
A Unique Case of a Child with Two Rare Hereditary Diseases: Familial Dilated Cardiomyopathy and Arterial Calcification


 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
Genetic Analysis
3. Results
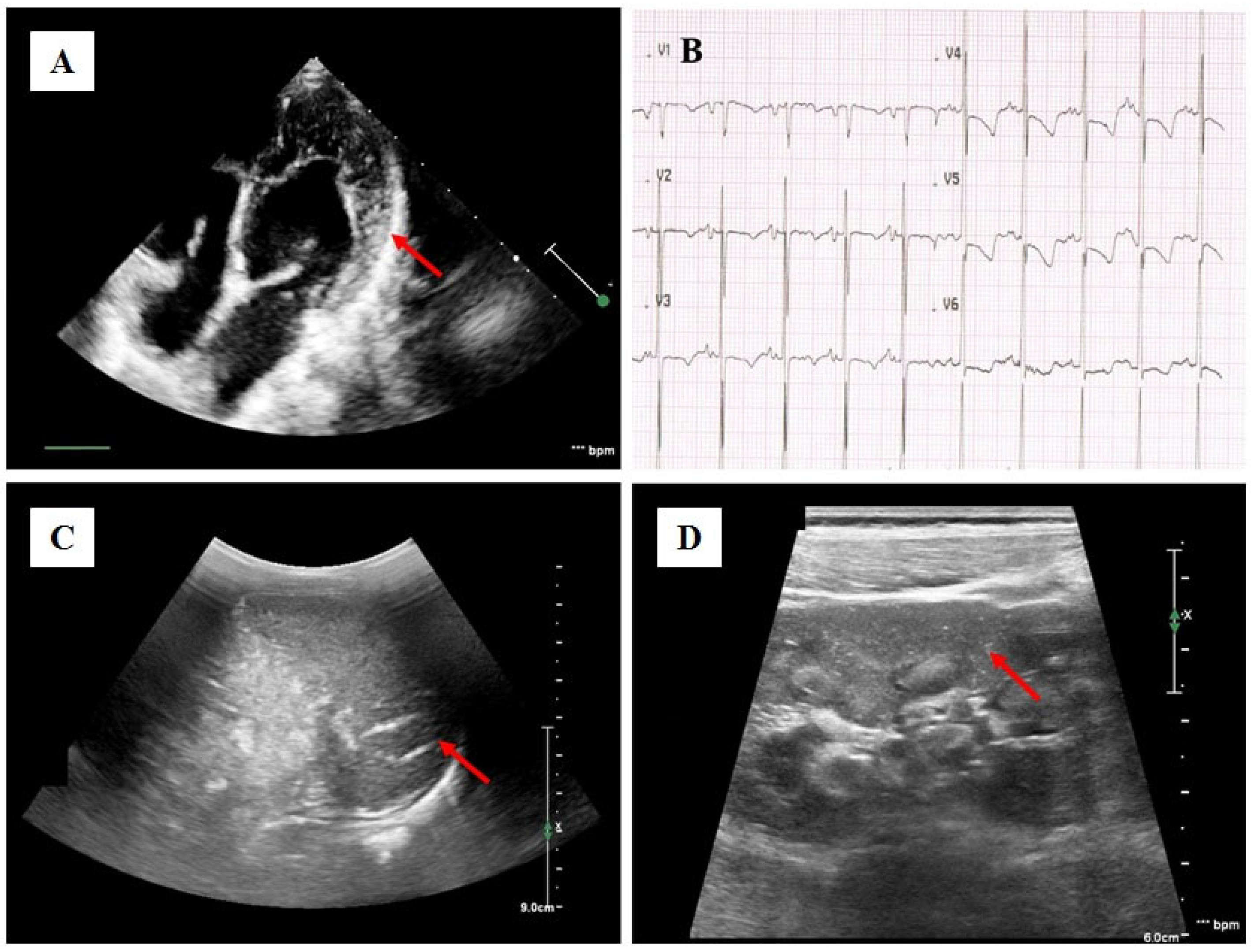
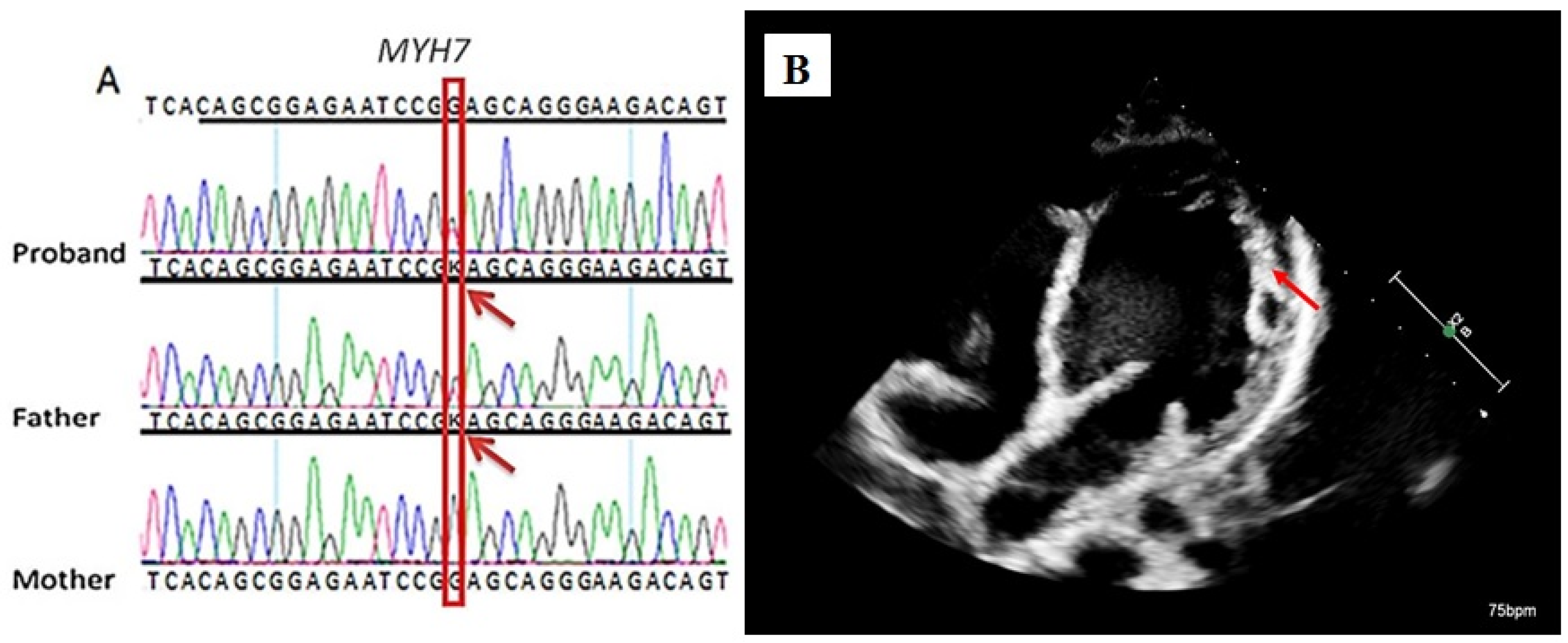
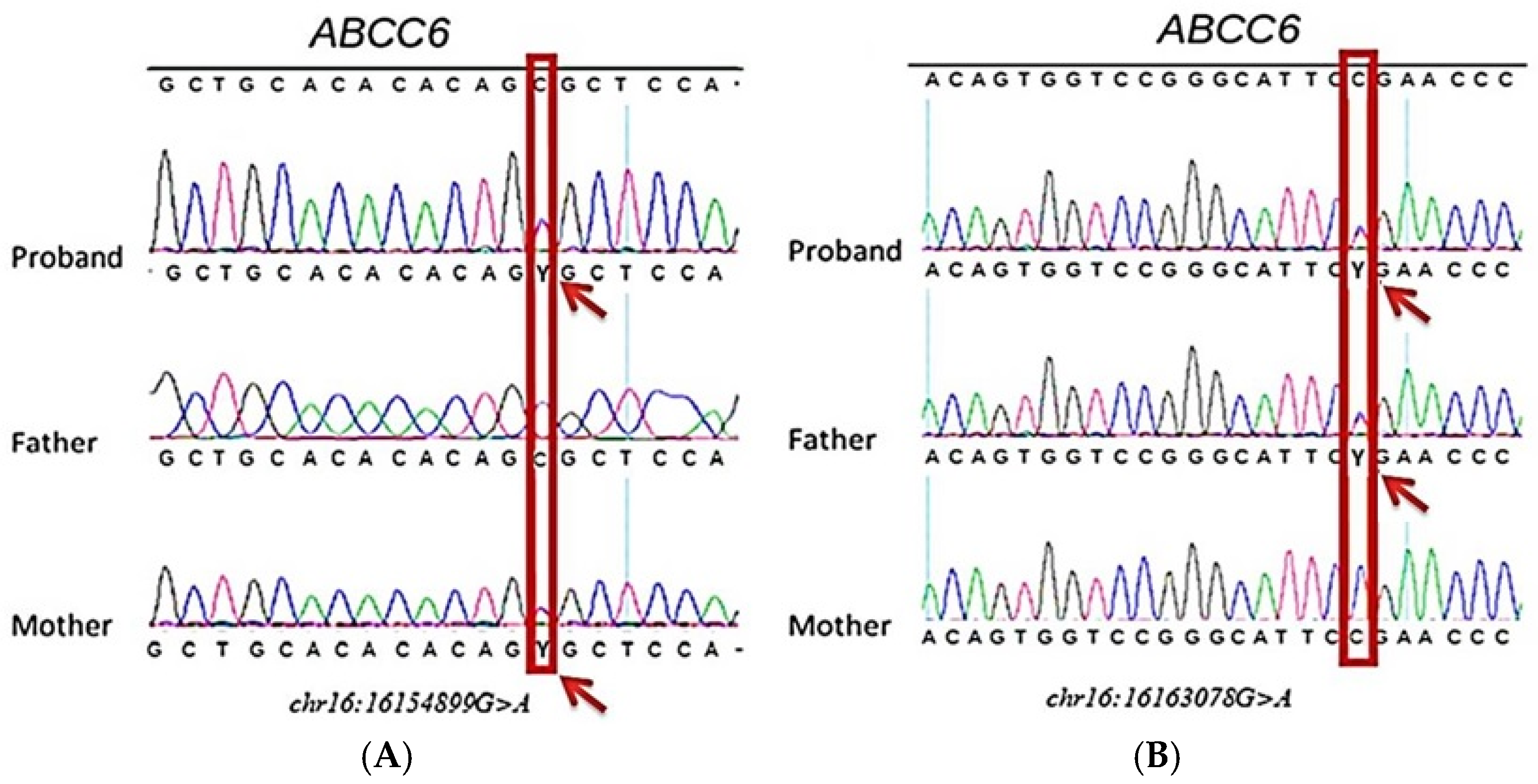
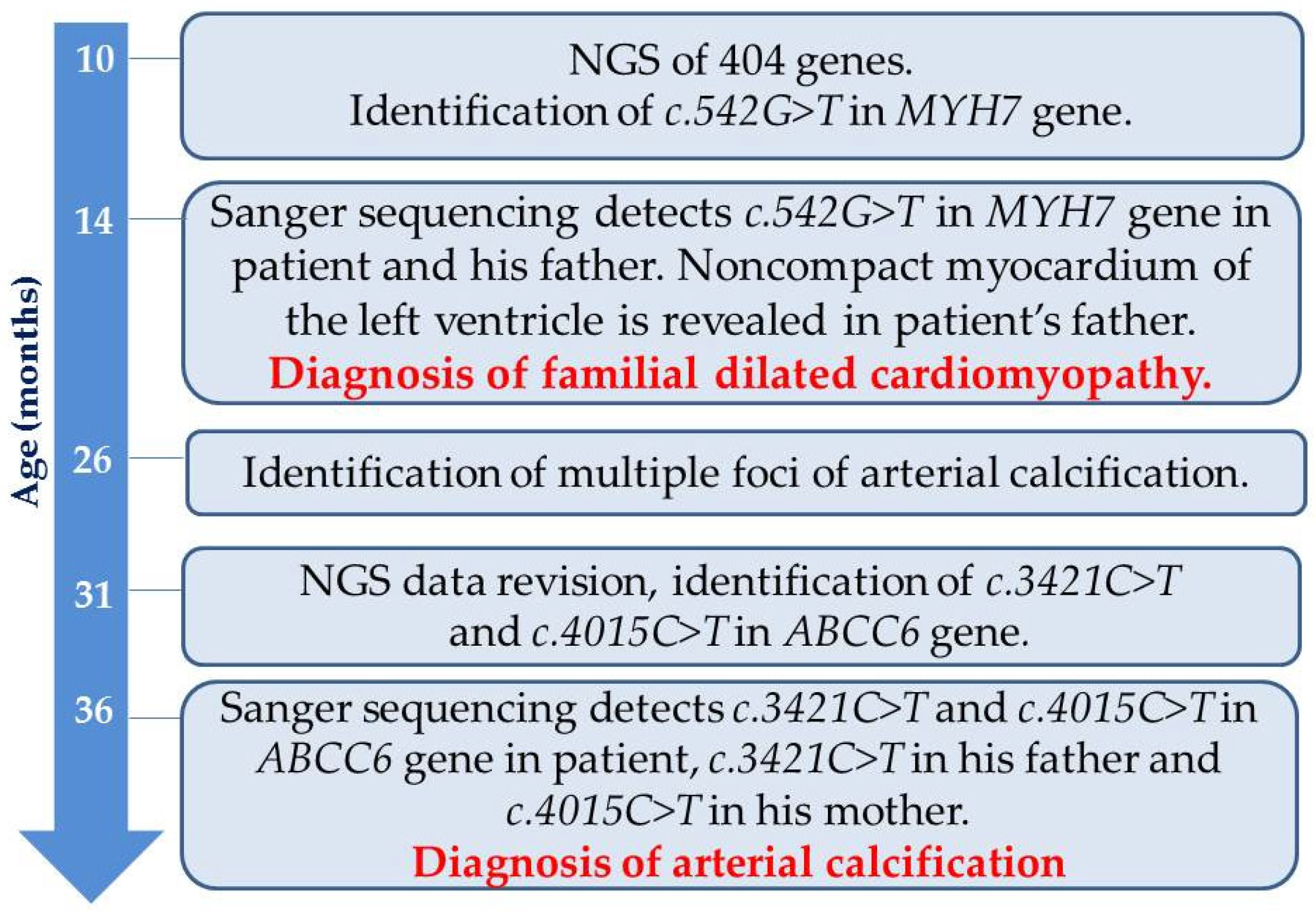
Case Presentation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABC | ATP binding cassette |
| AC | arterial calcification |
| CHF | chronic heart failure |
| DCM | dilated cardiomyopathy |
| ECG | electrocardiography |
| EchoCG | echocardiography |
| EF | ejection fraction |
| ESC | European Society of Cardiology |
| GACI | generalized arterial calcification of infancy |
| HM-ECG | Holter monitoring of ECG |
| IHD | ischemic heart disease |
| MSCT | multislice computed tomography |
| MRP6 | Multi-Drug Resistance-Associated Protein-6 |
| MRI | magnetic resonance imaging |
| NT | proBNP—N-terminal prohormone of brain natriuretic peptide |
| NV | nucleotide variant |
| NGS | next-generation sequencing |
| P/LP | pathogenic/likely pathogenic |
| PPi | inorganic pyrophosphate |
| PXE | pseudoxanthoma elasticum |
| RR | respiratory rate |
References
- Dellefave, L.; McNally, E.M. The genetics of dilated cardiomyopathy. Curr. Opin. Cardiol. 2010, 25, 198–204. [Google Scholar] [CrossRef]
- Rampersaud, E.; Siegfried, J.D.; Norton, N.; Li, D.; Martin, E.; Hershberger, R.E. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog. Pediatr. Cardiol. 2011, 31, 39–47. [Google Scholar] [CrossRef]
- Malinow, I.; Fong, D.C.; Miyamoto, M.; Badran, S.; Hong, C.C. Pediatric dilated cardiomyopathy: A review of current clinical approaches and pathogenesis. Front. Pediatr. 2024, 12, 1404942. [Google Scholar] [CrossRef]
- de Frutos, F.; Ochoa, J.P.; Navarro-Peñalver, M.; Baas, A.; Bjerre, J.V.; Zorio, E.; Méndez, I.; Lorca, R.; Verdonschot, J.A.J.; García-Granja, P.E.; et al. Natural History of MYH7-Related Dilated Cardiomyopathy. J. Am. Coll. Cardiol. 2022, 80, 1447–1461. [Google Scholar] [CrossRef]
- Pang, J.; Bao, Y.; Mitchell-Silbaugh, K.; Veevers, J.; Fang, X. Barth Syndrome Cardiomyopathy: An Update. Genes 2022, 13, 656. [Google Scholar] [CrossRef]
- Bea-Mascato, B.; Valverde, D. Genotype-phenotype associations in Alström syndrome: A systematic review and meta-analysis. J. Med. Genet. 2023, 61, 18–26. [Google Scholar] [CrossRef]
- Kaur, T.; Sriram, C.S.; Prasanna, P.; Kohli, U. Cardiovascular Phenotypic Spectrum of 1p36 Deletion Syndrome. J. Pediatr. Genet. 2021, 12, 329–334. [Google Scholar] [CrossRef]
- Boleti, O.; Norrish, G.; Field, E.; Dady, K.; Summers, K.; Nepali, G.; Bhole, V.; Uzun, O.; Wong, A.; Daubeney, P.E.F.; et al. Natural history and outcomes in paediatric RASopathy-associated hypertrophic cardiomyopathy. ESC Heart Fail. 2024, 11, 923–936. [Google Scholar] [CrossRef]
- Gandaeva, L.; Sonicheva-Paterson, N.; McKenna, W.J.; Savostyanov, K.; Myasnikov, R.; Pushkov, A.; Zhanin, I.; Barskiy, V.; Zharova, O.; Silnova, I.; et al. Clinical features of pediatric Danon disease and the importance of early diagnosis. Int. J. Cardiol. 2023, 389, 131189. [Google Scholar] [CrossRef]
- Savostyanov, K.; Pushkov, A.; Zhanin, I.; Mazanova, N.; Trufanov, S.; Pakhomov, A.; Alexeeva, A.; Sladkov, D.; Asanov, A.; Fisenko, A. The prevalence of Fabry disease among 1009 unrelated patients with hypertrophic cardiomyopathy: A Russian nationwide screening program using NGS technology. Orphanet J. Rare Dis. 2022, 17, 199. [Google Scholar] [CrossRef]
- Khayat, A.M.; Alshareef, B.G.; Alharbi, S.F.; AlZahrani, M.M.; Alshangity, B.A.; Tashkandi, N.F. Consanguineous Marriage and Its Association with Genetic Disorders in Saudi Arabia: A Review. Cureus 2024, 16, e53888. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ebbinghaus, H.; Ueberham, L.; Husser-Bollmann, D.; Bollmann, A.; Paetsch, I.; Jahnke, C.; Laufs, U.; Dinov, B. Corrigendum: Case Report: Four Cases of Cardiac Sarcoidosis in Patients with Inherited Cardiomyopathy—A Phenotypic Overlap, Co-Existence of Two Rare Cardiomyopathies or a Second-Hit Disease. Front. Cardiovasc. Med. 2024, 11, 1372782. [Google Scholar] [CrossRef]
- Bukaeva, A.; Myasnikov, R.; Kulikova, O.; Meshkov, A.; Kiseleva, A.; Petukhova, A.; Zotova, E.; Sparber, P.; Ershova, A.; Sotnikova, E.; et al. A Rare Coincidence of Three Inherited Diseases in a Family with Cardiomyopathy and Multiple Extracardiac Abnormalities. Int. J. Mol. Sci. 2024, 25, 7556. [Google Scholar] [CrossRef]
- Kablan, A.; Erturkmen Aru, E. Kabuki Syndrome and Charcot-Marie-Tooth Disease Co-Occurrence: Unique Case with Novel Variant. Mol. Syndromol. 2025, 16, 55–60. [Google Scholar] [CrossRef]
- Samuels, M.; Shields, K.; Hillman, P.; Farach, L. Rapid Whole Genome Sequencing Uncovers a Triple Diagnosis: X-Linked Chondrodysplasia Punctata, MECP2-Related Disorder, and Mosaic Jacobs Syndrome. Mol. Genet. Genom. Med. 2025, 13, e70061. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Guo, F.; Liu, R.; Pan, Y.; Colasanto, M.; Collins, C.; Hegde, M. Beyond Single Diagnosis: Exploring Multidiagnostic Realities in Pediatric Patients through Genome Sequencing. Hum. Mutat. 2024, 2024, 9115364. [Google Scholar] [CrossRef]
- Spedicati, B.; Morgan, A.; Pianigiani, G.; Musante, L.; Rubinato, E.; Santin, A.; Nardone, G.G.; Faletra, F.; Girotto, G. Challenging Occam’s Razor: Dual Molecular Diagnoses Explain Entangled Clinical Pictures. Genes 2022, 13, 2023. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ge, S.; Sun, L.; Wang, H.; Yang, W.; Xuan, Q.; Hua, D. Case report: Co-occurrence of Wilson’s and Alexander’s diseases revealed by genetic analysis. Front. Neurol. 2025, 16, 1514044. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Rayment, I.; Rypniewski, W.R.; Schmidt-Base, K.; Smith, R.; Tomchick, D.R.; Benning, M.M.; Winkelmann, D.A.; Wesenberg, G.; Holden, H.M. Three-dimensional structure of myosin subfragment-1: A molecular motor. Science 1993, 261, 50–58. [Google Scholar] [CrossRef]
- Alamo, L.; Ware, J.S.; Pinto, A.; Gillilan, R.E.; Seidman, J.G.; Seidman, C.E.; Padrón, R. Effects of myosin variants on interacting-heads motif explain distinct hypertrophic and dilated cardiomyopathy phenotypes. eLife 2017, 6, e24634. [Google Scholar] [CrossRef]
- Bundgaard, H.; Havndrup, O.; Andersen, P.S.; Larsen, L.A.; Brandt, N.J.; Vuust, J.; Kjeldsen, K.; Christiansen, M. Familial hypertrophic cardiomyopathy associated with a novel missense mutation affecting the ATP-binding region of the cardiac beta-myosin heavy chain. J. Mol. Cell Cardiol. 1999, 31, 745–750. [Google Scholar] [CrossRef]
- Villard, E.; Duboscq-Bidot, L.; Charron, P.; Benaiche, A.; Conraads, V.; Sylvius, N.; Komajda, M. Mutation screening in dilated cardiomyopathy: Prominent role of the beta myosin heavy chain gene. Eur. Heart J. 2005, 26, 794–803. [Google Scholar] [CrossRef]
- Khan, R.S.; Pahl, E.; Dellefave-Castillo, L.; Rychlik, K.; Ing, A.; Yap, K.L.; Brew, C.; Johnston, J.R.; McNally, E.M.; Webster, G. Genotype and cardiac outcomes in pediatric dilated cardiomyopathy. J. Am. Heart Assoc. 2022, 11, e022854. [Google Scholar] [CrossRef]
- Blair, E.; Redwood, C.; de Jesus Oliveira, M.; Moolman-Smook, J.C.; Brink, P.; Corfield, V.A.; Östman-Smith, I.; Watkins, H. Mutations of the light meromyosin domain of the β-myosin heavy chain rod in hypertrophic cardiomyopathy. Circ. Res. 2002, 90, 263–269. [Google Scholar] [CrossRef]
- Walsh, R.; Rutland, C.; Thomas, R.; Loughna, S. Cardiomyopathy: A systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2009, 115, 49–60. [Google Scholar] [CrossRef]
- Nitschke, Y.; Baujat, G.; Botschen, U.; Wittkampf, T.; du Moulin, M.; Stella, J.; Le Merrer, M.; Guest, G.; Lambot, K.; Tazarourte-Pinturier, M.F.; et al. Generalized arterial calcification of infancy and pseudoxanthoma elasticum can be caused by mutations in either ENPP1 or ABCC6. Am. J. Hum. Genet. 2012, 90, 25–39. [Google Scholar] [CrossRef]
- Jansen, R.S.; Duijst, S.; Mahakena, S.; Sommer, D.; Szeri, F.; Váradi, A.; Plomp, A.; Bergen, A.A.; Oude Elferink, R.P.; Borst, P.; et al. ABCC6–mediated ATP secretion by the liver is the main source of the mineralization inhibitor inorganic pyrophosphate in the systemic circulation—Brief report. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1985–1989. [Google Scholar] [CrossRef]
- Pfendner, E.G.; Uitto, J.; Gerard, G.F.; Terry, S.F. Pseudoxanthoma elasticum: Genetic diagnostic markers. Expert. Opin. Med. Diagn. 2008, 2, 63–79. [Google Scholar] [CrossRef]
- Schott, C.; Dilliott, A.A.; Wang, J.; McIntyre, A.D.; Son, S.; Colaiacovo, S.; Baker, C.; Gunaratnam, L.; House, A.A.; Susan Huang, S.H.; et al. Vascular calcification in chronic kidney disease associated with pathogenic variants in ABCC6. Gene 2024, 927, 148731. [Google Scholar] [CrossRef]
- Köblös, G.; Andrikovics, H.; Prohászka, Z.; Tordai, A.; Váradi, A.; Arányi, T. The R1141X loss-of-function mutation of the ABCC6 gene is a strong genetic risk factor for coronary artery disease. Genet. Test. Mol. Biomark. 2010, 14, 75–78. [Google Scholar] [CrossRef]
- Trip, M.D.; Smulders, Y.M.; Wegman, J.J.; Hu, X.; Boer, J.M.; ten Brink, J.B.; Zwinderman, A.H.; Kastelein, J.J.; Feskens, E.J.; Bergen, A.A. Frequent mutation in the ABCC6 gene (R1141X) is associated with a strong increase in the prevalence of coronary artery disease. Circulation 2002, 106, 773–775. [Google Scholar] [CrossRef] [PubMed]
- Hornstrup, L.S.; Tybjærg-Hansen, A.; Haase, C.L.; Nordestgaard, B.G.; Sillesen, H.; Grande, P.; Frikke-Schmidt, R. Heterozygosity for R1141X in ABCC6 and risk of ischemic vascular disease. Circ. Cardiovasc. Genet. 2011, 4, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Human Gene Mutation Database (HGMD) Professional. Expert-Curated Content to Streamline Variant Classification for Hereditary Workflows. Available online: https://digitalinsights.qiagen.com/products-overview/clinical-insights-portfolio/human-gene-mutation-database/ (accessed on 24 March 2025).

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Burykina, Y.; Chudakova, D.; Zharova, O.; Basargina, E.; Silnova, I.; Sdvigova, N.; Gandaeva, L.; Davydova, Y.; Kaverina, V.; Zhanin, I.; et al. A Unique Case of a Child with Two Rare Hereditary Diseases: Familial Dilated Cardiomyopathy and Arterial Calcification. Int. J. Mol. Sci. 2025, 26, 5900. https://doi.org/10.3390/ijms26125900
Burykina Y, Chudakova D, Zharova O, Basargina E, Silnova I, Sdvigova N, Gandaeva L, Davydova Y, Kaverina V, Zhanin I, et al. A Unique Case of a Child with Two Rare Hereditary Diseases: Familial Dilated Cardiomyopathy and Arterial Calcification. International Journal of Molecular Sciences. 2025; 26(12):5900. https://doi.org/10.3390/ijms26125900
Chicago/Turabian StyleBurykina, Yulia, Daria Chudakova, Olga Zharova, Elena Basargina, Irina Silnova, Natalia Sdvigova, Leila Gandaeva, Yulia Davydova, Valentina Kaverina, Ilya Zhanin, and et al. 2025. "A Unique Case of a Child with Two Rare Hereditary Diseases: Familial Dilated Cardiomyopathy and Arterial Calcification" International Journal of Molecular Sciences 26, no. 12: 5900. https://doi.org/10.3390/ijms26125900
APA StyleBurykina, Y., Chudakova, D., Zharova, O., Basargina, E., Silnova, I., Sdvigova, N., Gandaeva, L., Davydova, Y., Kaverina, V., Zhanin, I., Pushkov, A., Fisenko, A., & Savostyanov, K. (2025). A Unique Case of a Child with Two Rare Hereditary Diseases: Familial Dilated Cardiomyopathy and Arterial Calcification. International Journal of Molecular Sciences, 26(12), 5900. https://doi.org/10.3390/ijms26125900