
Progenitor Cell Dynamics in Androgenetic Alopecia: Insights from Spatially Resolved Transcriptomics

 , ,
, ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
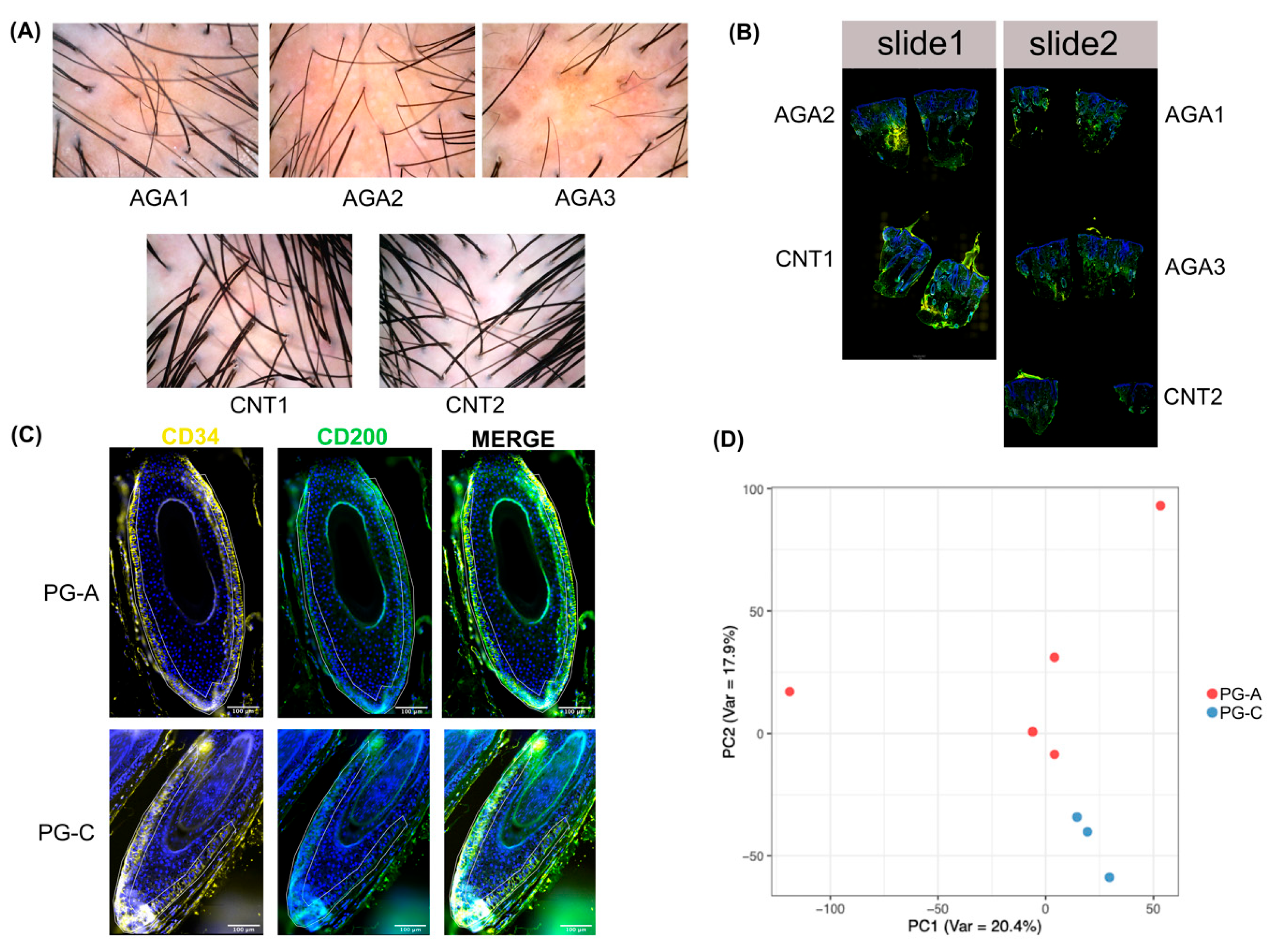
2.1. PG-A and PG-C Exhibit Distinct Transcriptomic Profiles
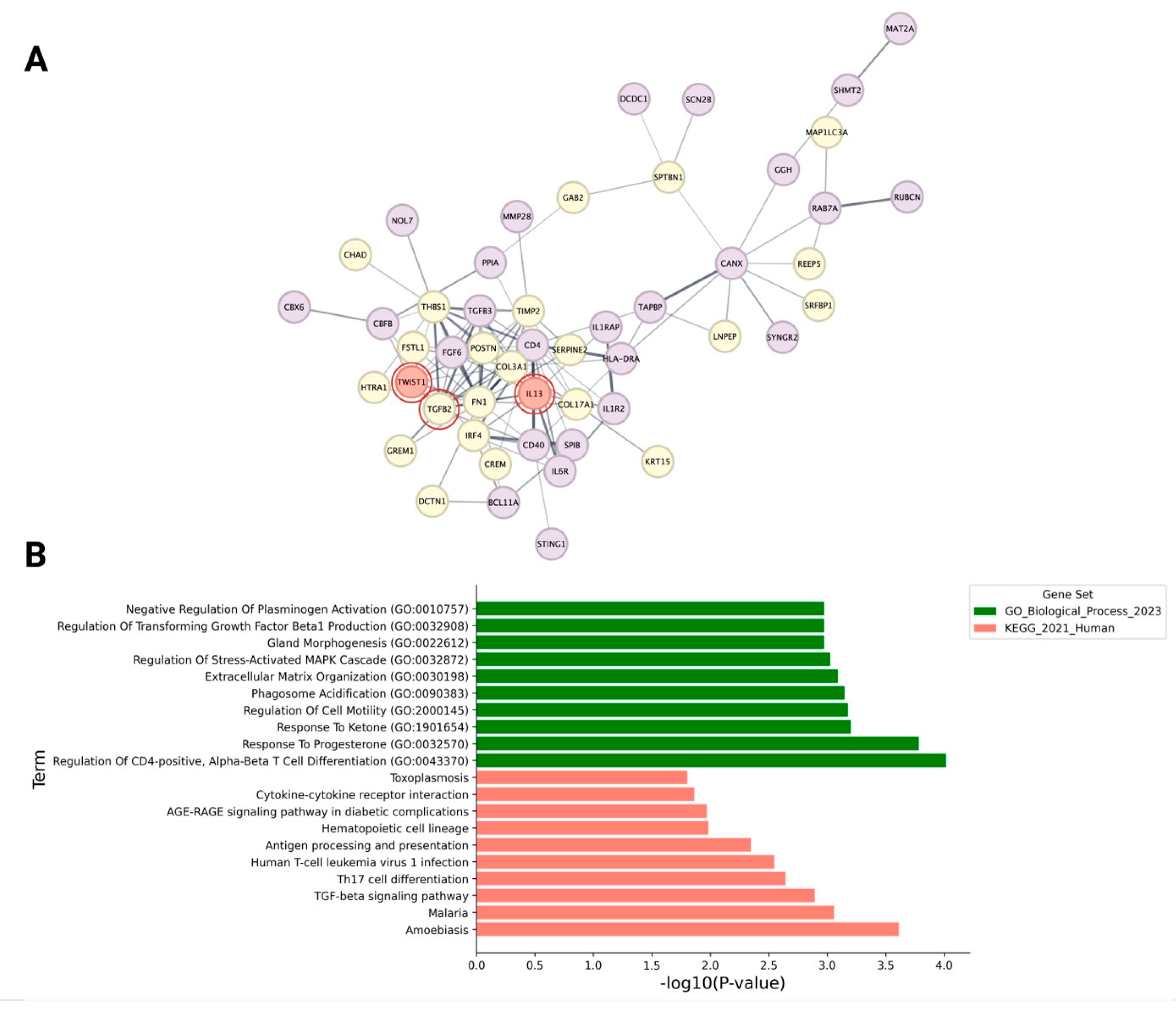
2.2. Transcriptional Signatures and Pathway Enrichment in HF Progenitor Cells Indicate Predilection for Epithelial–Mesenchymal Transition (EMT)
2.3. Influence of the Immune Microenvironment on the EMT and Fibrotic Processes in Progenitor Cells of AGA
3. Discussion
4. Materials and Methods
4.1. Biopsy Collection and Preparation
4.2. Slide Preparation for Spatial Transcriptome Profiling
4.3. Spatial Transcriptome Data Analysis
4.4. Overrepresentation Analysis
4.5. Protein–Protein Interaction Network Construction and Analysis
4.6. Gene Set Enrichment Analysis
4.7. Immunohistochemistry and Digital Image Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heilmann-Heimbach, S.; Herold, C.; Hochfeld, L.M.; Hillmer, A.M.; Nyholt, D.R.; Hecker, J.; Javed, A.; Chew, E.G.; Pechlivanis, S.; Drichel, D.; et al. Meta-analysis identifies novel risk loci and yields systematic insights into the biology of male-pattern baldness. Nat. Commun. 2017, 8, 14694. [Google Scholar] [CrossRef] [PubMed]
- Hagenaars, S.P.; Hill, W.D.; Harris, S.E.; Ritchie, S.J.; Davies, G.; Liewald, D.C.; Gale, C.R.; Porteous, D.J.; Deary, I.J.; Marioni, R.E. Genetic prediction of male pattern baldness. PLoS Genet. 2017, 13, e1006594. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Brockschmidt, F.F.; Kiefer, A.K.; Stefansson, H.; Nyholt, D.R.; Song, K.; Vermeulen, S.H.; Kanoni, S.; Glass, D.; Medland, S.E.; et al. Six novel susceptibility Loci for early-onset androgenetic alopecia and their unexpected association with common diseases. PLoS Genet. 2012, 8, e1002746. [Google Scholar] [CrossRef] [PubMed]
- Guarrera, M.; Rebora, A. The Higher Number and Longer Duration of Kenogen Hairs Are the Main Cause of the Hair Rarefaction in Androgenetic Alopecia. Ski. Appendage Disord. 2019, 5, 152–154. [Google Scholar] [CrossRef]
- Guarrera, M.; Rebora, A. Kenogen in female androgenetic alopecia. A longitudinal study. Dermatology 2005, 210, 18–20. [Google Scholar] [CrossRef]
- Courtois, M.; Loussouarn, G.; Hourseau, C.; Grollier, J.F. Hair cycle and alopecia. Ski. Pharmacol. 1994, 7, 84–89. [Google Scholar] [CrossRef]
- Yamauchi, K.; Kurosaka, A. Inhibition of glycogen synthase kinase-3 enhances the expression of alkaline phosphatase and insulin-like growth factor-1 in human primary dermal papilla cell culture and maintains mouse hair bulbs in organ culture. Arch. Dermatol. Res. 2009, 301, 357–365. [Google Scholar] [CrossRef]
- Leirós, G.J.; Ceruti, J.M.; Castellanos, M.L.; Kusinsky, A.G.; Balañá, M.E. Androgens modify Wnt agonists/antagonists expression balance in dermal papilla cells preventing hair follicle stem cell differentiation in androgenetic alopecia. Mol. Cell. Endocrinol. 2017, 439, 26–34. [Google Scholar] [CrossRef]
- Kwack, M.H.; Sung, Y.K.; Chung, E.J.; Im, S.U.; Ahn, J.S.; Kim, M.K.; Kim, J.C. Dihydrotestosterone-inducible dickkopf 1 from balding dermal papilla cells causes apoptosis in follicular keratinocytes. J. Investig. Dermatol. 2008, 128, 262–269. [Google Scholar] [CrossRef]
- Kwack, M.H.; Ahn, J.S.; Kim, M.K.; Kim, J.C.; Sung, Y.K. Dihydrotestosterone-inducible IL-6 inhibits elongation of human hair shafts by suppressing matrix cell proliferation and promotes regression of hair follicles in mice. J. Investig. Dermatol. 2012, 132, 43–49. [Google Scholar] [CrossRef]
- Inui, S.; Fukuzato, Y.; Nakajima, T.; Yoshikawa, K.; Itami, S. Androgen-inducible TGF-β1 from balding dermal papilla cells inhibits epithelial cell growth: A clue to understand paradoxical effects of androgen on human hair growth. FASEB J. 2002, 16, 1967–1969. [Google Scholar] [CrossRef]
- Yoo, H.G.; Kim, J.S.; Lee, S.R.; Pyo, H.K.; Moon, H.I.; Lee, J.H.; Kwon, O.S.; Chung, J.H.; Kim, K.H.; Eun, H.C.; et al. Perifollicular fibrosis: Pathogenetic role in androgenetic alopecia. Biol. Pharm. Bull. 2006, 29, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Hibino, T.; Nishiyama, T. Role of TGF-β2 in the human hair cycle. J. Dermatol. Sci. 2004, 35, 9–18. [Google Scholar] [CrossRef]
- Panteleyev, A.A. Functional anatomy of the hair follicle: The Secondary Hair Germ. Exp. Dermatol. 2018, 27, 701–720. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Aoi, N.; Sato, T.; Yamauchi, Y.; Suga, H.; Eto, H.; Kato, H.; Araki, J.; Yoshimura, K. Differential expression of stem-cell-associated markers in human hair follicle epithelial cells. Lab. Investig. 2009, 89, 844–856. [Google Scholar] [CrossRef]
- Garza, L.A.; Yang, C.C.; Zhao, T.; Blatt, H.B.; Lee, M.; He, H.; Stanton, D.C.; Carrasco, L.; Spiegel, J.H.; Tobias, J.W.; et al. Bald scalp in men with androgenetic alopecia retains hair follicle stem cells but lacks CD200-rich and CD34-positive hair follicle progenitor cells. J. Clin. Investig. 2011, 121, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhou, D.; Ma, T.; Liu, Q. Vascular Endothelial Growth Factor Protects CD200-Rich and CD34-Positive Hair Follicle Stem Cells Against Androgen-Induced Apoptosis Through the Phosphoinositide 3-Kinase/Akt Pathway in Patients With Androgenic Alopecia. Dermatol. Surg. 2020, 46, 358–368. [Google Scholar] [CrossRef]
- Huang, W.Y.; Lin, E.T.; Hsu, Y.C.; Lin, S.J. Anagen hair follicle repair: Timely regenerative attempts from plastic extra-bulge epithelial cells. Exp. Dermatol. 2019, 28, 406–412. [Google Scholar] [CrossRef]
- Bingham, G.C.; Lee, F.; Naba, A.; Barker, T.H. Spatial-omics: Novel approaches to probe cell heterogeneity and extracellular matrix biology. Matrix Biol. 2020, 91–92, 152–166. [Google Scholar] [CrossRef]
- Hasin, Y.; Seldin, M.; Lusis, A. Multi-omics approaches to disease. Genome Biol. 2017, 18, 83. [Google Scholar] [CrossRef]
- Hulsen, T.; Jamuar, S.S.; Moody, A.R.; Karnes, J.H.; Varga, O.; Hedensted, S.; Spreafico, R.; Hafler, D.A.; McKinney, E.F. From Big Data to Precision Medicine. Front. Med. 2019, 6, 34. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Qu, Q.; Jiang, W.; Liu, X.M.; Shi, P.L.; Fan, Z.X.; Du, L.J.; Wang, G.F.; Liu, X.N.; Guo, Z.H.; et al. Identification of Functional Patterns of Androgenetic Alopecia Using Transcriptome Profiling in Distinct Locations of Hair Follicles. J. Investig. Dermatol. 2018, 138, 972–975. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Zeisel, A.; Jacob, T.; Sun, X.; La Manno, G.; Lönnerberg, P.; Linnarsson, S.; Kasper, M. Single-Cell Transcriptomics Reveals that Differentiation and Spatial Signatures Shape Epidermal and Hair Follicle Heterogeneity. Cell Syst. 2016, 3, 221–237.e9. [Google Scholar] [CrossRef] [PubMed]
- Joost, S.; Annusver, K.; Jacob, T.; Sun, X.; Dalessandri, T.; Sivan, U.; Sequeira, I.; Sandberg, R.; Kasper, M. The Molecular Anatomy of Mouse Skin during Hair Growth and Rest. Cell Stem Cell 2020, 26, 441–457.e7. [Google Scholar] [CrossRef]
- Chovatiya, G.; Ghuwalewala, S.; Walter, L.D.; Cosgrove, B.D.; Tumbar, T. High-resolution single-cell transcriptomics reveals heterogeneity of self-renewing hair follicle stem cells. Exp. Dermatol. 2020, 30, 457–471. [Google Scholar] [CrossRef]
- Takahashi, R.; Grzenda, A.; Allison, T.F.; Rawnsley, J.; Balin, S.J.; Sabri, S.; Plath, K.; Lowry, W.E. Defining Transcriptional Signatures of Human Hair Follicle Cell States. J. Investig. Dermatol. 2020, 140, 764–773.e4. [Google Scholar] [CrossRef]
- Chen, S.; Luo, Y.; Gao, H.; Li, F.; Li, J.; Chen, Y.; You, R.; Lv, H.; Hua, K.; Jiang, R.; et al. Toward a unified information framework for cell atlas assembly. Natl. Sci. Rev. 2022, 9, nwab179. [Google Scholar] [CrossRef]
- Charoensuksira, S.; Tantiwong, S.; Pongklaokam, J.; Hanvivattanakul, S.; Surinlert, P.; Krajarng, A.; Thanasarnaksorn, W.; Hongeng, S.; Ponnikorn, S. Disturbance of Immune Microenvironment in Androgenetic Alopecia through Spatial Transcriptomics. Int. J. Mol. Sci. 2024, 25, 9031. [Google Scholar] [CrossRef]
- El-Domyati, M.; Attia, S.; Saleh, F.; Abdel-Wahab, H. Androgenetic alopecia in males: A histopathological and ultrastructural study. J. Cosmet. Dermatol. 2009, 8, 83–91. [Google Scholar] [CrossRef]
- Mahé, Y.F.; Michelet, J.F.; Billoni, N.; Jarrousse, F.; Buan, B.; Commo, S.; Saint-Léger, D.; Bernard, B.A. Androgenetic alopecia and microinflammation. Int. J. Dermatol. 2000, 39, 576–584. [Google Scholar] [CrossRef]
- Fu, W.; Chen, S.; Yang, R.; Li, C.; Gao, H.; Li, J.; Zhang, X. Cellular features of localized microenvironments in human meniscal degeneration: A single-cell transcriptomic study. Elife 2022, 11, e79585. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, F.; Sun, Y.; Gan, Y.; Zhu, D.; Wang, H.; Qu, Q.; Wang, J.; Chen, R.; Fan, Z.; et al. Association of fibrosis in the bulge portion with hair follicle miniaturization in androgenetic alopecia. J. Am. Acad. Dermatol. 2022, 86, 213–215. [Google Scholar] [CrossRef] [PubMed]
- Briggs, S.L. The role of fibronectin in fibroblast migration during tissue repair. J. Wound Care 2005, 14, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Muro, A.F.; Moretti, F.A.; Moore, B.B.; Yan, M.; Atrasz, R.G.; Wilke, C.A.; Flaherty, K.R.; Martinez, F.J.; Tsui, J.L.; Sheppard, D.; et al. An essential role for fibronectin extra type III domain A in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2008, 177, 638–645. [Google Scholar] [CrossRef]
- Park, J.; Schwarzbauer, J.E. Mammary epithelial cell interactions with fibronectin stimulate epithelial-mesenchymal transition. Oncogene 2014, 33, 1649–1657. [Google Scholar] [CrossRef]
- Jahoda, C.A.; Mauger, A.; Bard, S.; Sengel, P. Changes in fibronectin, laminin and type IV collagen distribution relate to basement membrane restructuring during the rat vibrissa follicle hair growth cycle. J. Anat. 1992, 181 Pt 1, 47–60. [Google Scholar]
- Couchman, J.R.; Gibson, W.T. Expression of basement membrane components through morphological changes in the hair growth cycle. Dev. Biol. 1985, 108, 290–298. [Google Scholar] [CrossRef]
- O’Keefe, E.J.; Woodley, D.T.; Falk, R.J.; Gammon, W.R.; Briggaman, R.A. Production of fibronectin by epithelium in a skin equivalent. J. Investig. Dermatol. 1987, 88, 634–639. [Google Scholar] [CrossRef]
- Wang, Z.; An, J.; Zhu, D.; Chen, H.; Lin, A.; Kang, J.; Liu, W.; Kang, X. Periostin: An emerging activator of multiple signaling pathways. J. Cell Commun. Signal. 2022, 16, 515–530. [Google Scholar] [CrossRef]
- Hong, Y.K.; Hwang, D.Y.; Yang, C.C.; Cheng, S.M.; Chen, P.C.; Aala, W.J.; Harn, I.C.H.; Evans, S.T.; Onoufriadis, A.; Liu, S.L.; et al. Profibrotic Subsets of SPP1+ Macrophages and POSTN+ Fibroblasts Contribute to Fibrotic Scarring in Acne Keloidalis. J. Investig. Dermatol. 2024, 144, 1491–1504.e10. [Google Scholar] [CrossRef]
- Xu, J.; Lamouille, S.; Derynck, R. TGF-β-induced epithelial to mesenchymal transition. Cell Res. 2009, 19, 156–172. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, L.; Soto, B.; Meier, R.; Geraghty, P. The Biology and Function of Tissue Inhibitor of Metalloproteinase 2 in the Lungs. Pulm. Med. 2022, 2022, 3632764. [Google Scholar] [CrossRef]
- Kuivaniemi, H.; Tromp, G. Type III collagen (COL3A1): Gene and protein structure, tissue distribution, and associated diseases. Gene 2019, 707, 151–171. [Google Scholar] [CrossRef]
- Jiang, D.; Guo, B.; Lin, F.; Hui, Q.; Tao, K. Effect of THBS1 on the Biological Function of Hypertrophic Scar Fibroblasts. BioMed Res. Int. 2020, 2020, 8605407. [Google Scholar] [CrossRef]
- Murphy-Ullrich, J.E.; Suto, M.J. Thrombospondin-1 regulation of latent TGF-β activation: A therapeutic target for fibrotic disease. Matrix Biol. 2018, 68–69, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Dey-Rao, R.; Sinha, A.A. A genomic approach to susceptibility and pathogenesis leads to identifying potential novel therapeutic targets in androgenetic alopecia. Genomics 2017, 109, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Mikheeva, S.A.; Mikheev, A.M.; Petit, A.; Beyer, R.; Oxford, R.G.; Khorasani, L.; Maxwell, J.P.; Glackin, C.A.; Wakimoto, H.; González-Herrero, I.; et al. TWIST1 promotes invasion through mesenchymal change in human glioblastoma. Mol. Cancer 2010, 9, 194. [Google Scholar] [CrossRef] [PubMed]
- Al-Hattab, D.S.; Safi, H.A.; Nagalingam, R.S.; Bagchi, R.A.; Stecy, M.T.; Czubryt, M.P. Scleraxis regulates Twist1 and Snai1 expression in the epithelial-to-mesenchymal transition. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H658–H668. [Google Scholar] [CrossRef]
- Ning, X.; Zhang, K.; Wu, Q.; Liu, M.; Sun, S. Emerging role of Twist1 in fibrotic diseases. J. Cell. Mol. Med. 2018, 22, 1383–1391. [Google Scholar] [CrossRef]
- Haglund, L.; Tillgren, V.; Addis, L.; Wenglén, C.; Recklies, A.; Heinegård, D. Identification and characterization of the integrin α2β1 binding motif in chondroadherin mediating cell attachment. J. Biol. Chem. 2011, 286, 3925–3934. [Google Scholar] [CrossRef]
- Haglund, L.; Ouellet, J.; Roughley, P. Variation in chondroadherin abundance and fragmentation in the human scoliotic disc. Spine 2009, 34, 1513–1518. [Google Scholar] [CrossRef] [PubMed]
- Turn, C.S.; Kolliputi, N. Two sides of a coin: The dual roles of chitinase 3-like 1 in idiopathic pulmonary fibrosis. Lung 2014, 192, 825–827. [Google Scholar] [CrossRef] [PubMed]
- Senna, M.M.; Peterson, E.; Jozic, I.; Chéret, J.; Paus, R. Frontiers in Lichen Planopilaris and Frontal Fibrosing Alopecia Research: Pathobiology Progress and Translational Horizons. JID Innov. 2022, 2, 100113. [Google Scholar] [CrossRef] [PubMed]
- Griggs, J.; Trüeb, R.M.; Gavazzoni Dias, M.F.R.; Hordinsky, M.; Tosti, A. Fibrosing alopecia in a pattern distribution. J. Am. Acad. Dermatol. 2021, 85, 1557–1564. [Google Scholar] [CrossRef]
- Gieseck, R.L., 3rd; Wilson, M.S.; Wynn, T.A. Type 2 immunity in tissue repair and fibrosis. Nat. Rev. Immunol. 2018, 18, 62–76. [Google Scholar] [CrossRef]
- Nguyen, J.K.; Austin, E.; Huang, A.; Mamalis, A.; Jagdeo, J. The IL-4/IL-13 axis in skin fibrosis and scarring: Mechanistic concepts and therapeutic targets. Arch. Dermatol. Res. 2020, 312, 81–92. [Google Scholar] [CrossRef]
- Armand, A.S.; Laziz, I.; Chanoine, C. FGF6 in myogenesis. Biochim. Biophys. Acta 2006, 1763, 773–778. [Google Scholar] [CrossRef]
- Qi, L.; Song, W.; Li, L.; Cao, L.; Yu, Y.; Song, C.; Wang, Y.; Zhang, F.; Li, Y.; Zhang, B.; et al. FGF4 induces epithelial-mesenchymal transition by inducing store-operated calcium entry in lung adenocarcinoma. Oncotarget 2016, 7, 74015–74030. [Google Scholar] [CrossRef]
- Schelch, K.; Wagner, C.; Hager, S.; Pirker, C.; Siess, K.; Lang, E.; Lin, R.; Kirschner, M.B.; Mohr, T.; Brcic, L.; et al. FGF2 and EGF induce epithelial-mesenchymal transition in malignant pleural mesothelioma cells via a MAPKinase/MMP1 signal. Carcinogenesis 2018, 39, 534–545. [Google Scholar] [CrossRef]
- Profyris, C.; Tziotzios, C.; Do Vale, I. Cutaneous scarring: Pathophysiology, molecular mechanisms, and scar reduction therapeutics Part I. The molecular basis of scar formation. J. Am. Acad. Dermatol. 2012, 66, 1–10, quiz 1–2. [Google Scholar] [CrossRef]
- Guo, J.; Liu, W.; Zeng, Z.; Lin, J.; Zhang, X.; Chen, L. Tgfb3 and Mmp13 regulated the initiation of liver fibrosis progression as dynamic network biomarkers. J. Cell. Mol. Med. 2021, 25, 867–879. [Google Scholar] [CrossRef]
- Sun, T.; Vander Heiden, J.A.; Gao, X.; Yin, J.; Uttarwar, S.; Liang, W.C.; Jia, G.; Yadav, R.; Huang, Z.; Mitra, M.; et al. Isoform-selective TGF-β3 inhibition for systemic sclerosis. Med 2024, 5, 132–147.e7. [Google Scholar] [CrossRef] [PubMed]
- Deng, Z.; Fan, T.; Xiao, C.; Tian, H.; Zheng, Y.; Li, C.; He, J. TGF-β signaling in health, disease, and therapeutics. Signal Transduct. Target. Ther. 2024, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Doncheva, N.T.; Morris, J.H.; Gorodkin, J.; Jensen, L.J. Cytoscape StringApp: Network Analysis and Visualization of Proteomics Data. J. Proteome Res. 2019, 18, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Li, C.Y.; Cai, J.H.; Tsai, J.J.P.; Wang, C.C.N. Identification of Hub Genes Associated With Development of Head and Neck Squamous Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef]
- Bankhead, P.; Loughrey, M.B.; Fernández, J.A.; Dombrowski, Y.; McArt, D.G.; Dunne, P.D.; McQuaid, S.; Gray, R.T.; Murray, L.J.; Coleman, H.G.; et al. QuPath: Open source software for digital pathology image analysis. Sci. Rep. 2017, 7, 16878. [Google Scholar] [CrossRef]
- Emmanuel, T.; Brent, M.B.; Iversen, L.; Johansen, C. Quantification of Immunohistochemically Stained Cells in Skin Biopsies. Dermatopathology 2022, 9, 82–93. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charoensuksira, S.; Surinlert, P.; Krajarng, A.; Nualsanit, T.; Payuhakrit, W.; Panpinyaporn, P.; Khumsri, W.; Thanasarnaksorn, W.; Suwanchinda, A.; Hongeng, S.; et al. Progenitor Cell Dynamics in Androgenetic Alopecia: Insights from Spatially Resolved Transcriptomics. Int. J. Mol. Sci. 2025, 26, 5792. https://doi.org/10.3390/ijms26125792
Charoensuksira S, Surinlert P, Krajarng A, Nualsanit T, Payuhakrit W, Panpinyaporn P, Khumsri W, Thanasarnaksorn W, Suwanchinda A, Hongeng S, et al. Progenitor Cell Dynamics in Androgenetic Alopecia: Insights from Spatially Resolved Transcriptomics. International Journal of Molecular Sciences. 2025; 26(12):5792. https://doi.org/10.3390/ijms26125792
Chicago/Turabian StyleCharoensuksira, Sasin, Piyaporn Surinlert, Aungkana Krajarng, Thararat Nualsanit, Witchuda Payuhakrit, Pimchanok Panpinyaporn, Wilunplus Khumsri, Wilai Thanasarnaksorn, Atchima Suwanchinda, Suradej Hongeng, and et al. 2025. "Progenitor Cell Dynamics in Androgenetic Alopecia: Insights from Spatially Resolved Transcriptomics" International Journal of Molecular Sciences 26, no. 12: 5792. https://doi.org/10.3390/ijms26125792
APA StyleCharoensuksira, S., Surinlert, P., Krajarng, A., Nualsanit, T., Payuhakrit, W., Panpinyaporn, P., Khumsri, W., Thanasarnaksorn, W., Suwanchinda, A., Hongeng, S., & Ponnikorn, S. (2025). Progenitor Cell Dynamics in Androgenetic Alopecia: Insights from Spatially Resolved Transcriptomics. International Journal of Molecular Sciences, 26(12), 5792. https://doi.org/10.3390/ijms26125792