic Clues of RMD
3.1. Brain Mineralopathies
3.1.1. Copper
3.1.2. Calcium
3.1.3. Iron
3.1.4. Manganese
3.2. Lysosomal Storage Diseases
3.3. Episodic Movement Disorders
3.4. Vitamin Deficiency-Related Disorders
3.5. Dopamine Responsive Disorders
4. Further Treatment Considerations
4.1. Conservative Treatments
4.2. Medical Treatments
4.3. Genetic Testing and Counselling
5. Conclusions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orphanet: Diseases. Available online: https://www.orpha.net/en/disease (accessed on 27 November 2024).
- Magrinelli, F.; Balint, B.; Bhatia, K.P. Challenges in Clinicogenetic Correlations: One Gene—Many Phenotypes. Mov. Disord. Clin. Pract. 2021, 8, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Gannamani, R.; van der Veen, S.; van Egmond, M.; de Koning, T.J.; Tijssen, M.A.J. Challenges in Clinicogenetic Correlations: One Phenotype—Many Genes. Mov. Disord. Clin. Pract. 2021, 8, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Farbman, E.S. The Case for Subspecialization in Neurology: Movement Disorders. Front. Neurol. 2011, 2, 22. [Google Scholar] [CrossRef]
- Mayor, S. Neurological patients experience long delays to see specialists, finds survey. BMJ 2019, 366, 14605. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Albanese, A.; Bhatia, K.P.; Cardoso, F.; Da Prat, G.; de Koning, T.J.; Espay, A.J.; Fung, V.; Garcia-Ruiz, P.J.; Gershanik, O.; et al. Treatable inherited rare movement disorders. Mov. Disord. 2018, 33, 21–35. [Google Scholar] [CrossRef]
- Khateeb, S.; Flusser, H.; Ofir, R.; Shelef, I.; Narkis, G.; Vardi, G.; Shorer, Z.; Levy, R.; Galil, A.; Elbedour, K.; et al. PLA2G6 mutation underlies infantile neuroaxonal dystrophy. Am. J. Hum. Genet. 2006, 79, 942–948. [Google Scholar] [CrossRef]
- Magrinelli, F.; Mehta, S.; Di Lazzaro, G.; Latorre, A.; Edwards, M.J.; Balint, B.; Basu, P.; Kobylecki, C.; Groppa, S.; Hegde, A.; et al. Dissecting the Phenotype and Genotype of PLA2G6-Related Parkinsonism. Mov. Disord. 2022, 37, 148–161. [Google Scholar] [CrossRef]
- Ajitkumar, A.; Lui, F.; De Jesus, O. Huntington Disease. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: http://www.ncbi.nlm.nih.gov/books/NBK559166/ (accessed on 29 May 2025).
- Schneider, S.A.; Bird, T. Huntington’s Disease, Huntington’s Disease Look-Alikes, and Benign Hereditary Chorea: What’s New? Mov. Disord. Clin. Pract. 2016, 3, 342–354. [Google Scholar] [CrossRef]
- Vitrikas, K.; Dalton, H.; Breish, D. Cerebral Palsy: An Overview. Am. Fam. Physician 2020, 101, 213–220. [Google Scholar]
- Pearson, T.S.; Pons, R.; Ghaoui, R.; Sue, C.M. Genetic mimics of cerebral palsy. Mov. Disord. 2019, 34, 625–636. [Google Scholar] [CrossRef]
- Lopriore, P.; Gomes, F.; Montano, V.; Siciliano, G.; Mancuso, M. Mitochondrial Epilepsy, a Challenge for Neurologists. Int. J. Mol. Sci. 2022, 23, 13216. [Google Scholar] [CrossRef] [PubMed]
- Edmonds, J.L.; Kirse, D.J.; Kearns, D.; Deutsch, R.; Spruijt, L.; Naviaux, R.K. The Otolaryngological Manifestations of Mitochondrial Disease and the Risk of Neurodegeneration With Infection. Arch. Otolaryngol. Neck Surg. 2002, 128, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Otto, P.A.; Horimoto, A.R.V.R. Penetrance rate estimation in autosomal dominant conditions. Genet. Mol. Biol. 2012, 35, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Kingdom, R.; Wright, C.F. Incomplete Penetrance and Variable Expressivity: From Clinical Studies to Population Cohorts. Front. Genet. 2022, 13, 920390. [Google Scholar] [CrossRef]
- Balint, B.; Bhatia, K.P. Isolated and combined dystonia syndromes—An update on new genes and their phenotypes. Eur. J. Neurol. 2015, 22, 610–617. [Google Scholar] [CrossRef]
- Saffari, A.; Lau, T.; Tajsharghi, H.; Karimiani, E.G.; Kariminejad, A.; Efthymiou, S.; Zifarelli, G.; Sultan, T.; Toosi, M.B.; Sedighzadeh, S.; et al. The clinical and genetic spectrum of autosomal-recessive TOR1A-related disorders. Brain 2023, 146, 3273–3288. [Google Scholar] [CrossRef]
- Xiromerisiou, G.; Houlden, H.; Scarmeas, N.; Stamelou, M.; Kara, E.; Hardy, J.; Lees, A.J.; Korlipara, P.; Limousin, P.; Paudel, R.; et al. THAP1 Mutations and Dystonia Phenotypes: Genotype Phenotype Correlations. Mov. Disord. 2012, 27, 1290–1294. [Google Scholar] [CrossRef]
- Raymond, D.; Saunders-Pullman, R.; Ozelius, L. SGCE Myoclonus-Dystonia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1414/ (accessed on 23 March 2025).
- Stamelou, M.; Charlesworth, G.; Cordivari, C.; Schneider, S.A.; Kägi, G.; Sheerin, U.-M.; Rubio-Agusti, I.; Batla, A.; Houlden, H.; Wood, N.W.; et al. The phenotypic spectrum of DYT24 due to ANO3 mutations. Mov. Disord. 2014, 29, 928–934. [Google Scholar] [CrossRef]
- Mencacci, N.E.; Rubio-Agusti, I.; Zdebik, A.; Asmus, F.; Ludtmann, M.H.R.; Ryten, M.; Plagnol, V.; Hauser, A.-K.; Bandres-Ciga, S.; Bettencourt, C.; et al. A Missense Mutation in KCTD17 Causes Autosomal Dominant Myoclonus-Dystonia. Am. J. Hum. Genet. 2015, 96, 938–947. [Google Scholar] [CrossRef]
- Martins, J.; Darling, A.; Garrido, C.; Espinós, C.; Martí, M.J.; Dueñas, B.P.; Temudo, T. Sensory Tricks in Pantothenate Kinase-Associated Neurodegeneration: Video-Analysis of 43 Patients. Mov. Disord. Clin. Pract. 2019, 6, 704–707. [Google Scholar] [CrossRef]
- Pandey, N.; Blair, K.; John, S. Kayser-Fleischer Ring. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2024. Available online: http://www.ncbi.nlm.nih.gov/books/NBK459187/ (accessed on 1 August 2024).
- Yiş, U.; Becker, K.; Yılmaz, Ş.; Çırak, S. Acanthocytosis and HyperCKemia. Turk. J. Hematol. 2018, 35, 296–297. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F. Primary Mitochondrial Disorders Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1224/ (accessed on 28 March 2025).
- Ostojic, S.M. Plasma creatine as a marker of mitochondrial dysfunction. Med. Hypotheses 2018, 113, 52–53. [Google Scholar] [CrossRef]
- Degtyareva, A.V.; Proshlyakova, T.Y.; Gautier, M.S.; Degtyarev, D.N.; Kamenets, E.A.; Baydakova, G.V.; Rebrikov, D.V.; Zakharova, E.Y. Oxysterol/chitotriosidase based selective screening for Niemann-Pick type C in infantile cholestasis syndrome patients. BMC Med. Genet. 2019, 20, 123. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Wevers, R.A.; Kamsteeg, E.-J.; Scheffer, H.; Verbeek, M.M.; Willemsen, M.A. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: A systematic review. JAMA Neurol. 2013, 70, 1440–1444. [Google Scholar] [CrossRef]
- Donzuso, G.; Mostile, G.; Nicoletti, A.; Zappia, M. Basal ganglia calcifications (Fahr’s syndrome): Related conditions and clinical features. Neurol. Sci. 2019, 40, 2251–2263. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Boddaert, N.; Schneider, S.A.; Houlden, H.; Bhatia, K.P.; Gregory, A.; Anderson, J.C.; Rooney, W.D.; Hogarth, P.; Hayflick, S.J. Neuroimaging Features of Neurodegeneration with Brain Iron Accumulation. AJNR Am. J. Neuroradiol. 2012, 33, 407–414. [Google Scholar] [CrossRef]
- Chang, C.-L.; Lin, C.-M. Eye-of-the-Tiger sign is not Pathognomonic of Pantothenate Kinase-Associated Neurodegeneration in Adult Cases. Brain Behav. 2011, 1, 55–56. [Google Scholar] [CrossRef]
- Herrero Hernandez, E.; Valentini, M.C.; Discalzi, G. T1-weighted hyperintensity in basal ganglia at brain magnetic resonance imaging: Are different pathologies sharing a common mechanism? Neurotoxicology 2002, 23, 669–674. [Google Scholar] [CrossRef]
- Kumar, M.; Gaharwar, U.; Paul, S.; Poojary, M.; Pandhare, K.; Scaria, V.; Bk, B. WilsonGen a comprehensive clinically annotated genomic variant resource for Wilson’s Disease. Sci. Rep. 2020, 10, 9037. [Google Scholar] [CrossRef]
- National Institute of Diabetes and Digestive and Kidney Diseases. Wilson Disease—NIDDK. Available online: https://www.niddk.nih.gov/health-information/liver-disease/wilson-disease (accessed on 1 August 2024).
- Stremmel, W.; Merle, U.; Weiskirchen, R. Clinical features of Wilson disease. Ann. Transl. Med. 2019, 7, S61. [Google Scholar] [CrossRef]
- Gupta, A.; Chakravarthi, S.; Goyal, M.K. ‘Face of giant panda’: A rare imaging sign in Wilson’s disease. QJM Int. J. Med. 2014, 107, 579. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, D.A.; Markowitz, C.E.; Liebeskind, D.S.; Galetta, S.L. The “double panda sign” in Wilson’s disease. Neurology 2003, 61, 969. [Google Scholar] [CrossRef] [PubMed]
- Kaur, A.; Kaur, R. Wilson’s Disease: A Brief Review with Neuroimaging Features. Indian J. Appl. Radiol. 2015, 1, 102. [Google Scholar]
- Rędzia-Ogrodnik, B.; Członkowska, A.; Antos, A.; Bembenek, J.; Kurkowska-Jastrzębska, I.; Przybyłkowski, A.; Skowrońska, M.; Smoliński, Ł.; Litwin, T. Pathognomonic neuroradiological signs in Wilson’s disease—Truth or myth? Park. Relat. Disord. 2023, 107, 105247. [Google Scholar] [CrossRef]
- Avan, A.; Członkowska, A.; Gaskin, S.; Granzotto, A.; Sensi, S.L.; Hoogenraad, T.U. The Role of Zinc in the Treatment of Wilson’s Disease. Int. J. Mol. Sci. 2022, 23, 9316. [Google Scholar] [CrossRef]
- Kıroğlu, Y.; Callı, C.; Karabulut, N.; Oncel, C. Intracranial calcifications on CT. Diagn. Interv. Radiol. Ank. Turk. 2010, 16, 263–269. [Google Scholar] [CrossRef]
- Valdés Hernández, M.d.C.; Maconick, L.C.; Tan, E.M.J.; Wardlaw, J.M. Identification of mineral deposits in the brain on radiological images: A systematic review. Eur. Radiol. 2012, 22, 2371–2381. [Google Scholar] [CrossRef]
- Magrinelli, F.; Jesuthasan, A.; Bhatia, K.P.; Batla, A. Basal ganglia calcification: ‘Fahr’s disease’. Pract. Neurol. 2025. published online first. [Google Scholar] [CrossRef]
- Monfrini, E.; Arienti, F.; Rinchetti, P.; Lotti, F.; Riboldi, G.M. Brain Calcifications: Genetic, Molecular, and Clinical Aspects. Int. J. Mol. Sci. 2023, 24, 8995. [Google Scholar] [CrossRef]
- Batla, A.; Gaddipati, C. Neurodegeneration with Brain Iron Accumulation. Ann. Indian Acad. Neurol. 2019, 22, 267–276. [Google Scholar] [CrossRef]
- Gregory, A.; Hayflick, S. Neurodegeneration with Brain Iron Accumulation Disorders Overview. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK121988/ (accessed on 1 August 2024).
- Gupta, P.R.; Gospe, S.M. Ophthalmic manifestations of MEPAN syndrome. Ophthalmic Genet. 2023, 44, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Bohlega, S.A.; Abusrair, A. Woodhouse-Sakati Syndrome. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK378974/ (accessed on 21 May 2025).
- Iankova, V.; Karin, I.; Klopstock, T.; Schneider, S.A. Emerging Disease-Modifying Therapies in Neurodegeneration With Brain Iron Accumulation (NBIA) Disorders. Front. Neurol. 2021, 12, 629414. [Google Scholar] [CrossRef] [PubMed]
- Miyajima, H.; Takahashi, Y.; Kamata, T.; Shimizu, H.; Sakai, N.; Gitlin, J.D. Use of desferrioxamine in the treatment of aceruloplasminemia. Ann. Neurol. 1997, 41, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S. Familial manganese-induced neurotoxicity due to mutations in SLC30A10 or SLC39A14. Neurotoxicology 2018, 64, 278–283. [Google Scholar] [CrossRef]
- Tuschl, K.; Meyer, E.; Valdivia, L.E.; Zhao, N.; Dadswell, C.; Abdul-Sada, A.; Hung, C.Y.; Simpson, M.A.; Chong, W.K.; Jacques, T.S.; et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism–dystonia. Nat. Commun. 2016, 7, 11601. [Google Scholar] [CrossRef]
- Choi, E.-K.; Aring, L.; Peng, Y.; Correia, A.B.; Lieberman, A.P.; Iwase, S.; Seo, Y.A. Neuronal SLC39A8 deficiency impairs cerebellar development by altering manganese homeostasis. JCI Insight 2024, 9, e168440. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Koller, W.C. The diagnosis of manganese-induced parkinsonism. Neurotoxicology 2006, 27, 340–346. [Google Scholar] [CrossRef]
- Kim, H.M.; Hu, S.-C.; Samii, A. Cock-walk. In Encyclopedia of Movement Disorders; Kompoliti, K., Metman, L.V., Eds.; Academic Press: Oxford, UK, 2010; pp. 228–230. ISBN 978-0-12-374105-9. Available online: https://www.sciencedirect.com/science/article/pii/B9780123741059004470 (accessed on 1 August 2024).
- Marti-Sanchez, L.; Ortigoza-Escobar, J.D.; Darling, A.; Villaronga, M.; Baide, H.; Molero-Luis, M.; Batllori, M.; Vanegas, M.I.; Muchart, J.; Aquino, L.; et al. Hypermanganesemia due to mutations in SLC39A14: Further insights into Mn deposition in the central nervous system. Orphanet J. Rare Dis. 2018, 13, 28. [Google Scholar] [CrossRef]
- Quadri, M.; Federico, A.; Zhao, T.; Breedveld, G.J.; Battisti, C.; Delnooz, C.; Severijnen, L.-A.; Di Toro Mammarella, L.; Mignarri, A.; Monti, L.; et al. Mutations in SLC30A10 Cause Parkinsonism and Dystonia with Hypermanganesemia, Polycythemia, and Chronic Liver Disease. Am. J. Hum. Genet. 2012, 90, 467–477. [Google Scholar] [CrossRef]
- Majewski, M.; Piwko, K.; Ordak, M.; Muszynska, E.; Nasierowski, T.; Bujalska-Zadrozny, M. Magnetic Resonance Imaging and Manganism: A Narrative Review and Laboratory Recommendations. J. Clin. Med. 2024, 13, 2823. [Google Scholar] [CrossRef]
- Avila, D.S.; Puntel, R.L.; Aschner, M. Manganese in health and disease. Met. Ions Life Sci. 2013, 13, 199–227. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi-Fakhari, D.; Hildebrandt, C.; Davis, P.E.; Rodan, L.H.; Anselm, I.; Bodamer, O. The Spectrum of Movement Disorders in Childhood-Onset Lysosomal Storage Diseases. Mov. Disord. Clin. Pract. 2018, 5, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Elendu, C.; Babawale, E.A.; Babarinde, F.O.; Babatunde, O.D.; Chukwu, C.; Chiegboka, S.F.; Shode, O.P.; Ngozi-ibeh, J.K.; Njoku, A.; Ikokwu, M.N.; et al. Neurological manifestations of lysosomal storage diseases. Ann. Med. Surg. 2024, 86, 6619–6635. [Google Scholar] [CrossRef]
- Del Grosso, A.; Parlanti, G.; Mezzena, R.; Cecchini, M. Current treatment options and novel nanotechnology-driven enzyme replacement strategies for lysosomal storage disorders. Adv. Drug Deliv. Rev. 2022, 188, 114464. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.W.; Brady, R.O.; Dambrosia, J.M.; Bisceglie, A.M.D.; Doppelt, S.H.; Hill, S.C.; Mankin, H.J.; Murray, G.J.; Parker, R.I.; Argoff, C.E.; et al. Replacement Therapy for Inherited Enzyme Deficiency—Macrophage-Targeted Glucocerebrosidase for Gaucher’s Disease. N. Engl. J. Med. 1991, 324, 1464–1470. [Google Scholar] [CrossRef]
- Beck, M. Treatment strategies for lysosomal storage disorders. Dev. Med. Child Neurol. 2018, 60, 13–18. [Google Scholar] [CrossRef]
- Coutinho, M.F.; Santos, J.I.; Alves, S. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065. [Google Scholar] [CrossRef]
- Erro, R.; Magrinelli, F.; Bhatia, K.P. Paroxysmal movement disorders: Paroxysmal dyskinesia and episodic ataxia. Handb. Clin. Neurol. 2023, 196, 347–365. [Google Scholar] [CrossRef]
- Harvey, S.; King, M.D.; Gorman, K.M. Paroxysmal Movement Disorders. Front. Neurol. 2021, 12, 659064. [Google Scholar] [CrossRef]
- Magrinelli, F.; Bhatia, K.P. Chapter 10—Paroxysmal movement disorders. In Handbook of Clinical Neurology; Neurologic Channelopathies; Hanna, M.G., Ed.; Elsevier: Amsterdam, The Netherlands, 2024; Volume 203, pp. 145–156. Available online: https://www.sciencedirect.com/science/article/pii/B9780323908207000100 (accessed on 29 May 2025).
- Demirkiran, M.; Jankovic, J. Paroxysmal dyskinesias: Clinical features and classification. Ann. Neurol. 1995, 38, 571–579. [Google Scholar] [CrossRef]
- Gras, D.; Roze, E.; Caillet, S.; Méneret, A.; Doummar, D.; Billette de Villemeur, T.; Vidailhet, M.; Mochel, F. GLUT1 deficiency syndrome: An update. Rev. Neurol. 2014, 170, 91–99. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ren, J.; Yang, T.; Xiong, W.; Qin, L.; An, D.; Hu, F.; Zhou, D. Genetic and phenotypic analyses of PRRT2 positive and negative paroxysmal kinesigenic dyskinesia. Ther. Adv. Neurol. Disord. 2024, 17, 17562864231224110. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.-J.; Li, H.-F.; Wu, Z.-Y. Paroxysmal Kinesigenic Dyskinesia: Genetics and Pathophysiological Mechanisms. Neurosci. Bull. 2023, 40, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Mounir Alaoui, O.; Charbonneau, P.-F.; Prin, P.; Mongin, M.; Choquer, M.; Damier, P.; Riant, F.; Degos, B. TMEM151A as an alternative to PRRT2 in paroxysmal kinesigenic dyskinesia: About three new cases. Park. Relat. Disord. 2023, 108, 105295. [Google Scholar] [CrossRef]
- Zorzi, G.; Zibordi, F.; Sorrentino, U.; Prokisch, H.; Garavaglia, B.; Zech, M. Potassium Channel Subunit Kir4.1 Mutated in Paroxysmal Kinesigenic Dyskinesia: Screening of an Italian Cohort. Mov. Disord. 2024, 39, 2302–2304. [Google Scholar] [CrossRef]
- De Gusmao, C.M.; Silveira-Moriyama, L. Paroxysmal movement disorders—Practical update on diagnosis and management. Expert Rev. Neurother. 2019, 19, 807–822. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Chen, D.-F.; Ke, H.-Z.; Zhao, S.-Y.; Li, H.-F.; Wu, Z.-Y. Paroxysmal Kinesigenic Dyskinesia Caused by 16p11.2 Microdeletion and Related Clinical Features. Neurol. Genet. 2022, 8, e659. [Google Scholar] [CrossRef]
- Shimojima, K.; Okumura, A.; Natsume, J.; Aiba, K.; Kurahashi, H.; Kubota, T.; Yokochi, K.; Yamamoto, T. Spinocerebellar ataxias type 27 derived from a disruption of the fibroblast growth factor 14 gene with mimicking phenotype of paroxysmal non-kinesigenic dyskinesia. Brain Dev. 2012, 34, 230–233. [Google Scholar] [CrossRef]
- Garone, G.; Capuano, A.; Travaglini, L.; Graziola, F.; Stregapede, F.; Zanni, G.; Vigevano, F.; Bertini, E.; Nicita, F. Clinical and Genetic Overview of Paroxysmal Movement Disorders and Episodic Ataxias. Int. J. Mol. Sci. 2020, 21, 3603. [Google Scholar] [CrossRef]
- de Gusmao, C.M.; Garcia, L.R.; Jesuthasan, A.; Muir, M.; Paciorkowski, A.; Mink, J.W.; Silveira-Moriyama, L. Kinesigenic Triggers in Episodic Ataxia Type 1. Mov. Disord. Clin. Pract. 2020, 7, 723–724. [Google Scholar] [CrossRef]
- Bhatia, K.P. The paroxysmal dyskinesias. J. Neurol. 1999, 246, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Leen, W.G.; Mewasingh, L.; Verbeek, M.M.; Kamsteeg, E.-J.; van de Warrenburg, B.P.; Willemsen, M.A. Movement disorders in GLUT1 deficiency syndrome respond to the modified Atkins diet. Mov. Disord. 2013, 28, 1439–1442. [Google Scholar] [CrossRef] [PubMed]
- de Gusmao, C.M.; Peixoto de Barcelos, I.; Pinto, A.L.R.; Silveira-Moriyama, L. Pearls & Oy-sters: Paroxysmal Exercise-Induced Dyskinesias Due to Pyruvate Dehydrogenase Deficiency. Neurology 2023, 101, 46–49. [Google Scholar] [CrossRef] [PubMed]
- Erro, R. Familial Paroxysmal Nonkinesigenic Dyskinesia. In GeneReviews®; Adam, M.P., Feldman, J., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. Available online: http://www.ncbi.nlm.nih.gov/books/NBK1221/ (accessed on 28 March 2025).
- Larsh, T. Alternative Medications for Paroxysmal Kinesigenic Dyskinesia (1972). Neurology 2021, 96, 1972. [Google Scholar] [CrossRef]
- Unterberger, I.; Trinka, E. Diagnosis and treatment of paroxysmal dyskinesias revisited. Ther. Adv. Neurol. Disord. 2008, 1, 4–11. [Google Scholar] [CrossRef]
- Sondhi, V.; Sharma, S. Vitamin-Responsive Movement Disorders in Children. Ann. Indian Acad. Neurol. 2020, 23, 325–331. [Google Scholar] [CrossRef]
- Ozand, P.T.; Gascon, G.G.; Al Essa, M.; Joshi, S.; Al Jishi, E.; Bakheet, S.; Al Watban, J.; Al-Kawi, M.Z.; Dabbagh, O. Biotin-responsive basal ganglia disease: A novel entity. Brain J. Neurol. 1998, 121 Pt 7, 1267–1279. [Google Scholar] [CrossRef]
- Manco, C.; Cortese, R.; Alberti, M.; Bianchi, S.; Monti, L.; De Stefano, N.; Battisti, C. FOLR1 Gene Variation With Adult-Onset Cerebral Folate Deficiency and Stable Clinical and MRI Features up to 2 Years. Neurol. Genet. 2023, 9, e200104. [Google Scholar] [CrossRef]
- Sofou, K.; Dahlin, M.; Hallböök, T.; Lindefeldt, M.; Viggedal, G.; Darin, N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef]
- Wijemanne, S.; Jankovic, J. Dopa-responsive dystonia—Clinical and genetic heterogeneity. Nat. Rev. Neurol. 2015, 11, 414–424. [Google Scholar] [CrossRef]
- Lee, W.-W.; Jeon, B.S. Clinical Spectrum of Dopa-Responsive Dystonia and Related Disorders. Curr. Neurol. Neurosci. Rep. 2014, 14, 461. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.-W.; Jeon, B.; Kim, R. Expanding the Spectrum of Dopa-Responsive Dystonia (DRD) and Proposal for New Definition: DRD, DRD-plus, and DRD Look-alike. J. Korean Med. Sci. 2018, 33, e184. [Google Scholar] [CrossRef] [PubMed]
- Hecht, F.; Hecht, B.K. Cancer in ataxia-telangiectasia patients. Cancer Genet. Cytogenet. 1990, 46, 9–19. [Google Scholar] [CrossRef]
- Zech, M.; Winkelmann, J. Next-generation sequencing and bioinformatics in rare movement disorders. Nat. Rev. Neurol. 2024, 20, 114–126. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Johnson, A.K.; Nelakuditi, V.; Guidugli, L.; Fischer, D.; Arndt, K.; Ma, L.; Sandford, E.; Shakkottai, V.; Boycott, K.; et al. Targeted exome analysis identifies the genetic basis of disease in over 50% of patients with a wide range of ataxia-related phenotypes. Genet. Med. 2019, 21, 195–206. [Google Scholar] [CrossRef]
- Feng, H.; Sjögren, B.; Karaj, B.; Shaw, V.; Gezer, A.; Neubig, R.R. Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology 2017, 89, 762–770. [Google Scholar] [CrossRef]
- Wirth, T.; Garone, G.; Kurian, M.A.; Piton, A.; Millan, F.; Telegrafi, A.; Drouot, N.; Rudolf, G.; Chelly, J.; Marks, W.; et al. Highlighting the Dystonic Phenotype Related to GNAO1. Mov. Disord. 2022, 37, 1547–1554. [Google Scholar] [CrossRef]
- FDA. FDA Approves First Gene Therapy for Treatment of Aromatic L-Amino Acid Decarboxylase Deficiency. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-gene-therapy-treatment-aromatic-l-amino-acid-decarboxylase-deficiency (accessed on 22 March 2025).

{kind=link}
| RMD Category | Diagnostic Clues and Relevant Investigations | Examples of Specific Conditions |
|---|---|---|
| Dopamine synthesis disorders | Oculogyric crises | TH deficiency, AADC deficiency Sepiapterin deficiency |
| Diurnal fluctuation of symptom severity Oculogyric crises | GCH deficiency (classic DRD—Segawa disease) | |
| Disorders with brain mineralisation | Kayser-Fleischer ring Sunflower cataract Low serum ceruloplasmin High 24 h urinary copper level Face of the giant panda sign on MRI | Wilson’s disease |
| Retinal degeneration/optic atrophy Retinitis pigmentosa Mantis sign Eye of the tiger sign on MRI | NBIA (PKAN and VAC14) (PKAN) (PKAN) | |
| Metabolic diseases | Vertical supranuclear gaze palsy (particularly downward) High plasma oxysterol levels | NP-C |
| Cherry-red spot on the fundus Low serum hexosaminidase A | Tay–Sachs disease | |
| Tendon xanthomas | Cerebrotendinous xanthomatosis | |
| Paroxysmal MD | Exercise-induced dyskinesia Low CSF-to-blood glucose ratio CSF lactate low to normal | PED (GLUT1 deficiency syndrome) |
| Dyskinesia (variable frequency of attacks, may be up to 100 per day, lasting <1 min) mainly triggered by sudden movement from rest | PKD | |
| Dyskinesia (1–3 attacks per day lasting minutes to hours) sometimes triggered by alcohol, caffeine, stress, menstruation, or sleep deprivation | PNKD | |
| Interictal myokymia | EA1 |
| Causative Genes | Inheritance Pattern | Phenotypic Features |
|---|---|---|
| SLC20A2 | Autosomal dominant | Movement disorders: Bradykinesia, rigidity, tremor, dystonia, ataxia, chorea, pyramidal Cognitive/psychiatric features: Cognitive deficits, depression, psychosis, anxiety Other symptoms/signs: Headache, speech disturbance, seizures, dysmorphic features Imaging: Basal ganglia calcification along with calcifications in the cerebellum (dentate nucleus), white matter, pons |
| PDGFB | Autosomal dominant | |
| PDGFRB | Autosomal dominant | |
| XPR1 | Autosomal dominant | |
| JAM2 | Autosomal recessive | |
| MYORG | Autosomal recessive | |
| NAA60 | Autosomal recessive |
| Disease and Causative Gene | Specific Clinical Features (Alongside Chorea, Dystonia, and Parkinsonism) | Potential MRI Findings |
|---|---|---|
| PKAN PANK2 | Oromandibular and truncal extensor dystonia (opisthotonus) | Eye of the tiger sign in the globus pallidus Mid-hypointensity in substantia nigra |
| PLAN PLA2G6 | Axial hypotonia (in early-childhood onset) Opisthotonus (in late-childhood onset) Seizures Children and young adults may also present with ataxia, speech regression, optic atrophy, neuropsychiatric disturbance, oculomotor abnormalities, and autonomic disturbance | Cerebellar and vermian atrophy with callosal thinning, vertical orientation, and claval hypertrophy |
| NEUROFERRITINOPATHY FTL | Triad of oromandibular dyskinesia, dysarthrophonia and action-specific facial dystonia | Cortical pencil sign Hypointense caudate, putamen, thalamus, globus pallidus, substantia nigra, and red nucleus |
| MPAN C19orf12 | Often presents in childhood with spastic paraparesis, behavioural disturbance, optic atrophy, and motor axonal neuropathy | Hypointensity in the globus pallidus and substantia nigra Hyperintense streaking of the medial medullary lamina of the globus pallidus Cortical and cerebellar atrophy |
| BPAN WDR45 | Biphasic disease, presenting in childhood with delayed speech and motor development, ataxic gait Cognitive decline and seizures in adulthood | Halo sign in substantia nigra on T1 Hyperintensity on T2 in the globus pallidus and cerebral peduncles |
| CoPAN COASY | Spasticity, axonal neuropathy, cognitive decline, and obsessive–compulsive behaviour in childhood | Eye of the tiger sign Hypointense substantia nigra and globus pallidus Swelling of the caudate and putamen |
| ACERULOPLASMINEMIA CP | Triad of retinal degeneration, diabetes mellitus, and anaemia Cerebellar dysfunction | Hypointense caudate, putamen, thalamus, globus pallidus, substantia nigra, cerebellum, and red nucleus |
| FAHN FA2H | Presents in childhood with cerebellar dysfunction, spasticity, optic atrophy, bristly hair, and seizures | Globus pallidus more hypointense than substantia nigra Confluent subcortical and periventricular cerebral T2 white matter hyperintensities Atrophy of cerebellum, medulla, and spinal cord |
| KRD/PARK9 ATP13A2 | Presents in adolescence with vertical supranuclear gaze palsy (particularly upward), facial-faucial–finger myoclonus and visual hallucinations | Diffuse cerebral, cerebellar, and brainstem atrophy Iron accumulation in putamen and caudate |
| WSS DCAF17 | Dysarthria, dysphagia, seizures, sensory polyneuropathy, and sensorineural deafness Craniofacial abnormalities and camptodactyly Alopecia, hypogonadism, hypothyroidism, and diabetes mellitus | Partially empty sella Progressive white matter lesions in the frontoparietal region Iron accumulation in the globus pallidus, substantia nigra, red nucleus |
| Disease | Exemplar Neurological Features |
|---|---|
| Abetalipoproteinemia | Tremor, nystagmus, peripheral neuropathy, delayed intellectual development, loss of deep tendon reflexes, retinitis pigmentosa |
| Aspartylglucosaminuria | Slowly progressive clumsiness, speech delay, hyperkinesia, facial dysmorphism |
| Ataxia with vitamin E deficiency | Progressive cerebellar syndrome, areflexia, positive Babinski sign, macular degeneration, pigmentary retinopathy |
| Biotinidase deficiency | Ataxia, seizures, hypotonia, developmental delay, optic atrophy, sensorineural hearing loss |
| Biotin-thiamine responsive basal ganglia disease | Recurrent subacute encephalopathy with confusion, seizures, ataxia, dystonia, cogwheel rigidity, supranuclear facial palsy, external ophthalmoplegia |
| Cerebral folate deficiency | Irritability, sleep disturbance, psychomotor retardation, dyskinesia, cerebellar ataxia, spastic diplegia, visual disturbance, sensorineural hearing loss |
| Cobalamin deficiency | Myelopathy, peripheral neuropathy with abnormal proprioception, cognitive impairment, dysesthesia, spastic paraparesis or tetraparesis, optic nerve atrophy |
| Coenzyme Q10 deficiency | Encephalopathy, seizures, dystonia, spasticity, intellectual disability, hypotonia, autonomic dysfunction, parkinsonism, cerebellar ataxia, pyramidal dysfunction, peripheral neuropathy |
| Glutaric acidaemia type 1 | Progressive macrocephaly, hypotonia, motor regression, seizures, headaches, vertigo, ataxia, dementia |
| Homocystinuria | Developmental delay, autism spectrum disorders, psychiatric disturbance, marfanoid appearance, ectopia lentis |
| Methylmalonic aciduria | Encephalopathy, failure to thrive, hypotonia, developmental delay, ataxia, dysarthria, spastic paraparesis, seizures |
| Phenylketonuria | Microcephaly, epilepsy, severe developmental delay, progressive supranuclear motor disturbances, tremor, cerebellar ataxia |
| Propionic acidaemia | Progressive encephalopathy, seizures, coma, hypotonia, developmental delay |
| Fabry Disease | Hemiparesis, diplopia, headaches, cerebellar syndrome, dysmetria, neuropsychiatric disturbance, dementia |
| Gaucher’s Disease | Oculomotor apraxia, seizures, progressive myoclonic epilepsy, bulbar and/or pyramidal signs, cognitive impairment |
| Mucopolysaccharidoses | Developmental delay, neurocognitive regression, behavioural changes, sleep disturbance, epilepsy, hydrocephalus, raised intracranial pressure symptoms, myelopathy |
| Neuronal Ceroid Lipofuscinoses | Progressive mental and motor deterioration, retinal degeneration causing blindness, epilepsy |
| Niemann–Pick Disease Type C | Hypotonia, developmental delay, vertical supranuclear gaze palsy, ataxia, seizures, gelastic cataplexy, dystonia, spasticity |
| Pyruvate dehydrogenase complex deficiency | Developmental delay and motor regression, growth retardation, hypotonia, seizures, ataxia, dystonia |
| Salla disease | Psychomotor delays, spasticity, athetosis, epileptic seizures, hypotonia, cerebellar syndrome |
| Tay–Sachs disease | Hypotonia, developmental delay and motor regression, decreased responsiveness over time, spasticity, seizures, ataxia, dyskinesia, sleep disturbance, screaming and irritability, macrocephaly, decerebrate posturing, dysphagia, dystonia, cognitive impairment |
| Disorder | Enzyme/Protein Deficiency | Clinical Features | Catecholamine Metabolite Levels |
|---|---|---|---|
| Biopterin synthesis/recycling defects | SPR | Dystonia, weakness, oculogyric crisis, axial hypotonia, motor, and speech delay | ↓ HVA ↓ 5-HIAA |
| AD-GTPCH1 | Gait disturbance due to foot dystonia with later development of generalised dystonia and parkinsonism | N/↓ HVA N/↓ 5-HIAA | |
| AR-GTPCH1 | ↓ HVA ↓ 5-HIAA | ||
| PTPS | Developmental delay, axial hypotonia, hypertonia of extremities, small for gestational age | ↓ HVA ↓ 5-HIAA | |
| DHPR | Developmental delay, axial hypotonia, hypertonia of extremities, epilepsy, microcephaly | ↓ HVA ↓ 5-HIAA | |
| PCD | Progressive hypotonia and delays in motor development, which are temporary | ↓ HVA ↓ 5-HIAA | |
| Primary neurotransmitter synthesis defects | TH | Gait disturbance due to lower-limb dystonia, which gradually progresses to generalised dystonia +/− parkinsonism with pyramidal signs | ↓ HVA N 5-HIAA |
| Monoamine transportopathies | DTDS SLC6A3 | Chorea, dystonia, ballismus, and orolingual dyskinesia, which progresses to involve parkinsonism–dystonia | ↑ HVA N 5-HIAA |
| VMAT2 SLC18A2 | Motor delay, eye deviation, severe parkinsonism, dystonia, ataxia, oculogyric crises, autonomic dysfunction and ptosis | N HVA N 5-HIAA | |
| Monoamine catabolism disorders | MAO-A/ MAO-B | Episodic hypotonia, intellectual disability, stereotyped movements, aggressive behaviour. | ↓ HVA ↓ 5-HIAA |
| DBH | Autonomic dysfunction, reduced exercise tolerance, hypoglycaemia, and ptosis | ↑ HVA ↑ 5-HIAA | |
| AADC | Truncal hypotonia, oculogyric crises, hypokinesia, athetosis, chorea, tremor, autonomic dysfunction | ↓ HVA ↓ 5-HIAA | |
| PNPO | High seizure frequency, developmental delay, hypotonia, dystonia | ↓ HVA ↓ 5-HIAA |
| Rare Movement Disorder | Treatment |
|---|---|
| Abetalipoproteinaemia | Dietary fat restriction, vitamin E and A |
| Aromatic L-amino acid decarboxylase deficiency | Dopamine agonists, monoamine oxidase inhibitors, pyridoxine |
| Ataxia with vitamin E deficiency | Vitamin E |
| Biotin–thiamine responsive basal ganglia disease | Biotin plus thiamine, avoid or treat triggers |
| Biotinidase deficiency | Biotin |
| Cerebral creatine deficiency | Creatine ± ornithine, dietary restriction of arginine |
| Cerebral folate deficiency | Folinic acid |
| Cerebrotendinous xanthomatosis | Chenodeoxycholic acid |
| Cobalamin deficiency | Cobalamin derivatives |
| Coenzyme Q10 deficiency | Coenzyme Q10 |
| Dopa-responsive dystonia | Levodopa, 5-hydroxytryptophan |
| Dystonia–parkinsonism with manganese accumulation | EDTA chelation therapy |
| Episodic ataxia type 2 | 4-aminopyridine, acetazolamide |
| Gaucher disease | Enzyme replacement therapy, N-butyl-deoxynojirimycin |
| GLUT1 deficiency syndrome | Ketogenic diet, triheptanoin |
| Glutaric aciduria type 1 | Avoid or treat triggers, dietary lysine restriction, L-carnitine |
| Homocystinuria | Vitamin B6, dietary restriction of methionine, betaine |
| Maple syrup urine disease | Avoid or treat triggers, dietary leucine restriction |
| Methylmalonic aciduria | Avoid or treat triggers, dietary protein restriction, L-carnitine |
| Molybdenum cofactor deficiency | Cyclic pyranopterin monophosphate |
| Niemann–Pick type C | Miglustat (N-butyl-deoxynojirimycin) |
| Paroxysmal kinesigenic dyskinesia | Carbamazepine, other anticonvulsants |
| Phenylketonuria | Dietary restriction of phenylalanine |
| Propionic acidaemia | Avoid or treat triggers, dietary protein restriction, L-carnitine |
| Pyruvate dehydrogenase complex deficiency | Thiamine, ketogenic diet, triheptanoin |
| Refsum disease | Dietary restriction of phytanic acids, plasmapharesis |
| Wilson’s disease | Penicillamine, trientine, zinc |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jesuthasan, A.; Magrinelli, F.; Batla, A.; Bhatia, K.P. Rare Movement Disorders—An Approach for Clinicians. Int. J. Mol. Sci. 2025, 26, 6024. https://doi.org/10.3390/ijms26136024
Jesuthasan A, Magrinelli F, Batla A, Bhatia KP. Rare Movement Disorders—An Approach for Clinicians. International Journal of Molecular Sciences. 2025; 26(13):6024. https://doi.org/10.3390/ijms26136024
Chicago/Turabian StyleJesuthasan, Aaron, Francesca Magrinelli, Amit Batla, and Kailash P. Bhatia. 2025. "Rare Movement Disorders—An Approach for Clinicians" International Journal of Molecular Sciences 26, no. 13: 6024. https://doi.org/10.3390/ijms26136024
APA StyleJesuthasan, A., Magrinelli, F., Batla, A., & Bhatia, K. P. (2025). Rare Movement Disorders—An Approach for Clinicians. International Journal of Molecular Sciences, 26(13), 6024. https://doi.org/10.3390/ijms26136024