
Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease

 , , , ,
, , , ,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
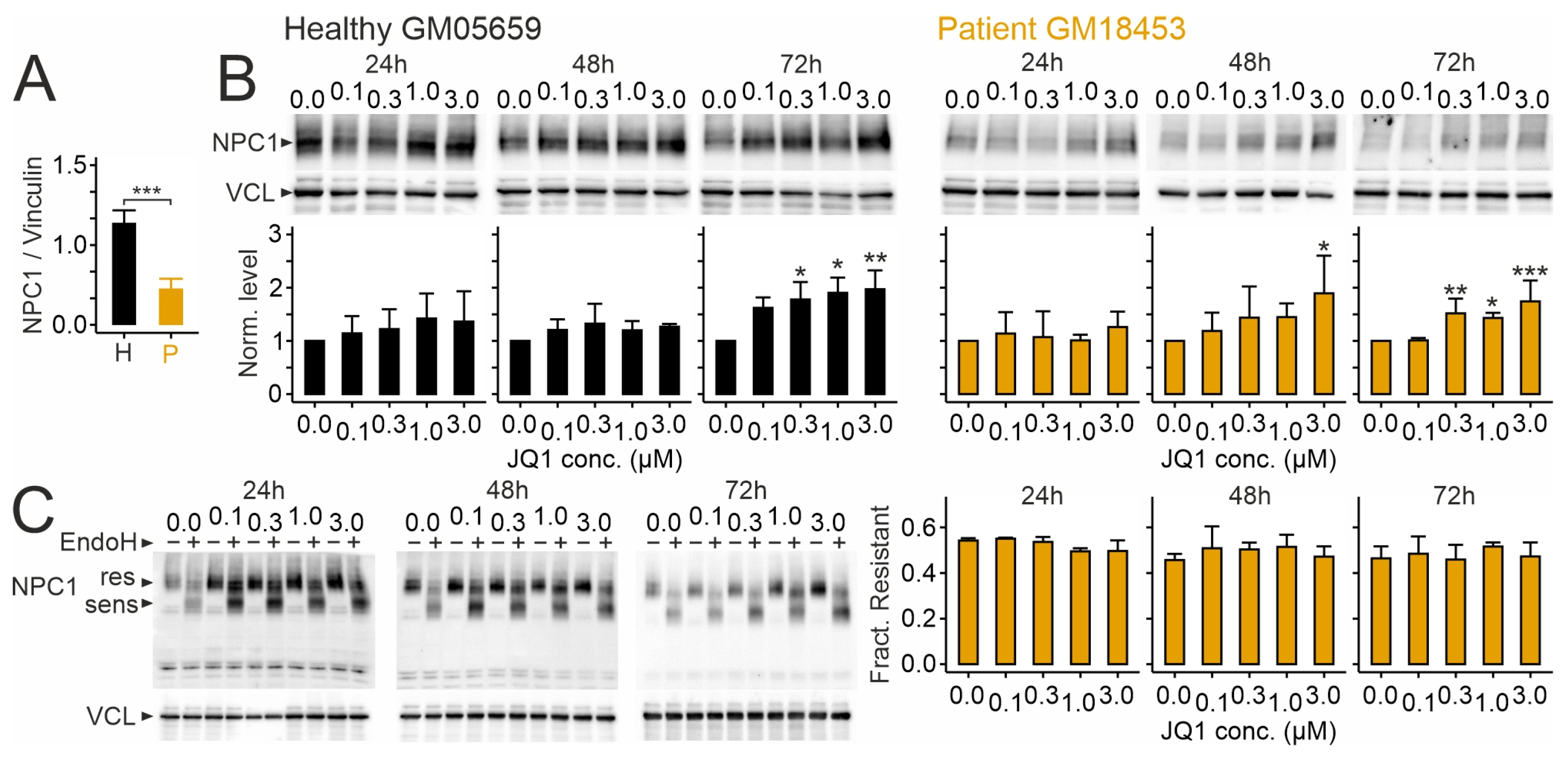
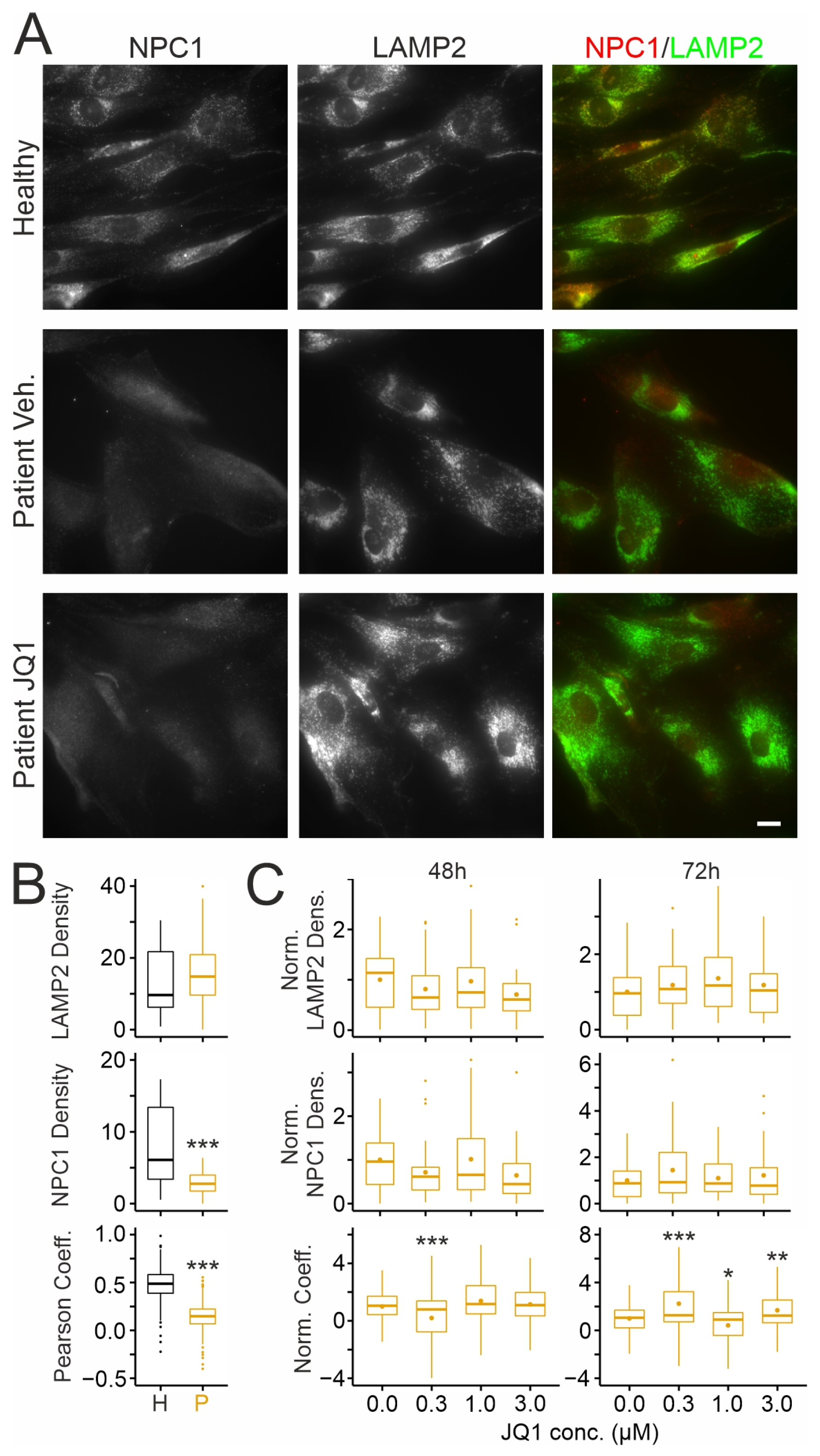
2.1. Effects of JQ1 on Viability, Protein Levels of NPC1, and Its Distribution in Human Skin Fibroblasts
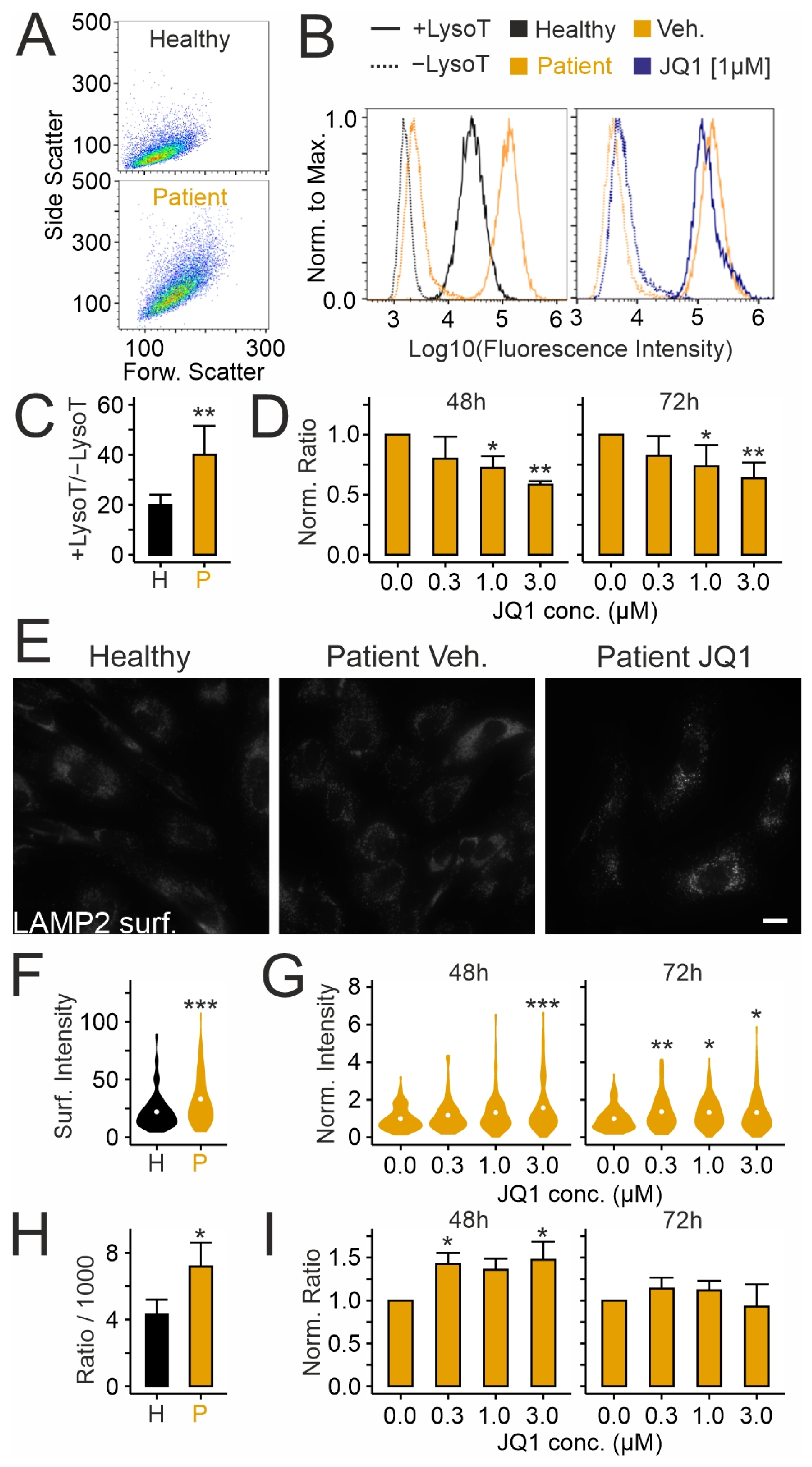
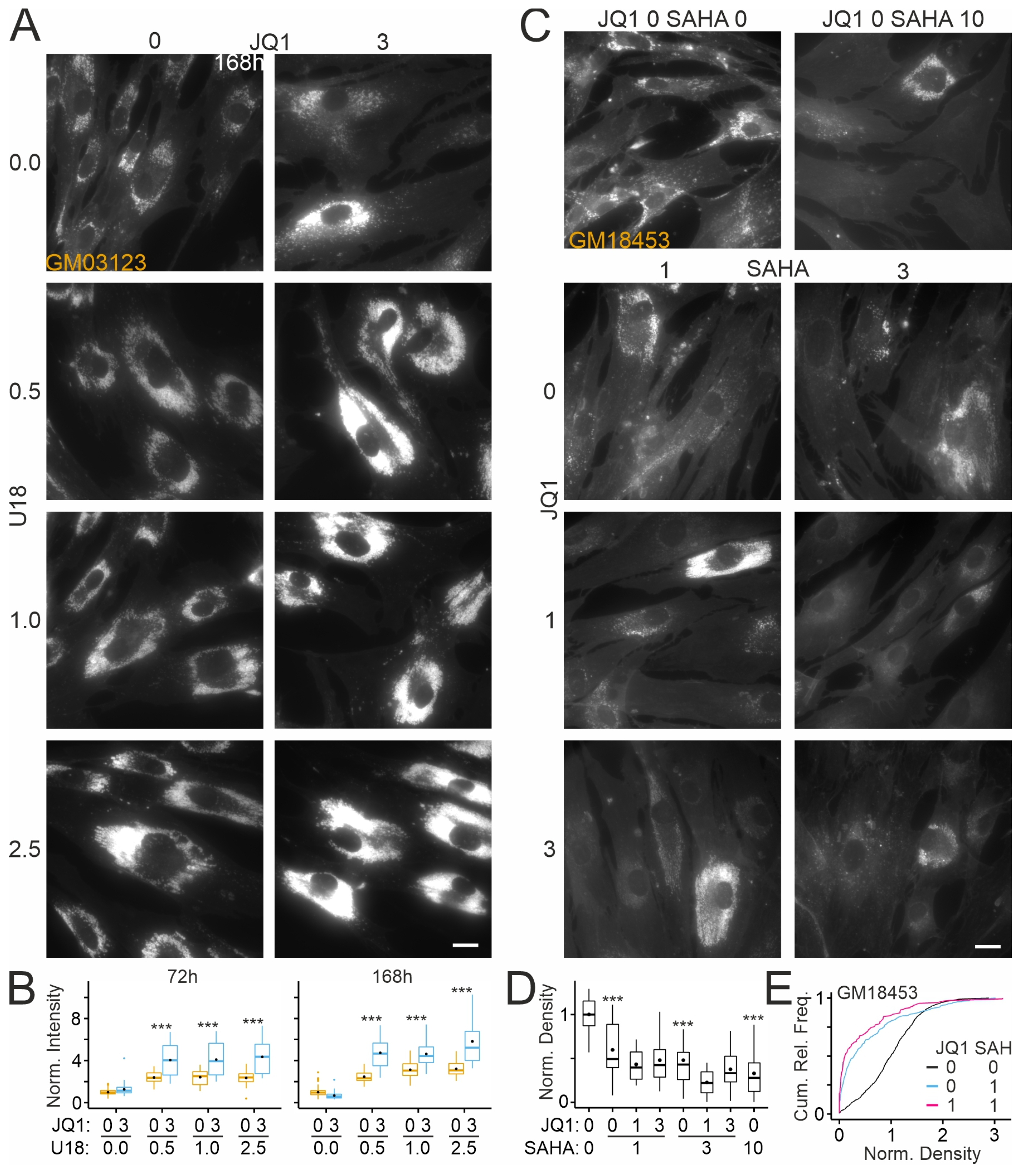
2.2. Effects of JQ1 on Lysosomes in Patient-Derived Skin Fibroblasts
2.3. Effects of JQ1 on Cholesterol Accumulation in Patient-Derived Skin Fibroblasts
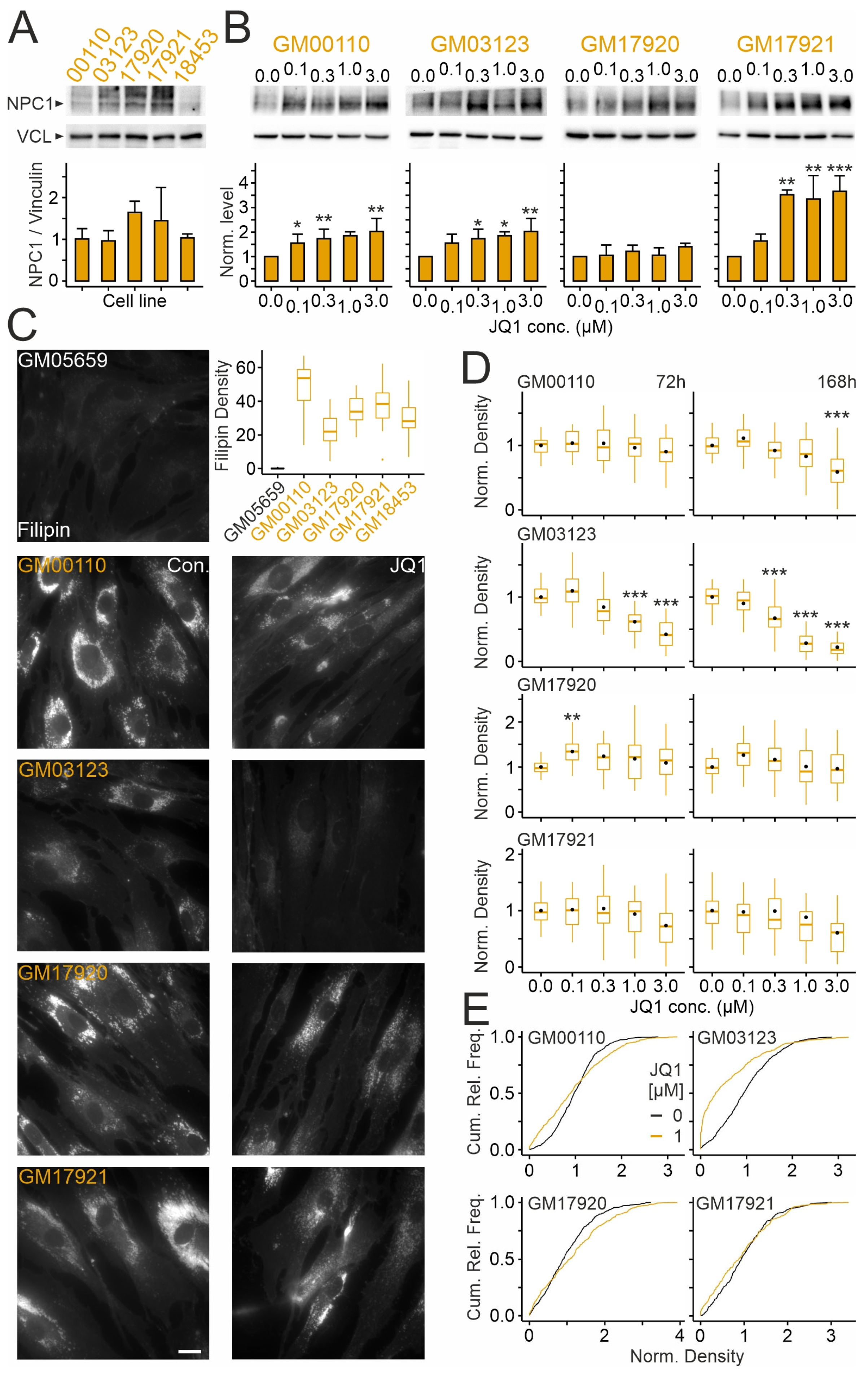
2.4. Effects of JQ1 on Skin Fibroblasts from Different NPCD Patients
2.5. Effects of JQ1 on Cholesterol Accumulation in Patient-Derived Fibroblasts in the Presence of an NPC1 Inhibitor
2.6. Effects of JQ1 on HDAC Inhibitor-Mediated Reduction in Cholesterol Accumulation in Patient-Derived Fibroblasts
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Drug Treatment
4.2. Cell Number and Viability
4.3. Lysate Preparation and Immunoblotting
4.4. Endoglycosidase H Assay
4.5. Immunocytochemical Staining
4.6. Cytochemical Staining
4.7. Lysotracker Staining and Cytometry
4.8. Hexosaminidase Activity Assay
4.9. Data Analysis and Visualization
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ANOVA | Analysis of variance |
| BET | Bromodomain and extraterminal domain |
| DMSO | Dimethylsulfoxide |
| EndoH | Endoglycosidase H |
| HDAC | Histone deacetylase |
| NPCD | Niemann–Pick type C disease |
| PBS | Phosphate-buffered saline |
| ROIs | Regions of interest |
| SAHA | Suberoylanilide hydroxamic acid |
| U18 | 3-beta-[2-(diethylamine)ethoxy]androst-5-en-17-one |
References
- Ebner, M.; Fröhlich, F.; Haucke, V. Mechanisms and functions of lysosomal lipid homeostasis. Cell Chem. Biol. 2025, 32, 392–407. [Google Scholar] [CrossRef] [PubMed]
- Ludlaim, A.M.; Waddington, S.N.; McKay, T.R. Unifying biology of neurodegeneration in lysosomal storage diseases. J. Inherit. Metab. Dis. 2025, 48, e12833. [Google Scholar] [CrossRef] [PubMed]
- Vanier, M.T. Niemann-pick disease type c. Orphanet J. Rare Dis. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Berry-Kravis, E. Niemann-pick disease, type c: Diagnosis, management and disease-targeted therapies in development. Semin. Pediatr. Neurol. 2021, 37, 100879. [Google Scholar] [CrossRef]
- Bolton, S.C.; Soran, V.; Marfa, M.P.; Imrie, J.; Gissen, P.; Jahnova, H.; Sharma, R.; Jones, S.; Santra, S.; Crushell, E.; et al. Clinical disease characteristics of patients with niemann-pick disease type c: Findings from the international niemann-pick disease registry (inpdr). Orphanet J. Rare Dis. 2022, 17, 51. [Google Scholar] [CrossRef]
- Yilmaz, S.B.; Baruteau, J.; Rahim, A.A.; Gissen, P. Clinical and molecular features of early infantile niemann pick type c disease. Int. J. Mol. Sci. 2020, 21, 5059. [Google Scholar] [CrossRef]
- Las Heras, M.; Szenfeld, B.; Ballout, R.A.; Buratti, E.; Zanlungo, S.; Dardis, A.; Klein, A.D. Understanding the phenotypic variability in niemann-pick disease type c (npc): A need for precision medicine. Npj Genom. Med. 2023, 8, 21. [Google Scholar] [CrossRef]
- Carstea, E.D.; Morris, J.A.; Coleman, K.G.; Loftus, S.K.; Zhang, D.; Cummings, C.; Gu, J.; Rosenfeld, M.A.; Pavan, W.J.; Krizman, D.B.; et al. Niemann-pick c1 disease gene: Homology to mediators of cholesterol homeostasis. Science 1997, 277, 228–231. [Google Scholar] [CrossRef]
- Loftus, S.K. Murine model of niemann-pick c disease: Mutation in a cholesterol homeostasis gene. Science 1997, 277, 232–235. [Google Scholar] [CrossRef]
- Naureckiene, S.; Sleat, D.; Lackland, H.; Fensom, A.; Vanier, M.T.; Wattiaux, R.; Jadot, M.; Lobel, P. Identification of he1 as the second gene of niemann-pick c disease. Science 2000, 290, 2298–2301. [Google Scholar] [CrossRef]
- Higgins, M.E.; Davies, J.P.; Chen, F.W.; Ioannou, Y.A. Niemann-pick c1 is a late endosome-resident protein that transiently associates with lysosomes and the trans-golgi network. Mol. Genet. Metab. 1999, 68, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Neufeld, E.B.; Wastney, M.; Patel, S.; Suresh, S.; Cooney, A.M.; Dwyer, N.K.; Roff, C.F.; Ohno, K.; Morris, J.A.; Carstea, E.D.; et al. The niemann-pick c1 protein resides in a vesicular compartment linked to retrograde transport of multiple lysosomal cargo. J. Biol. Chem. 1999, 274, 9627–9635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Sun, M.; Dwyer, N.K.; Comly, M.E.; Patel, S.C.; Sundaram, R.; Hanover, J.A.; Blanchette-Mackie, E.J. Differential trafficking of the niemann-pick c1 and 2 proteins highlights distinct roles in late endocytic lipid trafficking. Acta Paediatr. 2003, 92, 63–73. [Google Scholar] [CrossRef]
- Blom, T.S.; Linder, M.D.; Snow, K.; Pihko, H.; Hess, M.W.; Jokitalo, E.; Veckman, V.; Syvanen, A.C.; Ikonen, E. Defective endocytic trafficking of npc1 and npc2 underlying infantile niemann-pick type c disease. Hum. Mol. Genet. 2003, 12, 257–272. [Google Scholar] [CrossRef]
- Sleat, D.E.; Wiseman, J.A.; El-Banna, M.; Price, S.M.; Verot, L.; Shen, M.M.; Tint, G.S.; Vanier, M.T.; Walkley, S.U.; Lobel, P. Genetic evidence for nonredundant functional cooperativity between npc1 and npc2 in lipid transport. Proc. Natl. Acad. Sci. USA 2004, 101, 5886–5891. [Google Scholar] [CrossRef]
- Liou, H.L.; Dixit, S.S.; Xu, S.J.; Tint, G.S.; Stock, A.M.; Lobel, P. Npc2, the protein deficient in niemann-pick c2 disease, consists of multiple glycoforms that bind a variety of sterols. J. Biol. Chem. 2006, 281, 36710–36723. [Google Scholar] [CrossRef]
- Infante, R.E.; Wang, M.L.; Radhakrishnan, A.; Kwon, H.J.; Brown, M.S.; Goldstein, J.L. Npc2 facilitates bidirectional transfer of cholesterol between npc1 and lipid bilayers, a step in cholesterol egress from lysosomes. Proc. Natl. Acad. Sci. USA 2008, 105, 15287–15292. [Google Scholar] [CrossRef]
- Friedland, N.; Liou, H.-L.; Lobel, P.; Stock, A.M. Structure of a cholesterol-binding protein deficient in niemann-pick type c2 disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2512–2517. [Google Scholar] [CrossRef]
- Kwon, H.J.; Abi-Mosleh, L.; Wang, M.L.; Deisenhofer, J.; Goldstein, J.L.; Brown, M.S.; Infante, R.E. Structure of n-terminal domain of npc1 reveals distinct subdomains for binding and transfer of cholesterol. Cell 2009, 137, 1213–1224. [Google Scholar] [CrossRef]
- Li, X.; Saha, P.; Li, J.; Blobel, G.; Pfeffer, S.R. Clues to the mechanism of cholesterol transfer from the structure of npc1 middle lumenal domain bound to npc2. Proc. Natl. Acad. Sci. USA 2016, 113, 10079–10084. [Google Scholar] [CrossRef]
- Winkler, M.B.L.; Kidmose, R.T.; Szomek, M.; Thaysen, K.; Rawson, S.; Muench, S.P.; Wüstner, D.; Pedersen, B.P. Structural insight into eukaryotic sterol transport through niemann-pick type c proteins. Cell 2019, 179, 485–497.e418. [Google Scholar] [CrossRef] [PubMed]
- Qian, H.; Wu, X.; Du, X.; Yao, X.; Zhao, X.; Lee, J.; Yang, H.; Yan, N. Structural basis of low-ph-dependent lysosomal cholesterol egress by npc1 and npc2. Cell 2020, 182, 98–111.e118. [Google Scholar] [CrossRef] [PubMed]
- Frain, K.M.; Dedic, E.; Nel, L.; Bohush, A.; Olesen, E.; Thaysen, K.; Wüstner, D.; Stokes, D.L.; Pedersen, B.P. Conformational changes in the niemann-pick type c1 protein ncr1 drive sterol translocation. Proc. Natl. Acad. Sci. USA 2024, 121, e2315575121. [Google Scholar] [CrossRef]
- Pentchev, P.G.; Comly, M.E.; Kruth, H.S.; Vanier, M.T.; Wenger, D.A.; Patel, S.; Brady, R.O. A defect in cholesterol esterification in niemann-pick disease (type c) patients. Proc. Natl. Acad. Sci. USA 1985, 82, 8247–8251. [Google Scholar] [CrossRef]
- Vanier, M.T.; Rodriguez-Lafrasse, C.; Rousson, R.; Gazzah, N.; Juge, M.C.; Pentchev, P.G.; Revol, A.; Louisot, P. Type c niemann-pick disease: Spectrum of phenotypic variation in disruption of intracellular ldl-derived cholesterol processing. Biochim. Biophys. Acta 1991, 1096, 328–337. [Google Scholar] [CrossRef]
- Taniguchi, M.; Shinoda, Y.; Ninomiya, H.; Vanier, M.T.; Ohno, K. Sites and temporal changes of gangliosides gm1/gm2 storage in the niemann-pick disease type c mouse brain. Brain Dev. 2001, 23, 414–421. [Google Scholar] [CrossRef]
- Zervas, M.; Dobrenis, K.; Walkley, S.U. Neurons in niemann-pick disease type c accumulate gangliosides as well as unesterified cholesterol and undergo dendritic and axonal alterations. J. Neuropathol. Exp. Neurol. 2001, 60, 49–64. [Google Scholar] [CrossRef]
- te Vruchte, D.; Lloyd-Evans, E.; Veldman, R.J.; Neville, D.C.; Dwek, R.A.; Platt, F.M.; van Blitterswijk, W.J.; Sillence, D.J. Accumulation of glycosphingolipids in niemann-pick c disease disrupts endosomal transport. J. Biol. Chem. 2004, 279, 26167–26175. [Google Scholar] [CrossRef]
- Lloyd-Evans, E.; Morgan, A.J.; He, X.; Smith, D.A.; Elliot-Smith, E.; Sillence, D.J.; Churchill, G.C.; Schuchman, E.H.; Galione, A.; Platt, F.M. Niemann-pick disease type c1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nat. Med. 2008, 14, 1247–1255. [Google Scholar] [CrossRef]
- Altuzar, J.; Notbohm, J.; Stein, F.; Haberkant, P.; Hempelmann, P.; Heybrock, S.; Worsch, J.; Saftig, P.; Hoglinger, D. Lysosome-targeted multifunctional lipid probes reveal the sterol transporter npc1 as a sphingosine interactor. Proc. Natl. Acad. Sci. USA 2023, 120, e2213886120. [Google Scholar] [CrossRef]
- Zhang, M.; Dwyer, N.K.; Love, D.C.; Cooney, A.; Comly, M.; Neufeld, E.; Pentchev, P.G.; Blanchette-Mackie, E.J.; Hanover, J.A. Cessation of rapid late endosomal tubulovesicular trafficking in niemann-pick type c1 disease. Proc. Natl. Acad. Sci. USA 2001, 98, 4466–4471. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Wang, X.; Li, X.; Zhang, X.; Yao, Z.; Dibble, S.; Dong, X.P.; Yu, T.; Lieberman, A.P.; Showalter, H.D.; et al. Lipid storage disorders block lysosomal trafficking by inhibiting a trp channel and lysosomal calcium release. Nat. Commun. 2012, 3, 731. [Google Scholar] [CrossRef] [PubMed]
- Elrick, M.J.; Yu, T.; Chung, C.; Lieberman, A.P. Impaired proteolysis underlies autophagic dysfunction in niemann-pick type c disease. Hum. Mol. Genet. 2012, 21, 4876–4887. [Google Scholar] [CrossRef]
- Yambire, K.F.; Fernandez-Mosquera, L.; Steinfeld, R.; Muhle, C.; Ikonen, E.; Milosevic, I.; Raimundo, N. Mitochondrial biogenesis is transcriptionally repressed in lysosomal lipid storage diseases. Elife 2019, 8, e39598. [Google Scholar] [CrossRef]
- Pacheco, C.D.; Kunkel, R.; Lieberman, A.P. Autophagy in niemann-pick c disease is dependent upon beclin-1 and responsive to lipid trafficking defects. Hum. Mol. Genet. 2007, 16, 1495–1503. [Google Scholar] [CrossRef]
- Pfrieger, F.W. The niemann-pick type diseases—A synopsis of inborn errors in sphingolipid and cholesterol metabolism. Prog. Lipid Res. 2023, 90, 101225. [Google Scholar] [CrossRef]
- Freihuber, C.; Dahmani-Rabehi, B.; Brassier, A.; Broue, P.; Cances, C.; Chabrol, B.; Eyer, D.; Labarthe, F.; Latour, P.; Levade, T.; et al. Effects of miglustat therapy on neurological disorder and survival in early-infantile niemann-pick disease type c: A national french retrospective study. Orphanet J. Rare Dis. 2023, 18, 204. [Google Scholar] [CrossRef]
- Mengel, E.; Patterson, M.C.; Da Riol, R.M.; Del Toro, M.; Deodato, F.; Gautschi, M.; Grunewald, S.; Gronborg, S.; Harmatz, P.; Heron, B.; et al. Efficacy and safety of arimoclomol in niemann-pick disease type c: Results from a double-blind, randomised, placebo-controlled, multinational phase 2/3 trial of a novel treatment. J. Inherit. Metab. Dis. 2021, 44, 1463–1480. [Google Scholar] [CrossRef]
- Bremova-Ertl, T.; Ramaswami, U.; Brands, M.; Foltan, T.; Gautschi, M.; Gissen, P.; Gowing, F.; Hahn, A.; Jones, S.; Kay, R.; et al. Trial of n-acetyl-l-leucine in niemann-pick disease type c. New Engl. J. Med. 2024, 390, 421–431. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 339–358. [Google Scholar] [CrossRef]
- Eischer, N.; Arnold, M.; Mayer, A. Emerging roles of bet proteins in transcription and co-transcriptional rna processing. Wiley Interdiscip. Rev. -Rna 2023, 14, e1734. [Google Scholar] [CrossRef] [PubMed]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with bet inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef]
- Kulikowski, E.; Rakai, B.D.; Wong, N.C.W. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med. Res. Rev. 2021, 41, 223–245. [Google Scholar] [CrossRef]
- Martella, N.; Pensabene, D.; Varone, M.; Colardo, M.; Petraroia, M.; Sergio, W.; La Rosa, P.; Moreno, S.; Segatto, M. Bromodomain and extra-terminal proteins in brain physiology and pathology: Bet-ing on epigenetic regulation. Biomedicines 2023, 11, 750. [Google Scholar] [CrossRef]
- Segatto, M.; Fittipaldi, R.; Pin, F.; Sartori, R.; Ko, K.D.; Zare, H.; Fenizia, C.; Zanchettin, G.; Pierobon, E.S.; Hatakeyama, S.; et al. Epigenetic targeting of bromodomain protein brd4 counteracts cancer cachexia and prolongs survival. Nat. Commun. 2017, 8, 1707. [Google Scholar] [CrossRef]
- Segatto, M.; Szokoll, R.; Fittipaldi, R.; Bottino, C.; Nevi, L.; Mamchaoui, K.; Filippakopoulos, P.; Caretti, G. Bets inhibition attenuates oxidative stress and preserves muscle integrity in duchenne muscular dystrophy. Nat. Commun. 2020, 11, 6108. [Google Scholar] [CrossRef]
- Fu, L.; Wasiak, S.; Tsujikawa, L.M.; Rakai, B.D.; Stotz, S.C.; Wong, N.C.W.; Johansson, J.O.; Sweeney, M.; Mohan, C.M.; Khan, A.; et al. Inhibition of epigenetic reader proteins by apabetalone counters inflammation in activated innate immune cells from fabry disease patients receiving enzyme replacement therapy. Pharmacol. Res. Perspect. 2022, 10, e00949. [Google Scholar] [CrossRef]
- Zou, M.; Ke, Q.; Nie, Q.; Qi, R.L.; Zhu, X.F.; Liu, W.; Hu, X.B.; Sun, Q.; Fu, J.L.; Tang, X.C.; et al. Inhibition of cgas-sting by jq1 alleviates oxidative stress-induced retina inflammation and degeneration. Cell Death Differ. 2022, 29, 1816–1833. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of bet bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Tonini, C.; Colardo, M.; Colella, B.; Di Bartolomeo, S.; Berardinelli, F.; Caretti, G.; Pallottini, V.; Segatto, M. Inhibition of bromodomain and extraterminal domain (bet) proteins by jq1 unravels a novel epigenetic modulation to control lipid homeostasis. Int. J. Mol. Sci. 2020, 21, 1297. [Google Scholar] [CrossRef] [PubMed]
- Sakamaki, J.I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain protein brd4 is a transcriptional repressor of autophagy and lysosomal function. Mol. Cell 2017, 66, 517–532. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.E.; Eom, J.I.; Jeung, H.K.; Cheong, J.W.; Lee, J.Y.; Kim, J.S.; Min, Y.H. Targeting ampk-ulk1-mediated autophagy for combating bet inhibitor resistance in acute myeloid leukemia stem cells. Autophagy 2017, 13, 761–762. [Google Scholar] [CrossRef]
- Campbell, G.R.; Bruckman, R.S.; Herns, S.D.; Joshi, S.; Durden, D.L.; Spector, S.A. Induction of autophagy by pi3k/mtor and pi3k/mtor/brd4 inhibitors suppresses hiv-1 replication. J. Biol. Chem. 2018, 293, 5808–5820. [Google Scholar] [CrossRef]
- Sui, S.Y.; Zhang, J.; Xu, S.P.; Wang, Q.; Wang, P.Y.; Pang, D. Ferritinophagy is required for the induction of ferroptosis by the bromodomain protein brd4 inhibitor (+)-jq1 in cancer cells. Cell Death Dis. 2019, 10, 331. [Google Scholar] [CrossRef]
- Li, Y.; Xiang, J.; Zhang, J.; Lin, J.; Wu, Y.; Wang, X. Inhibition of brd4 by jq1 promotes functional recovery from spinal cord injury by activating autophagy. Front. Cell Neurosci. 2020, 14, 555591. [Google Scholar] [CrossRef]
- Shen, S.J.; Li, B.; Dai, J.J.; Wu, Z.K.; He, Y.; Wen, L.; Wang, X.P.; Hu, G.Y. Brd4 inhibition protects against acute pancreatitis through restoring impaired autophagic flux. Front. Pharmacol. 2020, 11, 618. [Google Scholar] [CrossRef]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A bet family protein degrader provokes senolysis by targeting nhej and autophagy in senescent cells. Nat. Commun. 2020, 11, 1935. [Google Scholar] [CrossRef]
- Gong, Z.G.; Zhao, Y.; Wang, Z.Y.; Fan, R.F.; Liu, Z.P.; Wang, L. Epigenetic regulator brd4 is involved in cadmium-induced acute kidney injury via contributing to lysosomal dysfunction, autophagy blockade and oxidative stress. J. Hazard. Mater. 2022, 423, 127110. [Google Scholar] [CrossRef]
- Yamamoto, T.; Ninomiya, H.; Matsumoto, M.; Ohta, Y.; Nanba, E.; Tsutsumi, Y.; Yamakawa, K.; Millat, G.; Vanier, M.T.; Pentchev, P.G.; et al. Genotype-phenotype relationship of niemann-pick disease type c: A possible correlation between clinical onset and levels of npc1 protein in isolated skin fibroblasts. J. Med. Genet. 2000, 37, 707–712. [Google Scholar] [CrossRef]
- Millat, G.; Marçais, C.; Tomasetto, C.; Chikh, K.; Fensom, A.H.; Harzer, K.; Wenger, D.A.; Ohno, K.; Vanier, M.T. Niemann-pick c1 disease: Correlations between npc1 mutations, levels of npc1 protein, and phenotypes emphasize the functional significance of the putative sterol-sensing domain and of the cysteine-rich luminal loop. Am. J. Hum. Genet. 2001, 68, 1373–1385. [Google Scholar] [CrossRef]
- Musalkova, D.; Majer, F.; Kuchar, L.; Luksan, O.; Asfaw, B.; Vlaskova, H.; Storkanova, G.; Reboun, M.; Poupetova, H.; Jahnova, H.; et al. Transcript, protein, metabolite and cellular studies in skin fibroblasts demonstrate variable pathogenic impacts of npc1 mutations. Orphanet J. Rare Dis. 2020, 15, 85. [Google Scholar] [CrossRef] [PubMed]
- Gelsthorpe, M.E.; Baumann, N.; Millard, E.; Gale, S.E.; Langmade, S.J.; Schaffer, J.E.; Ory, D.S. Niemann-pick type c1 i1061t mutant encodes a functional protein that is selected for endoplasmic reticulum-associated degradation due to protein misfolding. J. Biol. Chem. 2008, 283, 8229–8236. [Google Scholar] [CrossRef] [PubMed]
- Macías-Vidal, J.; Girós, M.; Guerrero, M.; Gascón, P.; Serratosa, J.; Bachs, O.; Coll, M.J. The proteasome inhibitor bortezomib reduced cholesterol accumulation in fibroblasts from niemann-pick type c patients carrying missense mutations. FEBS J. 2014, 281, 4450–4466. [Google Scholar] [CrossRef]
- Praggastis, M.; Tortelli, B.; Zhang, J.; Fujiwara, H.; Sidhu, R.; Chacko, A.; Chen, Z.; Chung, C.; Lieberman, A.P.; Sikora, J.; et al. A murine niemann-pick c1 i1061t knock-in model recapitulates the pathological features of the most prevalent human disease allele. J. Neurosci. 2015, 35, 8091–8106. [Google Scholar] [CrossRef]
- Schultz, M.L.; Krus, K.L.; Kaushik, S.; Dang, D.; Chopra, R.; Qi, L.; Shakkottai, V.G.; Cuervo, A.M.; Lieberman, A.P. Coordinate regulation of mutant npc1 degradation by selective er autophagy and march6-dependent erad. Nat. Commun. 2018, 9, 3671. [Google Scholar] [CrossRef]
- Feng, X.; Cozma, C.; Pantoom, S.; Hund, C.; Iwanov, K.; Petters, J.; Völkner, C.; Bauer, C.; Vogel, F.; Bauer, P.; et al. Determination of the pathological features of npc1 variants in a cellular complementation test. Int. J. Mol. Sci. 2019, 20, 5185. [Google Scholar] [CrossRef]
- Wang, C.; Scott, S.M.; Subramanian, K.; Loguercio, S.; Zhao, P.; Hutt, D.M.; Farhat, N.Y.; Porter, F.D.; Balch, W.E. Quantitating the epigenetic transformation contributing to cholesterol homeostasis using gaussian process. Nat. Commun. 2019, 10, 5052. [Google Scholar] [CrossRef]
- Pallottini, V.; Pfrieger, F.W. Understanding and treating niemann-pick type c disease: Models matter. Int. J. Mol. Sci. 2020, 21, 8979. [Google Scholar] [CrossRef]
- Vichaikul, S.; Gurrea-Rubio, M.; Amin, M.A.; Campbell, P.L.; Wu, Q.; Mattichak, M.N.; Brodie, W.D.; Palisoc, P.J.; Ali, M.; Muraoka, S.; et al. Inhibition of bromodomain extraterminal histone readers alleviates skin fibrosis in experimental models of scleroderma. JCI Insight 2022, 7, e150871. [Google Scholar] [CrossRef]
- Burmeister, A.; Stephan, A.; Alves Avelar, L.A.; Müller, M.R.; Seiwert, A.; Höfmann, S.; Fischer, F.; Torres-Gomez, H.; Hoffmann, M.J.; Niegisch, G.; et al. Establishment and evaluation of dual hdac/bet inhibitors as therapeutic options for germ cell tumors and other urological malignancies. Mol. Cancer Ther. 2022, 21, 1674–1688. [Google Scholar] [CrossRef] [PubMed]
- Pipalia, N.H.; Cosner, C.C.; Huang, A.; Chatterjee, A.; Bourbon, P.; Farley, N.; Helquist, P.; Wiest, O.; Maxfield, F.R. Histone deacetylase inhibitor treatment dramatically reduces cholesterol accumulation in niemann-pick type c1 mutant human fibroblasts. Proc. Natl. Acad. Sci. USA 2011, 108, 5620–5625. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Chung, C.; Shen, D.; Xu, H.; Lieberman, A.P. Ryanodine receptor antagonists adapt npc1 proteostasis to ameliorate lipid storage in niemann-pick type c disease fibroblasts. Hum. Mol. Genet. 2012, 21, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, J.L.; Larson, D.M.; Wassif, C.A.; Yanjanin, N.M.; Anderson, S.M.; Kirby, M.R.; Trivedi, N.S.; Porter, F.D.; Pavan, W.J. A somatic cell defect is associated with the onset of neurological symptoms in a lysosomal storage disease. Mol. Genet. Metab. 2013, 110, 188–190. [Google Scholar] [CrossRef]
- Xu, M.; Liu, K.; Swaroop, M.; Sun, W.; Dehdashti, S.J.; McKew, J.C.; Zheng, W. A phenotypic compound screening assay for lysosomal storage diseases. J. Biomol. Screen. 2014, 19, 168–175. [Google Scholar] [CrossRef]
- te Vruchte, D.; Speak, A.O.; Wallom, K.L.; Al Eisa, N.; Smith, D.A.; Hendriksz, C.J.; Simmons, L.; Lachmann, R.H.; Cousins, A.; Hartung, R.; et al. Relative acidic compartment volume as a lysosomal storage disorder-associated biomarker. J. Clin. Investig. 2014, 124, 1320–1328. [Google Scholar] [CrossRef]
- Samie, M.A.; Xu, H. Lysosomal exocytosis and lipid storage disorders. J. Lipid Res. 2014, 55, 995–1009. [Google Scholar] [CrossRef]
- Chen, F.W.; Li, C.; Ioannou, Y.A. Cyclodextrin induces calcium-dependent lysosomal exocytosis. PLoS ONE 2010, 5, e15054. [Google Scholar] [CrossRef]
- Demais, V.; Barthelemy, A.; Perraut, M.; Ungerer, N.; Keime, C.; Reibel, S.; Pfrieger, F.W. Reversal of pathologic lipid accumulation in npc1-deficient neurons by drug-promoted release of lamp1-coated lamellar inclusions. J. Neurosci. 2016, 36, 8012–8025. [Google Scholar] [CrossRef]
- Strauss, K.; Goebel, C.; Runz, H.; Mobius, W.; Weiss, S.; Feussner, I.; Simons, M.; Schneider, A. Exosome secretion ameliorates lysosomal storage of cholesterol in niemann-pick type c disease. J. Biol. Chem. 2010, 285, 26279–26288. [Google Scholar] [CrossRef]
- Bruno, F.; Camuso, S.; Capuozzo, E.; Canterini, S. The antifungal antibiotic filipin as a diagnostic tool of cholesterol alterations in lysosomal storage diseases and neurodegenerative disorders. Antibiotics 2023, 12, 122. [Google Scholar] [CrossRef] [PubMed]
- Lu, F.; Liang, Q.; Abi-Mosleh, L.; Das, A.; De Brabander, J.K.; Goldstein, J.L.; Brown, M.S. Identification of npc1 as the target of u18666a, an inhibitor of lysosomal cholesterol export and ebola infection. Elife 2015, 4, e12177. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Lee, B.H.; Lee, Y.S.; Kang, K.S. Defective cholesterol traffic and neuronal differentiation in neural stem cells of niemann-pick type c disease improved by valproic acid, a histone deacetylase inhibitor. Biochem. Biophys. Res. Commun. 2007, 360, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Munkacsi, A.B.; Chen, F.W.; Brinkman, M.A.; Higaki, K.; Gutiérrez, G.D.; Chaudhari, J.; Layer, J.V.; Tong, A.; Bard, M.; Boone, C.; et al. An “exacerbate-reverse” strategy in yeast identifies histone deacetylase inhibition as a correction for cholesterol and sphingolipid transport defects in human niemann-pick type c disease. J. Biol. Chem. 2011, 286, 23842–23851. [Google Scholar] [CrossRef]
- Wehrmann, Z.T.; Hulett, T.W.; Huegel, K.L.; Vaughan, K.T.; Wiest, O.; Helquist, P.; Goodson, H. Quantitative comparison of the efficacy of various compounds in lowering intracellular cholesterol levels in niemann-pick type c fibroblasts. PLoS ONE 2012, 7, e48561. [Google Scholar] [CrossRef]
- Gevry, N.Y.; Lalli, E.; Sassone-Corsi, P.; Murphy, B.D. Regulation of niemann-pick c1 gene expression by the 3′5′-cyclic adenosine monophosphate pathway in steroidogenic cells. Mol. Endocrinol. 2003, 17, 704–715. [Google Scholar] [CrossRef]
- Nunes, M.J.; Moutinho, M.; Gama, M.J.; Rodrigues, C.M.; Rodrigues, E. Histone deacetylase inhibition decreases cholesterol levels in neuronal cells by modulating key genes in cholesterol synthesis, uptake and efflux. PLoS ONE 2013, 8, e53394. [Google Scholar] [CrossRef]
- Newton, J.; Hait, N.C.; Maceyka, M.; Colaco, A.; Maczis, M.; Wassif, C.A.; Cougnoux, A.; Porter, F.D.; Milstien, S.; Platt, N.; et al. Fty720/fingolimod increases npc1 and npc2 expression and reduces cholesterol and sphingolipid accumulation in niemann-pick type c mutant fibroblasts. FASEB J. 2017, 31, 1719–1730. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Subramanian, K.; Mao, S.; Ralph, H.; Hutt, D.M.; Scott, S.M.; Balch, W.E.; Maxfield, F.R. Histone deacetylase inhibitors correct the cholesterol storage defect in most niemann-pick c1 mutant cells. J. Lipid Res. 2017, 58, 695–708. [Google Scholar] [CrossRef]
- Zampieri, S.; Bembi, B.; Rosso, N.; Filocamo, M.; Dardis, A. Treatment of human fibroblasts carrying npc1 missense mutations with mg132 leads to an improvement of intracellular cholesterol trafficking. JIMD Rep. 2012, 2, 59–69. [Google Scholar] [CrossRef]
- Ohgane, K.; Karaki, F.; Dodo, K.; Hashimoto, Y. Discovery of oxysterol-derived pharmacological chaperones for npc1: Implication for the existence of second sterol-binding site. Chem. Biol. 2013, 20, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Nakasone, N.; Nakamura, Y.S.; Higaki, K.; Oumi, N.; Ohno, K.; Ninomiya, H. Endoplasmic reticulum-associated degradation of niemann-pick c1: Evidence for the role of heat shock proteins and identification of lysine residues that accept ubiquitin. J. Biol. Chem. 2014, 289, 19714–19725. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, K.; Rauniyar, N.; Lavalleé-Adam, M.; Yates, J.R., 3rd; Balch, W.E. Quantitative analysis of the proteome response to the histone deacetylase inhibitor (hdaci) vorinostat in niemann-pick type c1 disease. Mol. Cell Proteom. 2017, 16, 1938–1957. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Scott, S.M.; Sun, S.; Zhao, P.; Hutt, D.M.; Shao, H.; Gestwicki, J.E.; Balch, W.E. Individualized management of genetic diversity in niemann-pick c1 through modulation of the hsp70 chaperone system. Hum. Mol. Genet. 2020, 29, 1–19. [Google Scholar] [CrossRef]
- Pipalia, N.H.; Saad, S.Z.; Subramanian, K.; Cross, A.; Al-Motawa, A.; Garg, K.; Blagg, B.S.J.; Neckers, L.; Helquist, P.; Wiest, O.; et al. Hsp90 inhibitors reduce cholesterol storage in niemann-pick type c1 mutant fibroblasts. J. Lipid Res. 2021, 62, 100114. [Google Scholar] [CrossRef]
- Völkner, C.; Pantoom, S.; Liedtke, M.; Lukas, J.; Hermann, A.; Frech, M.J. Assessment of fda-approved drugs as a therapeutic approach for niemann-pick disease type c1 using patient-specific ipsc-based model systems. Cells 2022, 11, 319. [Google Scholar] [CrossRef]
- Schultz, M.L.; Schache, K.J.; Azaria, R.D.; Kuiper, E.Q.; Erwood, S.; Ivakine, E.A.; Farhat, N.Y.; Porter, F.D.; Pathmasiri, K.C.; Cologna, S.M.; et al. Species-specific differences in npc1 protein trafficking govern therapeutic response in niemann-pick type c disease. JCI Insight 2022, 7, e160308. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (bet) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (p-tefb) from 7sk small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Fiskus, W.; Sharma, S.; Qi, J.; Valenta, J.A.; Schaub, L.J.; Shah, B.; Peth, K.; Portier, B.P.; Rodriguez, M.; Devaraj, S.G.T.; et al. Highly active combination of brd4 antagonist and histone deacetylase inhibitor against human acute myelogenous leukemia cells. Mol. Cancer Ther. 2014, 13, 1142–1154. [Google Scholar] [CrossRef]
- Bhadury, J.; Nilsson, L.M.; Veppil Muralidharan, S.; Green, L.C.; Li, Z.; Gesner, E.M.; Hansen, H.C.; Keller, U.B.; McLure, K.G.; Nilsson, J.A. Bet and hdac inhibitors induce similar genes and biological effects and synergize to kill in myc-induced murine lymphoma. Proc. Natl. Acad. Sci. USA 2014, 111, E2721–E2730. [Google Scholar] [CrossRef]
- Fu, R.; Yanjanin, N.M.; Elrick, M.J.; Ware, C.; Lieberman, A.P.; Porter, F.D. Apolipoprotein e genotype and neurological disease onset in niemann–pick disease, type c1. Am. J. Med. Genet. A 2012, 158A, 2775–2780. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, J.L.; Watkins-Chow, D.E.; Baxter, L.L.; Elliot, G.; Harper, U.L.; Wincovitch, S.M.; Wedel, J.C.; Incao, A.A.; Huebecker, M.; Boehm, F.J.; et al. Genetic background modifies phenotypic severity and longevity in a mouse model of niemann-pick disease type c1. Dis. Model. Mech. 2020, 13, dmm042614. [Google Scholar] [CrossRef] [PubMed]
- Farhat, N.Y.; Alexander, D.; McKee, K.; Iben, J.; Rodriguez-Gil, J.L.; Wassif, C.A.; Cawley, N.X.; Balch, W.E.; Porter, F.D. Sterol o-acyltransferase 1 (soat1): A genetic modifier of niemann-pick disease, type c1. Int. J. Mol. Sci. 2024, 25, 4217. [Google Scholar] [CrossRef]
- Pugach, E.K.; Feltes, M.; Kaufman, R.J.; Ory, D.S.; Bang, A.G. High-content screen for modifiers of niemann-pick type c disease in patient cells. Hum. Mol. Genet. 2018, 27, 2101–2112. [Google Scholar] [CrossRef]
- D’Angelo, G.; La Manno, G. The lipotype hypothesis. Nat. Rev. Mol. Cell Biol. 2023, 24, 1–2. [Google Scholar] [CrossRef]
- Vacca, F.; Vossio, S.; Mercier, V.; Moreau, D.; Johnson, S.; Scott, C.C.; Montoya, J.P.; Moniatte, M.; Gruenberg, J. Cyclodextrin triggers mcoln1-dependent endo-lysosome secretion in niemann-pick type c cells. J. Lipid Res. 2019, 60, 832–843. [Google Scholar] [CrossRef]
- Feltes, M.; Gale, S.E.; Moores, S.; Ory, D.S.; Schaffer, J.E. Monitoring the itinerary of lysosomal cholesterol in niemann-pick type c1-deficient cells after cyclodextrin treatment. J. Lipid Res. 2020, 61, 403–412. [Google Scholar] [CrossRef]
- Palmulli, R.; Couty, M.; Piontek, M.C.; Ponnaiah, M.; Dingli, F.; Verweij, F.J.; Charrin, S.; Tantucci, M.; Sasidharan, S.; Rubinstein, E.; et al. Cd63 sorts cholesterol into endosomes for storage and distribution via exosomes. Nat. Cell Biol. 2024, 26, 1093–1109. [Google Scholar] [CrossRef]
- Xu, M.; Liu, K.; Swaroop, M.; Porter, F.D.; Sidhu, R.; Firnkes, S.; Ory, D.S.; Marugan, J.J.; Xiao, J.; Southall, N.; et al. Δ-tocopherol reduces lipid accumulation in niemann-pick type c1 and wolman cholesterol storage disorders. J. Biol. Chem. 2012, 287, 39349–39360. [Google Scholar] [CrossRef]
- Cao, Q.; Zhong, X.Z.; Zou, Y.; Zhang, Z.; Toro, L.; Dong, X.P. Bk channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal ca2+ release. Dev. Cell 2015, 33, 427–441. [Google Scholar] [CrossRef]
- Arguello, G.; Balboa, E.; Tapia, P.J.; Castro, J.; Yanez, M.J.; Mattar, P.; Pulgar, R.; Zanlungo, S. Genistein activates transcription factor eb and corrects niemann-pick c phenotype. Int. J. Mol. Sci. 2021, 22, 4220. [Google Scholar] [CrossRef] [PubMed]
- Scotto Rosato, A.; Krogsaeter, E.K.; Jaślan, D.; Abrahamian, C.; Montefusco, S.; Soldati, C.; Spix, B.; Pizzo, M.T.; Grieco, G.; Böck, J.; et al. Tpc2 rescues lysosomal storage in mucolipidosis type iv, niemann–pick type c1, and batten disease. EMBO Mol. Med. 2022, 14, e15377. [Google Scholar] [CrossRef] [PubMed]
- Cougnoux, A.; Pergande, M.R.; Serna-Perez, F.; Cologna, S.M. Investigation of 2-hydroxypropyl-β-cyclodextrin treatment in a neuronal-like cell model of niemann–pick type c using quantitative proteomics. J. Am. Soc. Mass. Spectrom. 2023, 34, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Barthelemy, A.; Demais, V.; Stancu, I.-C.; Vasile, E.; Houben, T.; Reber, M.; Pallottini, V.; Perraut, M.; Reibel, S.; Pfrieger, F.W. Glial contribution to cyclodextrin-mediated reversal of cholesterol accumulation in murine npc1-deficient neurons in vivo. Neurobiol. Dis. 2021, 158, 105469. [Google Scholar] [CrossRef]
- Du, K.; Chen, H.; Pan, Z.; Zhao, M.; Cheng, S.; Luo, Y.; Zhang, W.; Li, D. Small-molecule activation of tfeb alleviates niemann–pick disease type c via promoting lysosomal exocytosis and biogenesis. eLife 2025, 13, RP103137. [Google Scholar] [CrossRef]
- Garcia, G.; Bar-Ziv, R.; Averbukh, M.; Dasgupta, N.; Dutta, N.; Zhang, H.; Fan, W.; Moaddeli, D.; Tsui, C.K.; Castro Torres, T.; et al. Large-scale genetic screens identify bet-1 as a cytoskeleton regulator promoting actin function and life span. Aging Cell 2023, 22, e13742. [Google Scholar] [CrossRef]
- Shen, H.; Li, J.; Xie, X.; Yang, H.; Zhang, M.; Wang, B.; Kent, K.C.; Plutzky, J.; Guo, L.-W. Brd2 regulation of sigma-2 receptor upon cholesterol deprivation. Life Sci. Alliance 2021, 4, e201900540. [Google Scholar] [CrossRef]
- McLure, K.G.; Gesner, E.M.; Tsujikawa, L.; Kharenko, O.A.; Attwell, S.; Campeau, E.; Wasiak, S.; Stein, A.; White, A.; Fontano, E.; et al. Rvx-208, an inducer of apoa-i in humans, is a bet bromodomain antagonist. PLoS ONE 2014, 8, e83190. [Google Scholar] [CrossRef]
- Schultz, M.L.; Fawaz, M.V.; Azaria, R.D.; Hollon, T.C.; Liu, E.A.; Kunkel, T.J.; Halseth, T.A.; Krus, K.L.; Ming, R.; Morin, E.E.; et al. Synthetic high-density lipoprotein nanoparticles for the treatment of niemann-pick diseases. BMC Med. 2019, 17, 200. [Google Scholar] [CrossRef]
- Kunkel, T.J.; Townsend, A.; Sullivan, K.A.; Merlet, J.; Schuchman, E.H.; Jacobson, D.A.; Lieberman, A.P. The cholesterol transporter npc1 is essential for epigenetic regulation and maturation of oligodendrocyte lineage cells. Nat. Commun. 2023, 14, 3964. [Google Scholar] [CrossRef]
- Cruz, J.C.; Chang, T.Y. Fate of endogenously synthesized cholesterol in niemann-pick type c1 cells. J. Biol. Chem. 2000, 275, 41309–41316. [Google Scholar] [CrossRef] [PubMed]
- Watabe, K.; Ida, H.; Uehara, K.; Oyanagi, K.; Sakamoto, T.; Tanaka, J.; Garver, W.S.; Miyawaki, S.; Ohno, K.; Eto, Y. Establishment and characterization of immortalized schwann cells from murine model of niemann-pick disease type c (spm/spm). J. Peripher. Nerv. Syst. 2001, 6, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Stein, V.M.; Crooks, A.; Ding, W.; Prociuk, M.; O’Donnell, P.; Bryan, C.; Sikora, T.; Dingemanse, J.; Vanier, M.T.; Walkley, S.U.; et al. Miglustat improves purkinje cell survival and alters microglial phenotype in feline niemann-pick disease type c. J. Neuropathol. Exp. Neurol. 2012, 71, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Millat, G.; Marçais, C.; Rafi, M.A.; Yamamoto, T.; Morris, J.A.; Pentchev, P.G.; Ohno, K.; Wenger, D.A.; Vanier, M.T. Niemann-pick c1 disease: The i1061t substitution is a frequent mutant allele in patients of western european descent and correlates with a classic juvenile phenotype. Am. J. Hum. Genet. 1999, 65, 1321–1329. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nanba, E.; Ninomiya, H.; Higaki, K.; Taniguchi, M.; Zhang, H.D.; Akaboshi, S.; Watanabe, Y.; Takeshima, T.; Inui, K.; et al. Npc1 gene mutations in japanese patients with niemann-pick disease type c. Hum. Genet. 1999, 105, 10–16. [Google Scholar] [CrossRef]
- Park, W.D.; O’Brien, J.F.; Lundquist, P.A.; Kraft, D.L.; Vockley, C.W.; Karnes, P.S.; Patterson, M.C.; Snow, K. Identification of 58 novel mutations in niemann-pick disease type c: Correlation with biochemical phenotype and importance of ptc1 -like domains in npc1. Hum. Mutat. 2003, 22, 313–325. [Google Scholar] [CrossRef]
- Team, R.C. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Parente, M.; Barthelemy, A.; Tonini, C.; Caputo, S.; Sacchi, A.; Leone, S.; Segatto, M.; Pfrieger, F.W.; Pallottini, V. Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease. Int. J. Mol. Sci. 2025, 26, 5769. https://doi.org/10.3390/ijms26125769
Parente M, Barthelemy A, Tonini C, Caputo S, Sacchi A, Leone S, Segatto M, Pfrieger FW, Pallottini V. Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease. International Journal of Molecular Sciences. 2025; 26(12):5769. https://doi.org/10.3390/ijms26125769
Chicago/Turabian StyleParente, Martina, Amélie Barthelemy, Claudia Tonini, Sara Caputo, Alessandra Sacchi, Stefano Leone, Marco Segatto, Frank W. Pfrieger, and Valentina Pallottini. 2025. "Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease" International Journal of Molecular Sciences 26, no. 12: 5769. https://doi.org/10.3390/ijms26125769
APA StyleParente, M., Barthelemy, A., Tonini, C., Caputo, S., Sacchi, A., Leone, S., Segatto, M., Pfrieger, F. W., & Pallottini, V. (2025). Exploration of Bromodomain Proteins as Drug Targets for Niemann–Pick Type C Disease. International Journal of Molecular Sciences, 26(12), 5769. https://doi.org/10.3390/ijms26125769