
Advancing Treatment in Pediatric Multiple Sclerosis: The Promise of B-Cell-Targeting Therapies


Abstract
1. Introduction
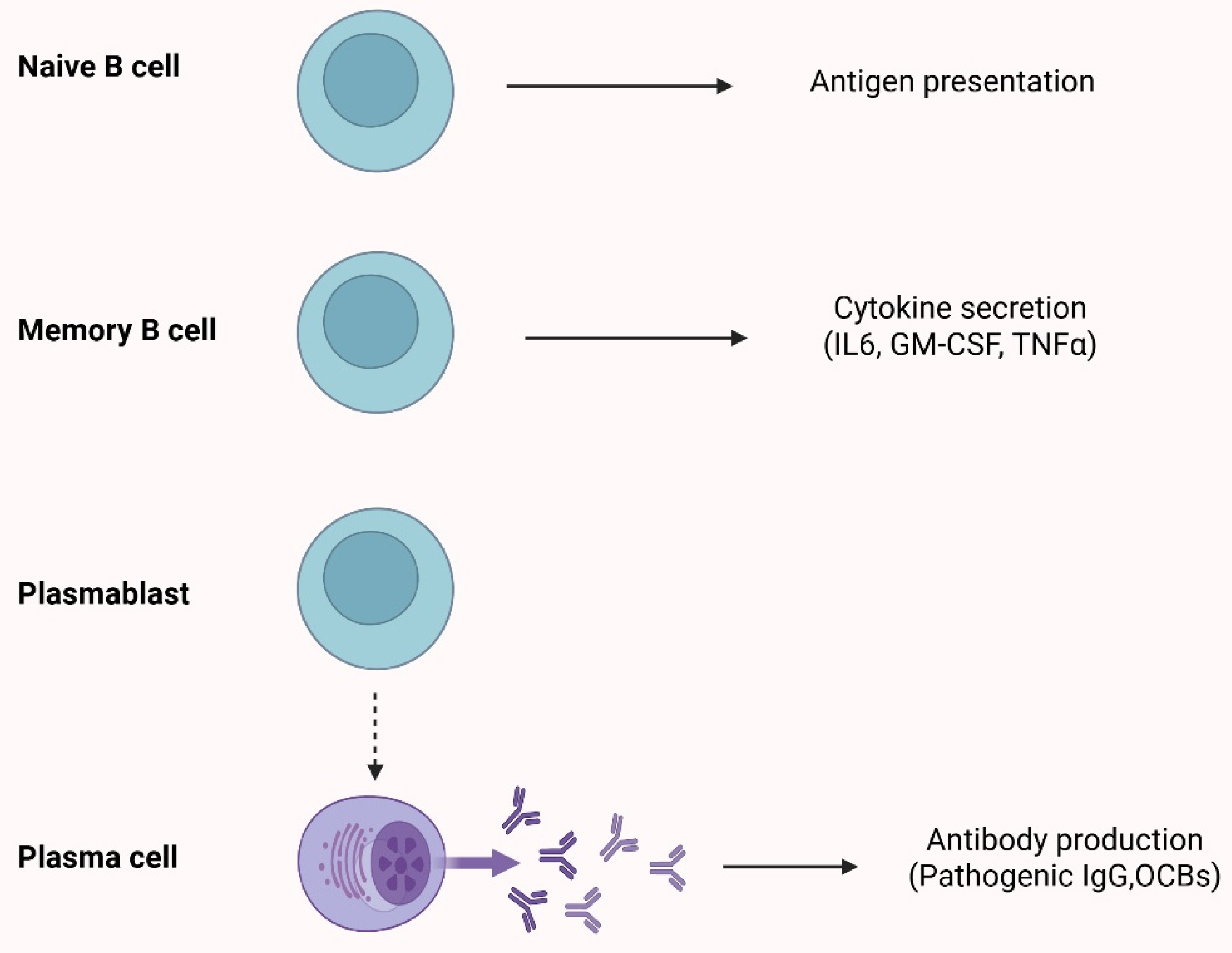
2. The Role of B-Cells in Multiple Sclerosis
3. Methodology
4. Therapeutic Agents Targeting B-Cells and Their Use in the Treatment of POMS
4.1. Anti-CD20 Agents: Mechanism of Action
4.2. Rituximab
4.3. Ocrelizumab
4.4. Ofatumumab
5. Anti-CD25 Agents: Daclizumab
6. Anti-CD52 Agents: Alemtuzumab
7. Future Perspectives for B-Cells Targeting Therapies in the Context of MS
7.1. Targeting CD19
7.2. Targeting BAFF Axis
7.3. Bruton’s Tyrosine Kinase Inhibitors (BTKis)
8. Conclusions
Funding
Conflicts of Interest
References
- Chitnis, T. Pediatric Central Nervous System Demyelinating Diseases. Continuum 2019, 25, 793–814. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.A.; Chitnis, T.; Krupp, L.; Ness, J.; Chabas, D.; Kuntz, N.; Waubant, E. Pediatric Multiple Sclerosis. Nat. Rev. Neurol. 2009, 5, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Yan, K.; Balijepalli, C.; Desai, K.; Gullapalli, L.; Druyts, E. Epidemiology of Pediatric Multiple Sclerosis: A Systematic Literature Review and Meta-Analysis. Mult. Scler. Relat. Disord. 2020, 44, 102260. [Google Scholar] [CrossRef]
- Brola, W.; Steinborn, B. Pediatric Multiple Sclerosis—Current Status of Epidemiology, Diagnosis and Treatment. Neurol. Neurochir. Pol. 2020, 54, 508–517. [Google Scholar] [CrossRef]
- Willer, C.J.; Dyment, D.A.; Risch, N.J.; Sadovnick, A.D.; Ebers, G.C.; Canadian Collaborative Study Group. Twin Concordance and Sibling Recurrence Rates in Multiple Sclerosis. Proc. Natl. Acad. Sci. USA 2003, 100, 12877–12882. [Google Scholar] [CrossRef]
- Dobson, R.; Giovannoni, G. Multiple Sclerosis—A Review. Eur. J. Neurol. 2019, 26, 27–40. [Google Scholar] [CrossRef]
- Jeong, A.; Oleske, D.M.; Holman, J. Epidemiology of Pediatric-Onset Multiple Sclerosis: A Systematic Review of the Literature. J. Child Neurol. 2019, 34, 705–712. [Google Scholar] [CrossRef]
- Ysrraelit, M.C.; Correale, J. Impact of Sex Hormones on Immune Function and Multiple Sclerosis Development. Immunology 2019, 156, 9–22. [Google Scholar] [CrossRef]
- Waubant, E. Effect of Puberty on Multiple Sclerosis Risk and Course. Mult. Scler. 2018, 24, 32–35. [Google Scholar] [CrossRef]
- Banwell, B.; Bar-Or, A.; Arnold, D.L.; Sadovnick, D.; Narayanan, S.; McGowan, M.; O’Mahony, J.; Magalhaes, S.; Hanwell, H.; Vieth, R.; et al. Clinical, Environmental, and Genetic Determinants of Multiple Sclerosis in Children with Acute Demyelination: A Prospective National Cohort Study. Lancet Neurol. 2011, 10, 436–445. [Google Scholar] [CrossRef]
- Skarlis, C.; Anagnostouli, M. The Role of Melatonin in Multiple Sclerosis. Neurol. Sci. 2020, 41, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Lowy, D.; Chitnis, T. Pathogenesis of Pediatric Multiple Sclerosis. J. Child. Neurol. 2012, 27, 1394–1407. [Google Scholar] [CrossRef] [PubMed]
- Gontika, M.; Skarlis, C.; Artemiadis, A.; Pons, R.; Mastroyianni, S.; Vartzelis, G.; Theodorou, V.; Kilindireas, K.; Stefanis, L.; Dalakas, M.; et al. HLA-DRB1 Allele Impact on Pediatric Multiple Sclerosis in a Hellenic Cohort. Mult. Scler. J. Exp. Transl. Clin. 2020, 6, 2055217320908046. [Google Scholar] [CrossRef] [PubMed]
- Skarlis, C.; Markoglou, N.; Gontika, M.; Artemiadis, A.; Pons, M.-R.; Stefanis, L.; Dalakas, M.; Chrousos, G.; Anagnostouli, M. The Impact of HLA-DRB1 Alleles in a Hellenic, Pediatric-Onset Multiple Sclerosis Cohort: Implications on Clinical and Neuroimaging Profile. Neurol. Sci. 2024, 45, 5405–5411. [Google Scholar] [CrossRef]
- Prapas, P.; Anagnostouli, M. Macrophages and HLA-Class II Alleles in Multiple Sclerosis: Insights in Therapeutic Dynamics. Int. J. Mol. Sci. 2024, 25, 7354. [Google Scholar] [CrossRef]
- Skarlis, C.; Markoglou, N.; Artemiadis, A.; Gontika, M.; Koutsis, G.; Chrousos, G.; Anagnostouli, M. Implication of Apolipoprotein E Gene Variants in Pediatric-Onset Multiple Sclerosis: Possible Association with Disease Susceptibility and Its Clinical Characteristics, in a Hellenic Cohort. Mult. Scler. Relat. Disord. 2024, 90, 105797. [Google Scholar] [CrossRef]
- Munger, K.L.; Chitnis, T.; Ascherio, A. Body Size and Risk of MS in Two Cohorts of US Women. Neurology 2009, 73, 1543–1550. [Google Scholar] [CrossRef]
- Mikaeloff, Y.; Caridade, G.; Tardieu, M.; Suissa, S.; KIDSEP Study Group. Parental Smoking at Home and the Risk of Childhood-Onset Multiple Sclerosis in Children. Brain 2007, 130, 2589–2595. [Google Scholar] [CrossRef]
- Banwell, B.; Krupp, L.; Kennedy, J.; Tellier, R.; Tenembaum, S.; Ness, J.; Belman, A.; Boiko, A.; Bykova, O.; Waubant, E.; et al. Clinical Features and Viral Serologies in Children with Multiple Sclerosis: A Multinational Observational Study. Lancet Neurol. 2007, 6, 773–781. [Google Scholar] [CrossRef]
- Hedström, A.K.; Sundqvist, E.; Bäärnhielm, M.; Nordin, N.; Hillert, J.; Kockum, I.; Olsson, T.; Alfredsson, L. Smoking and Two Human Leukocyte Antigen Genes Interact to Increase the Risk for Multiple Sclerosis. Brain 2011, 134, 653–664. [Google Scholar] [CrossRef]
- Renoux, C.; Vukusic, S.; Mikaeloff, Y.; Edan, G.; Clanet, M.; Dubois, B.; Debouverie, M.; Brochet, B.; Lebrun-Frenay, C.; Pelletier, J.; et al. Natural History of Multiple Sclerosis with Childhood Onset. N. Engl. J. Med. 2007, 356, 2603–2613. [Google Scholar] [CrossRef] [PubMed]
- McKay, K.A.; Hillert, J.; Manouchehrinia, A. Long-Term Disability Progression of Pediatric-Onset Multiple Sclerosis. Neurology 2019, 92, e2764–e2773. [Google Scholar] [CrossRef] [PubMed]
- Alroughani, R.; Boyko, A. Pediatric Multiple Sclerosis: A Review. BMC Neurol. 2018, 18, 27. [Google Scholar] [CrossRef]
- Pfeifenbring, S.; Bunyan, R.F.; Metz, I.; Röver, C.; Huppke, P.; Gärtner, J.; Lucchinetti, C.F.; Brück, W. Extensive Acute Axonal Damage in Pediatric Multiple Sclerosis Lesions. Ann. Neurol. 2015, 77, 655–667. [Google Scholar] [CrossRef]
- Bar-Or, A.; Fawaz, L.; Fan, B.; Darlington, P.J.; Rieger, A.; Ghorayeb, C.; Calabresi, P.A.; Waubant, E.; Hauser, S.L.; Zhang, J.; et al. Abnormal B-Cell Cytokine Responses a Trigger of T-Cell-Mediated Disease in MS? Ann. Neurol. 2010, 67, 452–461. [Google Scholar] [CrossRef]
- Li, R.; Rezk, A.; Miyazaki, Y.; Hilgenberg, E.; Touil, H.; Shen, P.; Moore, C.S.; Michel, L.; Althekair, F.; Rajasekharan, S.; et al. Proinflammatory GM-CSF-Producing B Cells in Multiple Sclerosis and B Cell Depletion Therapy. Sci. Transl. Med. 2015, 7, 310ra166. [Google Scholar] [CrossRef]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct Effector Cytokine Profiles of Memory and Naive Human B Cell Subsets and Implication in Multiple Sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef]
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The Compartmentalized Inflammatory Response in the Multiple Sclerosis Brain Is Composed of Tissue-Resident CD8+ T Lymphocytes and B Cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef]
- Magliozzi, R.; Howell, O.; Vora, A.; Serafini, B.; Nicholas, R.; Puopolo, M.; Reynolds, R.; Aloisi, F. Meningeal B-Cell Follicles in Secondary Progressive Multiple Sclerosis Associate with Early Onset of Disease and Severe Cortical Pathology. Brain 2007, 130, 1089–1104. [Google Scholar] [CrossRef]
- Carlson, A.K.; Amin, M.; Cohen, J.A. Drugs Targeting CD20 in Multiple Sclerosis: Pharmacology, Efficacy, Safety, and Tolerability. Drugs 2024, 84, 285–304. [Google Scholar] [CrossRef]
- de Sèze, J.; Maillart, E.; Gueguen, A.; Laplaud, D.A.; Michel, L.; Thouvenot, E.; Zephir, H.; Zimmer, L.; Biotti, D.; Liblau, R. Anti-CD20 Therapies in Multiple Sclerosis: From Pathology to the Clinic. Front. Immunol. 2023, 14, 1004795. [Google Scholar] [CrossRef] [PubMed]
- Skarlis, C.; Papadopoulos, V.; Raftopoulou, S.; Mavragani, C.P.; Evangelopoulos, M.-E. B-Cell Activating Factor Gene Variants in Multiple Sclerosis: Possible Associations with Disease Susceptibility among Females. Clin. Immunol. 2023, 257, 109847. [Google Scholar] [CrossRef] [PubMed]
- Ntellas, P.; Dardiotis, E.; Sevdali, E.; Siokas, V.; Aloizou, A.-M.; Tsinti, G.; Germenis, A.E.; Hadjigeorgiou, G.M.; Eibel, H.; Speletas, M. TNFRSF13C/BAFFR P21R and H159Y Polymorphisms in Multiple Sclerosis. Mult. Scler. Relat. Disord. 2020, 37, 101422. [Google Scholar] [CrossRef] [PubMed]
- Cencioni, M.T.; Mattoscio, M.; Magliozzi, R.; Bar-Or, A.; Muraro, P.A. B Cells in Multiple Sclerosis—From Targeted Depletion to Immune Reconstitution Therapies. Nat. Rev. Neurol. 2021, 17, 399–414. [Google Scholar] [CrossRef]
- Margoni, M.; Rinaldi, F.; Perini, P.; Gallo, P. Therapy of Pediatric-Onset Multiple Sclerosis: State of the Art, Challenges, and Opportunities. Front. Neurol. 2021, 12, 676095. [Google Scholar] [CrossRef]
- Margoni, M.; Preziosa, P.; Filippi, M.; Rocca, M.A. Anti-CD20 Therapies for Multiple Sclerosis: Current Status and Future Perspectives. J. Neurol. 2022, 269, 1316–1334. [Google Scholar] [CrossRef]
- Crickx, E.; Weill, J.-C.; Reynaud, C.-A.; Mahévas, M. Anti-CD20–Mediated B-Cell Depletion in Autoimmune Diseases: Successes, Failures and Future Perspectives. Kidney Int. 2020, 97, 885–893. [Google Scholar] [CrossRef]
- Baecher-Allan, C.; Kaskow, B.J.; Weiner, H.L. Multiple Sclerosis: Mechanisms and Immunotherapy. Neuron 2018, 97, 742–768. [Google Scholar] [CrossRef]
- Simon, A.K.; Hollander, G.A.; McMichael, A. Evolution of the Immune System in Humans from Infancy to Old Age. Proc. Biol. Sci. 2015, 282, 20143085. [Google Scholar] [CrossRef]
- Santana-Sánchez, P.; Vaquero-García, R.; Legorreta-Haquet, M.V.; Chávez-Sánchez, L.; Chávez-Rueda, A.K. Hormones and B-Cell Development in Health and Autoimmunity. Front. Immunol. 2024, 15, 1385501. [Google Scholar] [CrossRef]
- Ghezzi, A. Old and New Strategies in the Treatment of Pediatric Multiple Sclerosis: A Personal View for a New Treatment Approach. Neurol. Ther. 2024, 13, 949–963. [Google Scholar] [CrossRef] [PubMed]
- Airas, L.; Bermel, R.A.; Chitnis, T.; Hartung, H.-P.; Nakahara, J.; Stuve, O.; Williams, M.J.; Kieseier, B.C.; Wiendl, H. A Review of Bruton’s Tyrosine Kinase Inhibitors in Multiple Sclerosis. Ther. Adv. Neurol. Disord. 2024, 17, 17562864241233041. [Google Scholar] [CrossRef] [PubMed]
- Al-Hawary, S.I.S.; Jasim, S.A.; Hjazi, A.; Ullah, H.; Bansal, P.; Deorari, M.; Sapaev, I.B.; Ami, A.A.; Mohmmed, K.H.; Abosaoda, M.K. A New Perspective on Therapies Involving B-Cell Depletion in Autoimmune Diseases. Mol. Biol. Rep. 2024, 51, 629. [Google Scholar] [CrossRef]
- Beers, S.A.; Chan, C.H.T.; French, R.R.; Cragg, M.S.; Glennie, M.J. CD20 as a Target for Therapeutic Type I and II Monoclonal Antibodies. Semin. Hematol. 2010, 47, 107–114. [Google Scholar] [CrossRef]
- Etemadifar, M.; Nouri, H.; Sedaghat, N.; Ramezani, A.; Kargaran, P.K.; Salari, M.; Kaveyee, H. Anti-CD20 Therapies for Pediatric-Onset Multiple Sclerosis: A Systematic Review. Mult. Scler. Relat. Disord. 2024, 91, 105849. [Google Scholar] [CrossRef]
- Boross, P.; Leusen, J.H.W. Mechanisms of Action of CD20 Antibodies. Am. J. Cancer Res. 2012, 2, 676–690. [Google Scholar]
- Meyer, S.; Evers, M.; Jansen, J.H.M.; Buijs, J.; Broek, B.; Reitsma, S.E.; Moerer, P.; Amini, M.; Kretschmer, A.; Ten Broeke, T.; et al. New Insights in Type I and II CD20 Antibody Mechanisms-of-Action with a Panel of Novel CD20 Antibodies. Br. J. Haematol. 2018, 180, 808–820. [Google Scholar] [CrossRef]
- Goldman, S.; Smith, L.; Anderson, J.R.; Perkins, S.; Harrison, L.; Geyer, M.B.; Gross, T.G.; Weinstein, H.; Bergeron, S.; Shiramizu, B.; et al. Rituximab and FAB/LMB 96 Chemotherapy in Children with Stage III/IV B-Cell Non-Hodgkin Lymphoma: A Children’s Oncology Group Report. Leukemia 2013, 27, 1174–1177. [Google Scholar] [CrossRef]
- Minard-Colin, V.; Aupérin, A.; Pillon, M.; Burke, G.A.A.; Barkauskas, D.A.; Wheatley, K.; Delgado, R.F.; Alexander, S.; Uyttebroeck, A.; Bollard, C.M.; et al. Rituximab for High-Risk, Mature B-Cell Non-Hodgkin’s Lymphoma in Children. N. Engl. J. Med. 2020, 382, 2207–2219. [Google Scholar] [CrossRef]
- Cohen, S.B.; Emery, P.; Greenwald, M.W.; Dougados, M.; Furie, R.A.; Genovese, M.C.; Keystone, E.C.; Loveless, J.E.; Burmester, G.-R.; Cravets, M.W.; et al. Rituximab for Rheumatoid Arthritis Refractory to Anti-Tumor Necrosis Factor Therapy: Results of a Multicenter, Randomized, Double-Blind, Placebo-Controlled, Phase III Trial Evaluating Primary Efficacy and Safety at Twenty-Four Weeks. Arthritis Rheum. 2006, 54, 2793–2806. [Google Scholar] [CrossRef]
- Stone, J.H.; Merkel, P.A.; Spiera, R.; Seo, P.; Langford, C.A.; Hoffman, G.S.; Kallenberg, C.G.M.; St Clair, E.W.; Turkiewicz, A.; Tchao, N.K.; et al. Rituximab versus Cyclophosphamide for ANCA-Associated Vasculitis. N. Engl. J. Med. 2010, 363, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.A.; Kamat, S.N.; Lalkaka, J.A.; Singhal, B. Efficacy and Safety of Rituximab in Central Nervous System Demyelinating Disorders. Ann. Indian. Acad. Neurol. 2021, 24, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Karenfort, M.; Kieseier, B.C.; Tibussek, D.; Assmann, B.; Schaper, J.; Mayatepek, E. Rituximab as a Highly Effective Treatment in a Female Adolescent with Severe Multiple Sclerosis. Dev. Med. Child. Neurol. 2009, 51, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Malani Shukla, N.; Casper, T.C.; Ness, J.; Wheeler, Y.; Chitnis, T.; Lotze, T.; Gorman, M.; Benson, L.; Weinstock-Guttmann, B.; Aaen, G.; et al. Demographic Features and Clinical Course of Patients With Pediatric-Onset Multiple Sclerosis on Newer Disease-Modifying Treatments. Pediatr. Neurol. 2023, 145, 125–131. [Google Scholar] [CrossRef]
- Krysko, K.M.; Graves, J.S.; Rensel, M.; Weinstock-Guttman, B.; Rutatangwa, A.; Aaen, G.; Belman, A.; Benson, L.; Chitnis, T.; Gorman, M.; et al. Real-World Effectiveness of Initial Disease-Modifying Therapies in Pediatric Multiple Sclerosis. Ann. Neurol. 2020, 88, 42–55. [Google Scholar] [CrossRef]
- Salzer, J.; Lycke, J.; Wickström, R.; Naver, H.; Piehl, F.; Svenningsson, A. Rituximab in Paediatric Onset Multiple Sclerosis: A Case Series. J. Neurol. 2016, 263, 322–326. [Google Scholar] [CrossRef]
- Ghezzi, A.; Banwell, B.; Bar-Or, A.; Chitnis, T.; Dale, R.C.; Gorman, M.; Kornek, B.; Krupp, L.; Krysko, K.M.; Nosadini, M.; et al. Rituximab in Patients with Pediatric Multiple Sclerosis and Other Demyelinating Disorders of the CNS: Practical Considerations. Mult. Scler. 2021, 27, 1814–1822. [Google Scholar] [CrossRef]
- Breu, M.; Sandesjö, F.; Milos, R.-I.; Svoboda, J.; Salzer, J.; Schneider, L.; Reichelt, J.B.; Bertolini, A.; Blaschek, A.; Fink, K.; et al. Rituximab Treatment in Pediatric-Onset Multiple Sclerosis. Eur. J. Neurol. 2024, 31, e16228. [Google Scholar] [CrossRef]
- McAtee, C.L.; Lubega, J.; Underbrink, K.; Curry, K.; Msaouel, P.; Barrow, M.; Muscal, E.; Lotze, T.; Srivaths, P.; Forbes, L.R.; et al. Association of Rituximab Use With Adverse Events in Children, Adolescents, and Young Adults. JAMA Netw. Open 2021, 4, e2036321. [Google Scholar] [CrossRef]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef]
- Nicotera, A.G.; Spoto, G.; Saia, M.C.; Midiri, M.; Turriziani, L.; Amore, G.; Di Rosa, G. Treatment of Multiple Sclerosis in Children: A Brief Overview. Clin. Immunol. 2022, 237, 108947. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.-P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Mar, S.; Valeriani, M.; Steinborn, B.; Schreiner, T.; Waubant, E.; Filippi, M.; Kotulska, K.; Mazurkiewicz-Beldzinska, M.; El Azzouzi, B.; Lin, C.-J.; et al. Ocrelizumab Dose Selection for Treatment of Pediatric Relapsing–Remitting Multiple Sclerosis: Results of the OPERETTA I Study. J. Neurol. 2025, 272, 137. [Google Scholar] [CrossRef]
- Hoffmann-La Roche. A Phase III Multicenter, Randomized, Double-Blind, Double-Dummy Study to Evaluate Safety and Efficacy of Ocrelizumab in Comparison with Fingolimod in Children and Adolescents with Relapsing-Remitting Multiple Sclerosis; Hoffmann-La Roche: Basel, Switzerland, 2025. Available online: https://clinicaltrials.gov/ (accessed on 6 April 2025).
- Masoud, S.; McAdoo, S.P.; Bedi, R.; Cairns, T.D.; Lightstone, L. Ofatumumab for B Cell Depletion in Patients with Systemic Lupus Erythematosus Who Are Allergic to Rituximab. Rheumatology 2018, 57, 1156–1161. [Google Scholar] [CrossRef]
- Ofatumumab. Am. J. Health-Syst. Pharm. 2020, 77, 2025–2028. [CrossRef]
- Hagenbeek, A.; Gadeberg, O.; Johnson, P.; Pedersen, L.M.; Walewski, J.; Hellmann, A.; Link, B.K.; Robak, T.; Wojtukiewicz, M.; Pfreundschuh, M.; et al. First Clinical Use of Ofatumumab, a Novel Fully Human Anti-CD20 Monoclonal Antibody in Relapsed or Refractory Follicular Lymphoma: Results of a Phase 1/2 Trial. Blood 2008, 111, 5486–5495. [Google Scholar] [CrossRef]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Simpson, A.; Mowry, E.M.; Newsome, S.D. Early Aggressive Treatment Approaches for Multiple Sclerosis. Curr. Treat. Options Neurol. 2021, 23, 19. [Google Scholar] [CrossRef]
- Gärtner, J.; Hauser, S.L.; Bar-Or, A.; Montalban, X.; Cohen, J.A.; Cross, A.H.; Deiva, K.; Ganjgahi, H.; Häring, D.A.; Li, B.; et al. Efficacy and Safety of Ofatumumab in Recently Diagnosed, Treatment-Naive Patients with Multiple Sclerosis: Results from ASCLEPIOS I and II. Mult. Scler. 2022, 28, 1562–1575. [Google Scholar] [CrossRef]
- Hauser, S.L.; Cross, A.H.; Winthrop, K.; Wiendl, H.; Nicholas, J.; Meuth, S.G.; Giacomini, P.S.; Saccà, F.; Mancione, L.; Zielman, R.; et al. Safety Experience with Continued Exposure to Ofatumumab in Patients with Relapsing Forms of Multiple Sclerosis for up to 3.5 Years. Mult. Scler. 2022, 28, 1576–1590. [Google Scholar] [CrossRef]
- Kuzminykh, E.D.; Lebedev, V.M.; Senchenko, V.E.; Cherepyansky, M.S.; Gonchar, V.A.; Korobko, D.S. The first experience with the use of ofatumumab for the treatment of pediatric multiple sclerosis in real-life clinical practice in Russia. A case series. Neurol. Neuropsychiatry Psychosom. 2025, 17, 72–77. [Google Scholar] [CrossRef]
- Novartis Pharmaceuticals. A 2-Year Randomized, 3-Arm, Double-Blind, Non-Inferiority Study Comparing the Efficacy and Safety of Ofatumumab and Siponimod Versus Fingolimod in Pediatric Patients with Multiple Sclerosis Followed by an Open-Label Extension; Novartis Pharmaceuticals: Tokyo, Japan, 2025. Available online: https://clinicaltrials.gov/ (accessed on 6 April 2025).
- Martin, R. Anti-CD25 (Daclizumab) Monoclonal Antibody Therapy in Relapsing-Remitting Multiple Sclerosis. Clin. Immunol. 2012, 142, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Baldassari, L.E.; Rose, J.W. Daclizumab: Development, Clinical Trials, and Practical Aspects of Use in Multiple Sclerosis. Neurotherapeutics 2017, 14, 842–858. [Google Scholar] [CrossRef] [PubMed]
- Gorman, M.P.; Tillema, J.-M.; Ciliax, A.M.; Guttmann, C.R.G.; Chitnis, T. Daclizumab Use in Patients with Pediatric Multiple Sclerosis. Arch. Neurol. 2012, 69, 78–81. [Google Scholar] [CrossRef]
- Cohen, J.A.; Coles, A.J.; Arnold, D.L.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; Fisher, E.; et al. Alemtuzumab versus Interferon Beta 1a as First-Line Treatment for Patients with Relapsing-Remitting Multiple Sclerosis: A Randomised Controlled Phase 3 Trial. Lancet 2012, 380, 1819–1828. [Google Scholar] [CrossRef]
- Coles, A.J.; Twyman, C.L.; Arnold, D.L.; Cohen, J.A.; Confavreux, C.; Fox, E.J.; Hartung, H.-P.; Havrdova, E.; Selmaj, K.W.; Weiner, H.L.; et al. Alemtuzumab for Patients with Relapsing Multiple Sclerosis after Disease-Modifying Therapy: A Randomised Controlled Phase 3 Trial. Lancet 2012, 380, 1829–1839. [Google Scholar] [CrossRef]
- Berger, T.; Elovaara, I.; Fredrikson, S.; McGuigan, C.; Moiola, L.; Myhr, K.-M.; Oreja-Guevara, C.; Stoliarov, I.; Zettl, U.K. Alemtuzumab Use in Clinical Practice: Recommendations from European Multiple Sclerosis Experts. CNS Drugs 2017, 31, 33–50. [Google Scholar] [CrossRef]
- Chitnis, T.; Arnold, D.L.; Quartier, P.; Chirieac, M.; Hu, W.; Jurgensen, S.; Havrdova, E.K. Safety, Efficacy, and Tolerability of Alemtuzumab in Pediatric Patients with Active Relapsing-Remitting Multiple Sclerosis: The LemKids Study. Mult. Scler. 2025, 31, 23–35. [Google Scholar] [CrossRef]
- Margoni, M.; Rinaldi, F.; Miante, S.; Franciotta, S.; Perini, P.; Gallo, P. Alemtuzumab Following Natalizumab in Highly Active Paediatric-Onset Multiple Sclerosis. Mult. Scler. J. Exp. Transl. Clin. 2019, 5, 2055217319875471. [Google Scholar] [CrossRef]
- Jure Hunt, D.; Traboulsee, A. Short-Term Outcomes of Pediatric Multiple Sclerosis Patients Treated with Alemtuzumab at a Canadian University Multiple Sclerosis Clinic. Mult. Scler. J. Exp. Transl. Clin. 2020, 6, 2055217320926613. [Google Scholar] [CrossRef]
- Zmira, O.; Halpern, A.I.; Abraham, L.; Achiron, A. Efficacy and Safety of Alemtuzumab Treatment in a Real-World Cohort of Patients with Multiple Sclerosis. Acta Neurol. Belg. 2021, 121, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Frau, J.; Coghe, G.; Lorefice, L.; Fenu, G.; Musu, L.; Cocco, E. Efficacy and Safety of Alemtuzumab in a Real-Life Cohort of Patients with Multiple Sclerosis. J. Neurol. 2019, 266, 1405–1411. [Google Scholar] [CrossRef] [PubMed]
- Lemtrada-Referral|European Medicines Agency (EMA). Available online: https://www.ema.europa.eu/en/medicines/human/referrals/lemtrada (accessed on 27 May 2025).
- Tedder, T.F. CD19: A Promising B Cell Target for Rheumatoid Arthritis. Nat. Rev. Rheumatol. 2009, 5, 572–577. [Google Scholar] [CrossRef] [PubMed]
- Kanatas, P.; Stouras, I.; Stefanis, L.; Stathopoulos, P. B-Cell-Directed Therapies: A New Era in Multiple Sclerosis Treatment. Can. J. Neurol. Sci. 2023, 50, 355–364. [Google Scholar] [CrossRef]
- Chen, D.; Gallagher, S.; Monson, N.L.; Herbst, R.; Wang, Y. Inebilizumab, a B Cell-Depleting Anti-CD19 Antibody for the Treatment of Autoimmune Neurological Diseases: Insights from Preclinical Studies. J. Clin. Med. 2016, 5, 107. [Google Scholar] [CrossRef]
- Schuh, E.; Berer, K.; Mulazzani, M.; Feil, K.; Meinl, I.; Lahm, H.; Krane, M.; Lange, R.; Pfannes, K.; Subklewe, M.; et al. Features of Human CD3+CD20+ T Cells. J. Immunol. 2016, 197, 1111–1117. [Google Scholar] [CrossRef]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-Cell Depletion in Vitro and in Vivo with an Afucosylated Anti-CD19 Antibody. J. Pharmacol. Exp. Ther. 2010, 335, 213–222. [Google Scholar] [CrossRef]
- Agius, M.A.; Klodowska-Duda, G.; Maciejowski, M.; Potemkowski, A.; Li, J.; Patra, K.; Wesley, J.; Madani, S.; Barron, G.; Katz, E.; et al. Safety and Tolerability of Inebilizumab (MEDI-551), an Anti-CD19 Monoclonal Antibody, in Patients with Relapsing Forms of Multiple Sclerosis: Results from a Phase 1 Randomised, Placebo-Controlled, Escalating Intravenous and Subcutaneous Dose Study. Mult. Scler. 2019, 25, 235–245. [Google Scholar] [CrossRef]
- Eilertsen, G.Ø.; Van Ghelue, M.; Strand, H.; Nossent, J.C. Increased Levels of BAFF in Patients with Systemic Lupus Erythematosus Are Associated with Acute-Phase Reactants, Independent of BAFF Genetics: A Case-Control Study. Rheumatology 2011, 50, 2197–2205. [Google Scholar] [CrossRef]
- Flessa, C.-M.; Zampeli, E.; Evangelopoulos, M.-E.; Natsis, V.; Bodewes, I.L.A.; Huijser, E.; Versnel, M.A.; Moutsopoulos, H.M.; Mavragani, C.P. Genetic Variants of the BAFF Gene and Risk of Fatigue Among Patients With Primary Sjögren’s Syndrome. Front. Immunol. 2022, 13, 836824. [Google Scholar] [CrossRef]
- Steri, M.; Orrù, V.; Idda, M.L.; Pitzalis, M.; Pala, M.; Zara, I.; Sidore, C.; Faà, V.; Floris, M.; Deiana, M.; et al. Overexpression of the Cytokine BAFF and Autoimmunity Risk. N. Engl. J. Med. 2017, 376, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Skarlis, C.; Marketos, N.; Mavragani, C.P. Biologics in Sjögren’s Syndrome. Pharmacol. Res. 2019, 147, 104389. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, K.; Zhong, X.; Qiu, W.; Dai, Y.; Wu, A.; Hu, X. Cerebrospinal Fluid BAFF and APRIL Levels in Neuromyelitis Optica and Multiple Sclerosis Patients during Relapse. J. Clin. Immunol. 2012, 32, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Skarlis, C.; Papadopoulos, V.; Raftopoulou, S.; Mavragani, C.P.; Evangelopoulos, M.-E. Association of B-Cell Activating Factor Gene Variants with Serum Anti-JCV Antibody Positivity in Male Patients with Multiple Sclerosis under Natalizumab Treatment: Implications for Progressive Multifocal Leukoencephalopathy Risk Stratification. J. Neurol. Sci. 2024, 461, 123046. [Google Scholar] [CrossRef]
- Magliozzi, R.; Marastoni, D.; Calabrese, M. The BAFF / APRIL System as Therapeutic Target in Multiple Sclerosis. Expert. Opin. Ther. Targets 2020, 24, 1135–1145. [Google Scholar] [CrossRef]
- Benson, M.J.; Dillon, S.R.; Castigli, E.; Geha, R.S.; Xu, S.; Lam, K.-P.; Noelle, R.J. Cutting Edge: The Dependence of Plasma Cells and Independence of Memory B Cells on BAFF and APRIL. J. Immunol. 2008, 180, 3655–3659. [Google Scholar] [CrossRef]
- Kappos, L.; Hartung, H.-P.; Freedman, M.S.; Boyko, A.; Radü, E.W.; Mikol, D.D.; Lamarine, M.; Hyvert, Y.; Freudensprung, U.; Plitz, T.; et al. Atacicept in Multiple Sclerosis (ATAMS): A Randomised, Placebo-Controlled, Double-Blind, Phase 2 Trial. Lancet Neurol. 2014, 13, 353–363. [Google Scholar] [CrossRef]
- Dingjan, G.M.; Middendorp, S.; Dahlenborg, K.; Maas, A.; Grosveld, F.; Hendriks, R.W. Bruton’s Tyrosine Kinase Regulates the Activation of Gene Rearrangements at the Lambda Light Chain Locus in Precursor B Cells in the Mouse. J. Exp. Med. 2001, 193, 1169–1178. [Google Scholar] [CrossRef]
- Krämer, J.; Bar-Or, A.; Turner, T.J.; Wiendl, H. Bruton Tyrosine Kinase Inhibitors for Multiple Sclerosis. Nat. Rev. Neurol. 2023, 19, 289–304. [Google Scholar] [CrossRef]
- Martin, E.; Aigrot, M.-S.; Grenningloh, R.; Stankoff, B.; Lubetzki, C.; Boschert, U.; Zalc, B. Bruton’s Tyrosine Kinase Inhibition Promotes Myelin Repair. Brain Plast. 2019, 5, 123–133. [Google Scholar] [CrossRef]
- Montalban, X.; Arnold, D.L.; Weber, M.S.; Staikov, I.; Piasecka-Stryczynska, K.; Willmer, J.; Martin, E.C.; Dangond, F.; Syed, S.; Wolinsky, J.S.; et al. Placebo-Controlled Trial of an Oral BTK Inhibitor in Multiple Sclerosis. N. Engl. J. Med. 2019, 380, 2406–2417. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Vermersch, P.; Arnold, D.L.; Bar-Or, A.; Cree, B.A.C.; Cross, A.H.; Kubala Havrdova, E.; Kappos, L.; Stuve, O.; Wiendl, H.; et al. Safety and Efficacy of Evobrutinib in Relapsing Multiple Sclerosis (evolutionRMS1 and evolutionRMS2): Two Multicentre, Randomised, Double-Blind, Active-Controlled, Phase 3 Trials. Lancet Neurol. 2024, 23, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.J.; Bar-Or, A.; Traboulsee, A.; Oreja-Guevara, C.; Giovannoni, G.; Vermersch, P.; Syed, S.; Li, Y.; Vargas, W.S.; Turner, T.J.; et al. Tolebrutinib in Nonrelapsing Secondary Progressive Multiple Sclerosis. N. Engl. J. Med. 2025, 392, 1883–1892. [Google Scholar] [CrossRef]
- Oh, J.; Arnold, D.L.; Cree, B.A.C.; Ionete, C.; Kim, H.J.; Sormani, M.P.; Syed, S.; Chen, Y.; Maxwell, C.R.; Benoit, P.; et al. Tolebrutinib versus Teriflunomide in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2025, 392, 1893–1904. [Google Scholar] [CrossRef]
- Haselmayer, P.; Camps, M.; Liu-Bujalski, L.; Nguyen, N.; Morandi, F.; Head, J.; O’Mahony, A.; Zimmerli, S.C.; Bruns, L.; Bender, A.T.; et al. Efficacy and Pharmacodynamic Modeling of the BTK Inhibitor Evobrutinib in Autoimmune Disease Models. J. Immunol. 2019, 202, 2888–2906. [Google Scholar] [CrossRef]
- Skarlis, C.; Markoglou, N.; Gontika, M.; Bougea, A.; Katsavos, S.; Artemiadis, A.; Chrousos, G.; Dalakas, M.; Stefanis, L.; Anagnostouli, M. First-Line Disease Modifying Treatments in Pediatric-Onset Multiple Sclerosis in Greece: Therapy Initiation at More Advanced Age Is the Main Cause of Treatment Failure, in a Retrospective Observational Study, with a Cohort from a Single Multiple Sclerosis Center. Neurol. Sci. 2023, 44, 693–701. [Google Scholar] [CrossRef]
- Gontika, M.; Skarlis, C.; Markoglou, N.; Evangelopoulos, M.-E.; Velonakis, G.; Chrousos, G.P.; Dalakas, M.; Stefanis, L.; Anagnostouli, M. Fingolimod as a First- or Second-Line Treatment in a Mini-Series of Young Hellenic Patients with Adolescent-Onset Multiple Sclerosis: Focus on Immunological Data. Neurol. Sci. 2022, 43, 2641–2649. [Google Scholar] [CrossRef]
- Gontika, M.; Skarlis, C.; Markoglou, N.; Tzanetakos, D.; Vakrakou, A.; Toulas, P.; Koutsis, G.; Evangelopoulos, M.-E.; Pons, R.; Dardiotis, E.; et al. Natalizumab Therapy in Patients with Pediatric-Onset Multiple Sclerosis in Greece: Clinical and Immunological Insights of Time-Long Administration and Future Directions-a Single-Center Retrospective Observational Study. Naunyn Schmiedebergs Arch. Pharmacol. 2022, 395, 933–943. [Google Scholar] [CrossRef]
- Jakimovski, D.; Awan, S.; Eckert, S.P.; Farooq, O.; Weinstock-Guttman, B. Multiple Sclerosis in Children: Differential Diagnosis, Prognosis, and Disease-Modifying Treatment. CNS Drugs 2022, 36, 45–59. [Google Scholar] [CrossRef]
- Sharmin, S.; Roos, I.; Malpas, C.B.; Iaffaldano, P.; Simone, M.; Filippi, M.; Kubala Havrdova, E.; Ozakbas, S.; Brescia Morra, V.; Alroughani, R.; et al. Disease-Modifying Therapies in Managing Disability Worsening in Paediatric-Onset Multiple Sclerosis: A Longitudinal Analysis of Global and National Registries. Lancet Child Adolesc. Health 2024, 8, 348–357. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Feature | POMS | AOMS | References |
|---|---|---|---|
| Age at Disease Onset | <18 years | ≥18 years | [21,22,23,24] |
| Initial Disease Course | Almost exclusively RRMS | Relapsing or progressive | [21,22,23,24] |
| Inflammatory Activity | Higher—more aggressive inflammatory onset | Often less inflammatory at the onset | [21,22,23,24] |
| Relapse Rate | Higher (2.3–2.8 times more frequent) | Lower | [21,22,23,24] |
| Number of Demyelinating Lesions | Increased | Fewer | [21,22,23,24] |
| Axonal/Neuroaxonal Damage | More pronounced | Less pronounced | [24] |
| Disability Accumulation Rate | Slower initially | Faster initial progression | [21,22,23,24] |
| Transition to SPMS | Approximately 10 years earlier than in AOMS | Later transition | [22] |
| Final Disability Milestone | Reached at a younger age due to early onset | Reached at older age | [21,22,23,24] |
| HLA-DRB1* | Predominantly HLA-DRB1*03 and classically HLA-DRB1*15:0 in some populations | HLA-DRB1*15:01 | [14] |
| Therapy | Mechanism of Action | Efficacy in POMS | Safety Concerns | Current Clinical Trials Status in POMS |
|---|---|---|---|---|
| Rituximab | A chimeric anti-CD20 monoclonal antibody that depletes B-cells through ADCC, CDC, and apoptosis | Shown to reduce relapse rates, MRI activity, and disability progression in retrospective studies | Infusion reactions, infections, rare risk of PML | No approved trials in the pediatric MS population |
| Ocrelizumab | Humanized anti-CD20 monoclonal antibody with enhanced ADCC activity | Promising preliminary results from the OPERETTA trials; sustained B-cell depletion and reduced relapses | Infusion-related reactions, infections, rare risk of malignancies | Active, not recruiting |
| Ofatumumab | Fully human anti-CD20 monoclonal antibody with higher CDC activity than rituximab | Shown to reduce ARR and MRI lesions in adults; limited POMS data | Injection-site reactions, infections, possible immune suppression | Ongoing phase II trial in POMS (NCT04926818, NEOS) |
| Alemtuzumab | Anti-CD52 monoclonal antibody leading to depletion of T- and B-cells | Small studies suggest effectiveness, but safety concerns limit the use | Autoimmune disorders (thyroid, ITP), infusion reactions | Early termination of the NCT03368664 LemKIds study |
| BTK Inhibitors | BTK, reducing B-cell activation and microglia-mediated inflammation | Early trials show promise in adult MS; no POMS-specific trials yet | Mild liver enzyme elevations, infections | No approved pediatric trials yet |
| Anti-CD19 therapies (Inebilizumab) | Targets CD19, affecting a broader range of B-cell subsets than anti-CD20 therapies | Early-phase adult MS trials are ongoing; no pediatric data yet | Immunosuppression, infection risk | No approved pediatric trials yet |
| Anti-CD25 therapies (Daclizumab) | Monoclonal antibody targeting interleukin (IL)-2 receptor alpha (IL-2Rα) chain (CD25), leading to a reduction in activated T-cells | No clinical trials, only case series | Fatal encephalitis in adult clinical trials | No approved pediatric trials |
| Clinical Trial | Therapy | Study Design | Primary Outcomes | Status |
|---|---|---|---|---|
| OPERETTA I (NCT04075266) | Ocrelizumab | Phase II open-label | Pharmacokinetics, safety, ARR reduction | Active, not recruiting |
| OPERETTA II (NCT05123703) | Ocrelizumab vs. Fingolimod | Phase III randomized | ARR reduction, MRI outcomes | Active, not recruiting |
| NEOS (NCT04926818) | Ofatumumab vs. Siponimod vs. Fingolimod | Phase II randomized | ARR reduction, MRI lesion burden | Active, not recruiting |
| LemKids (NCT03368664) | Alemtuzumab | Open-label | Reduction in MRI activity, ARR reduction | Early termination |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skarlis, C.; Kotsari, M.; Anagnostouli, M. Advancing Treatment in Pediatric Multiple Sclerosis: The Promise of B-Cell-Targeting Therapies. Int. J. Mol. Sci. 2025, 26, 5989. https://doi.org/10.3390/ijms26135989
Skarlis C, Kotsari M, Anagnostouli M. Advancing Treatment in Pediatric Multiple Sclerosis: The Promise of B-Cell-Targeting Therapies. International Journal of Molecular Sciences. 2025; 26(13):5989. https://doi.org/10.3390/ijms26135989
Chicago/Turabian StyleSkarlis, Charalampos, Maria Kotsari, and Maria Anagnostouli. 2025. "Advancing Treatment in Pediatric Multiple Sclerosis: The Promise of B-Cell-Targeting Therapies" International Journal of Molecular Sciences 26, no. 13: 5989. https://doi.org/10.3390/ijms26135989
APA StyleSkarlis, C., Kotsari, M., & Anagnostouli, M. (2025). Advancing Treatment in Pediatric Multiple Sclerosis: The Promise of B-Cell-Targeting Therapies. International Journal of Molecular Sciences, 26(13), 5989. https://doi.org/10.3390/ijms26135989