
The Originally Established PBE Cell Line as a Reliable In Vitro Model for Investigating SIV Infection and Immunity

 ,
, 
 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
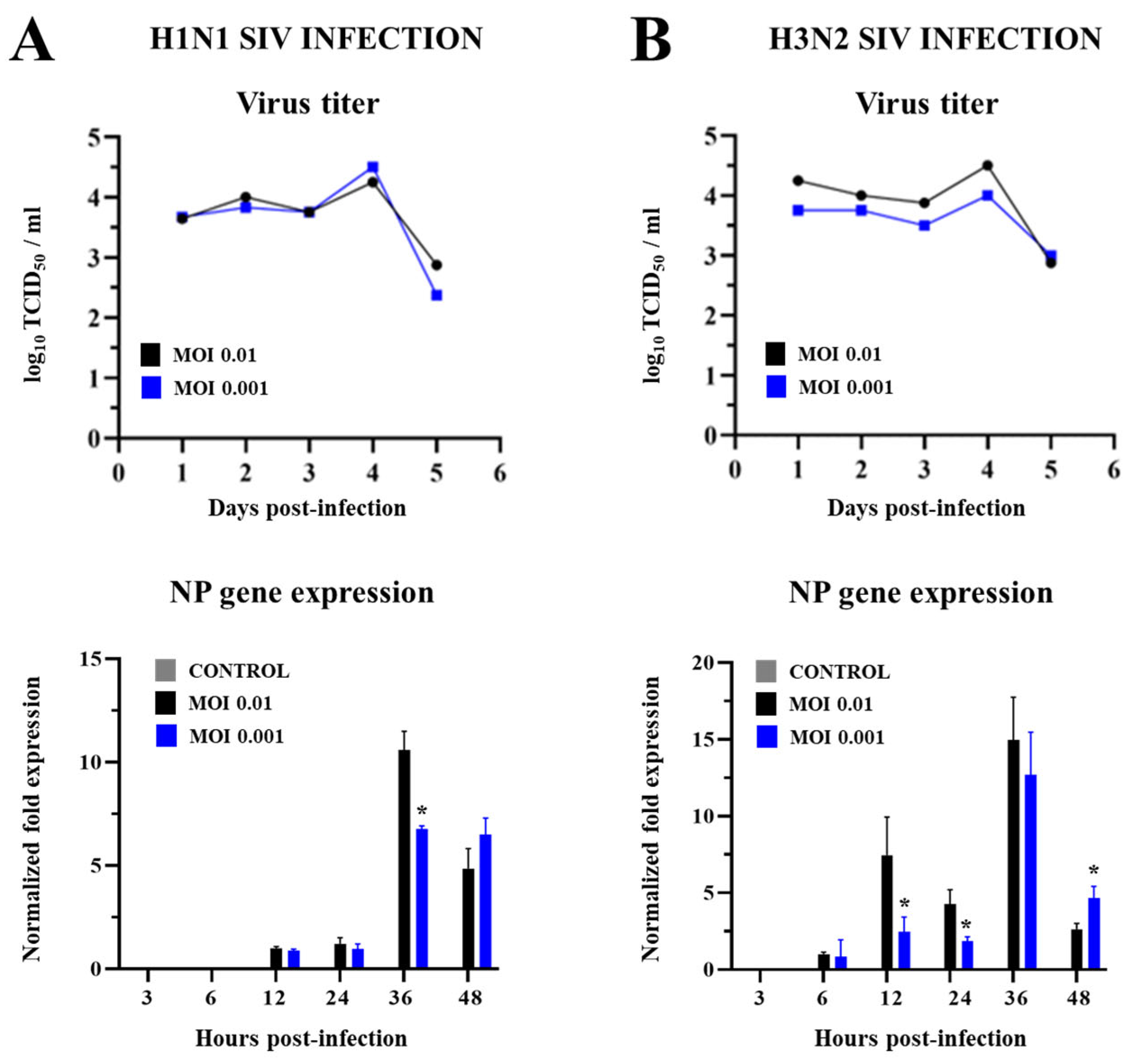
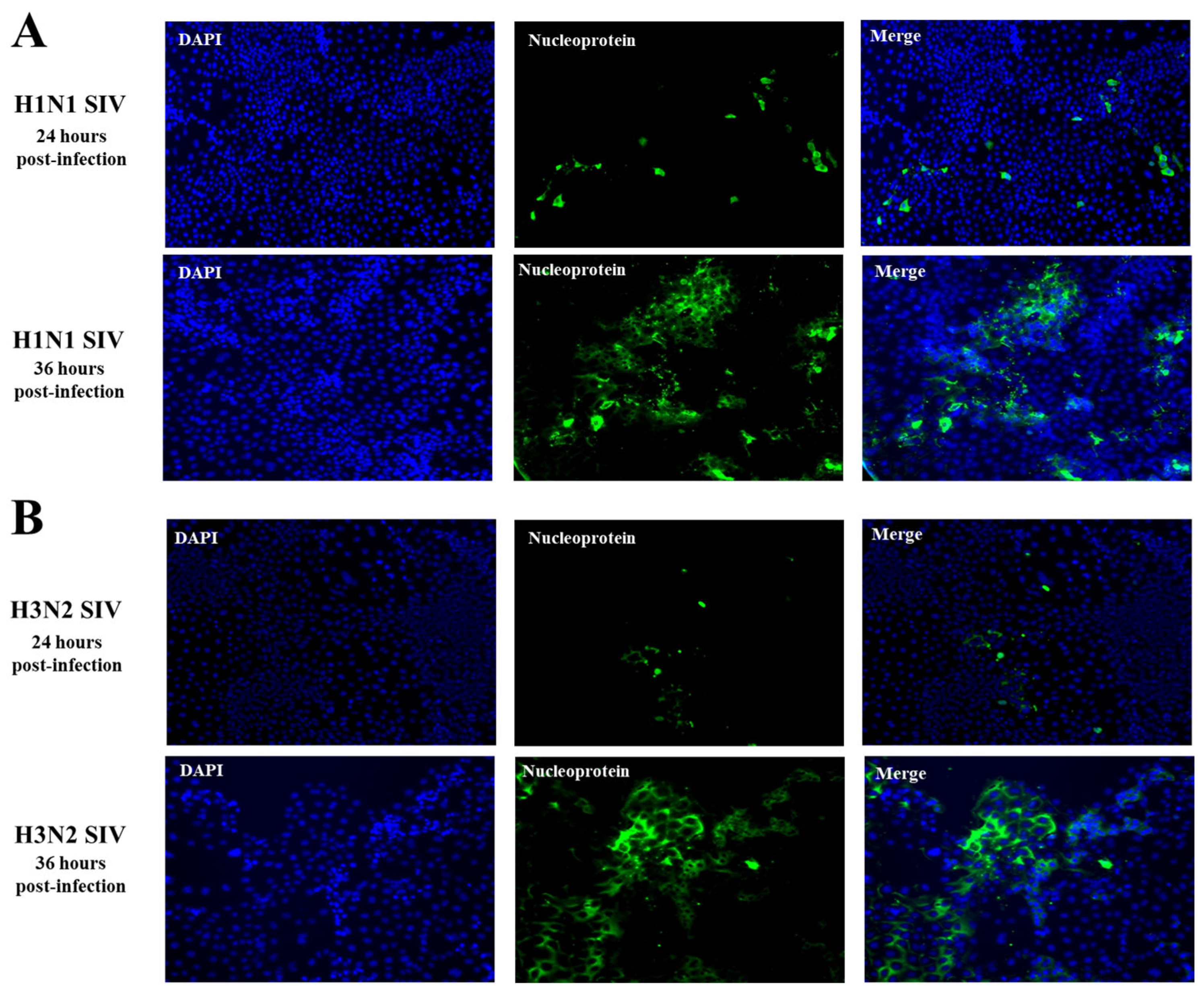
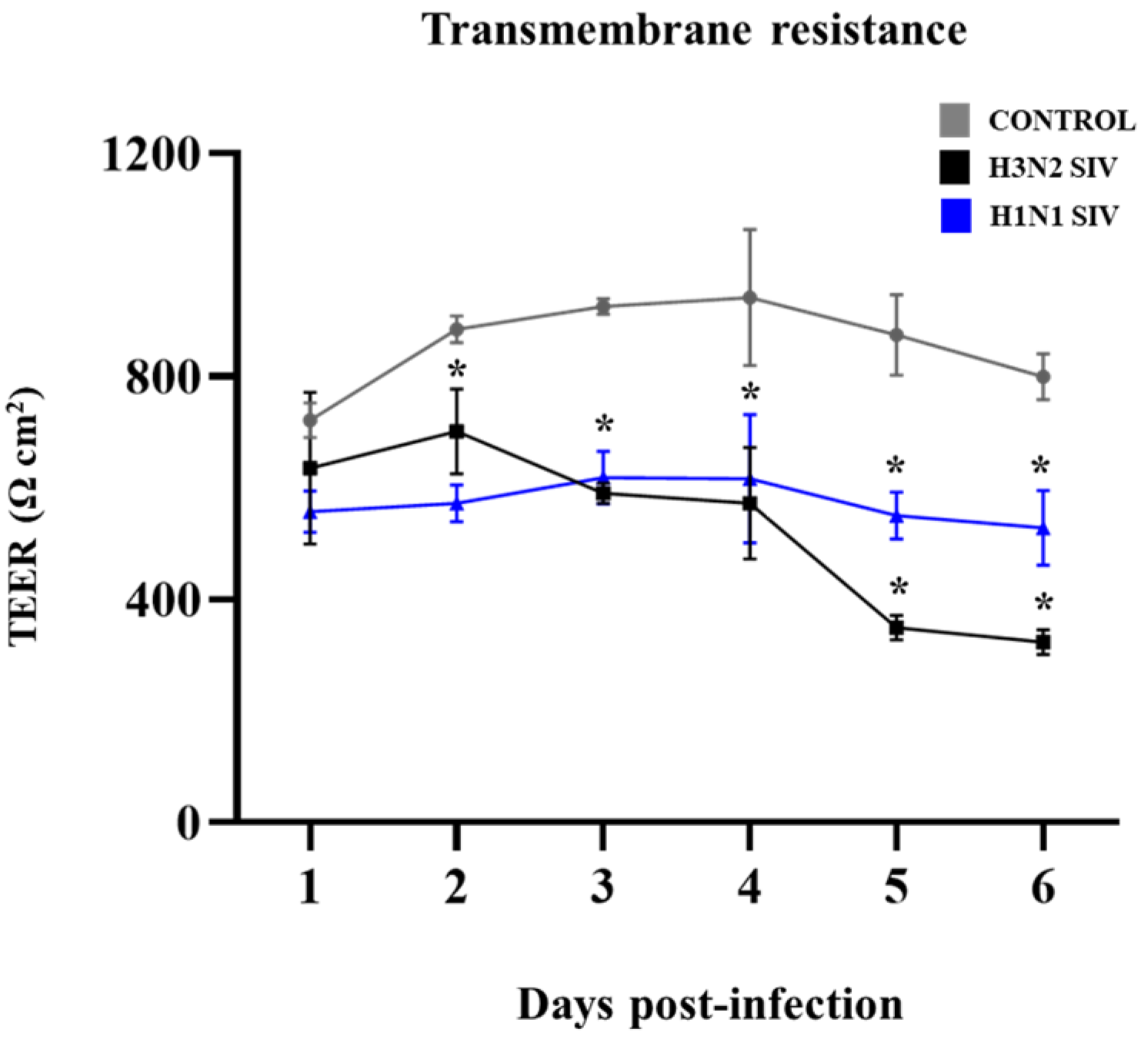
2.1. SIV Efficiently Infects PBE Cells
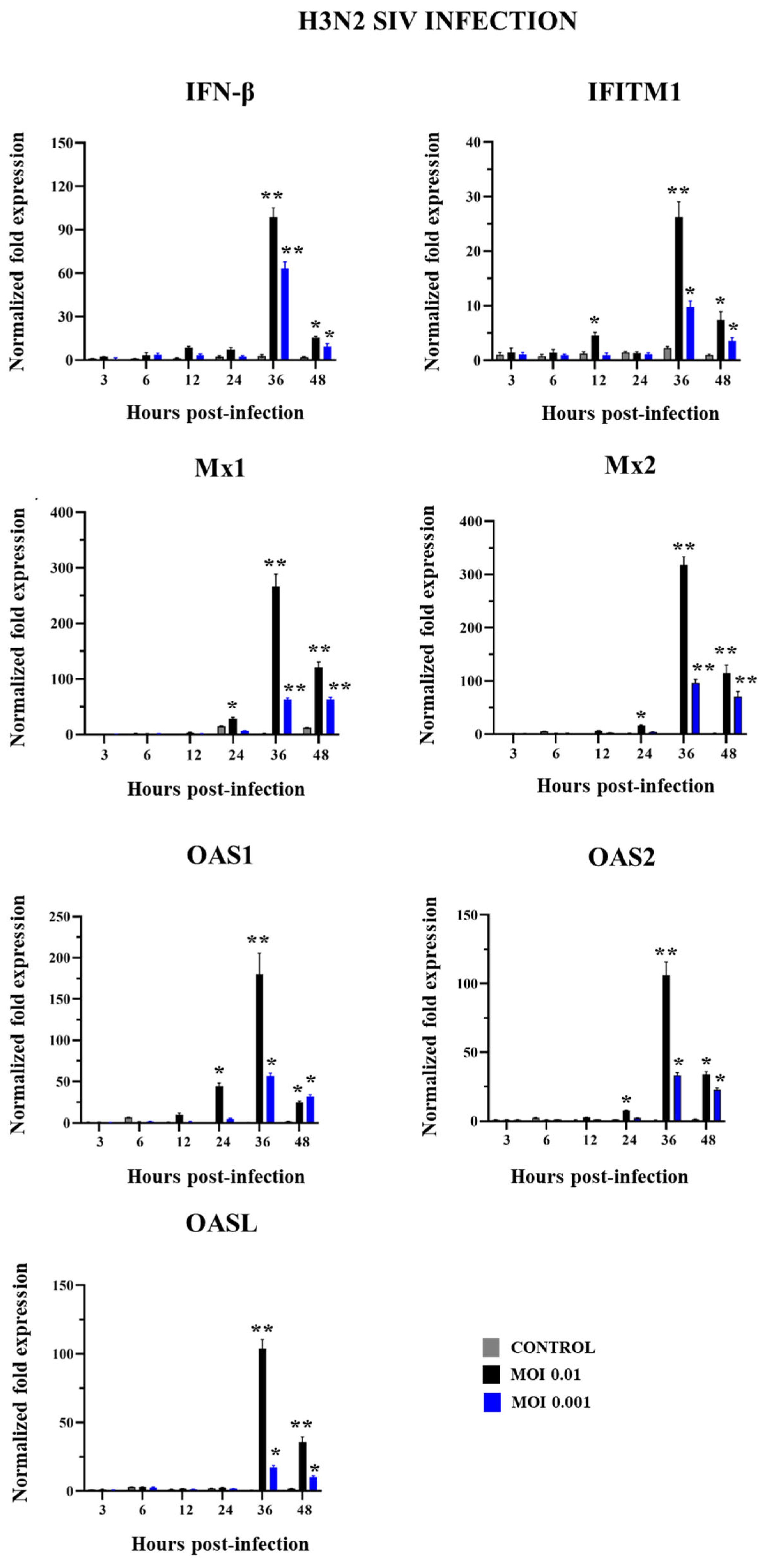
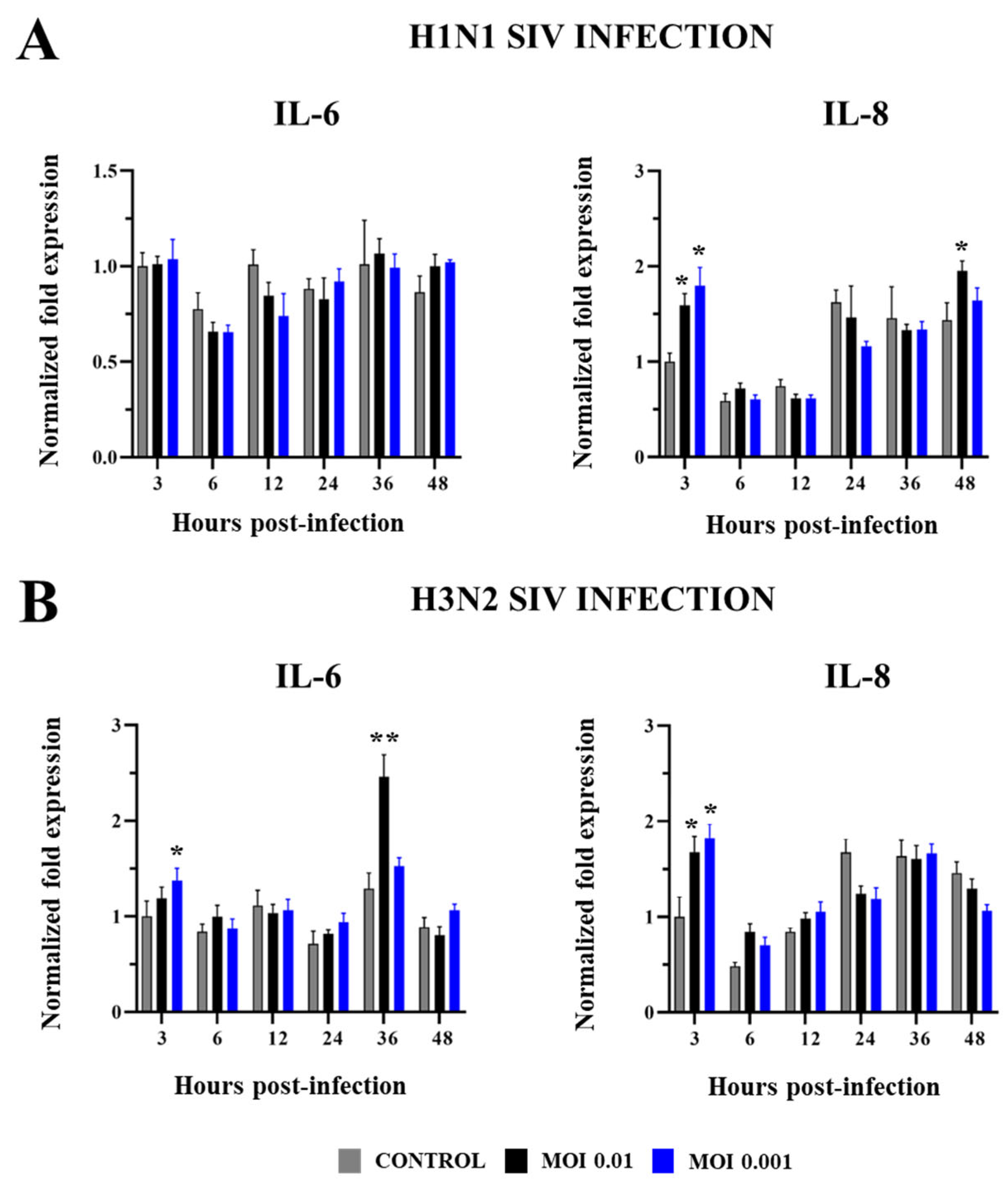
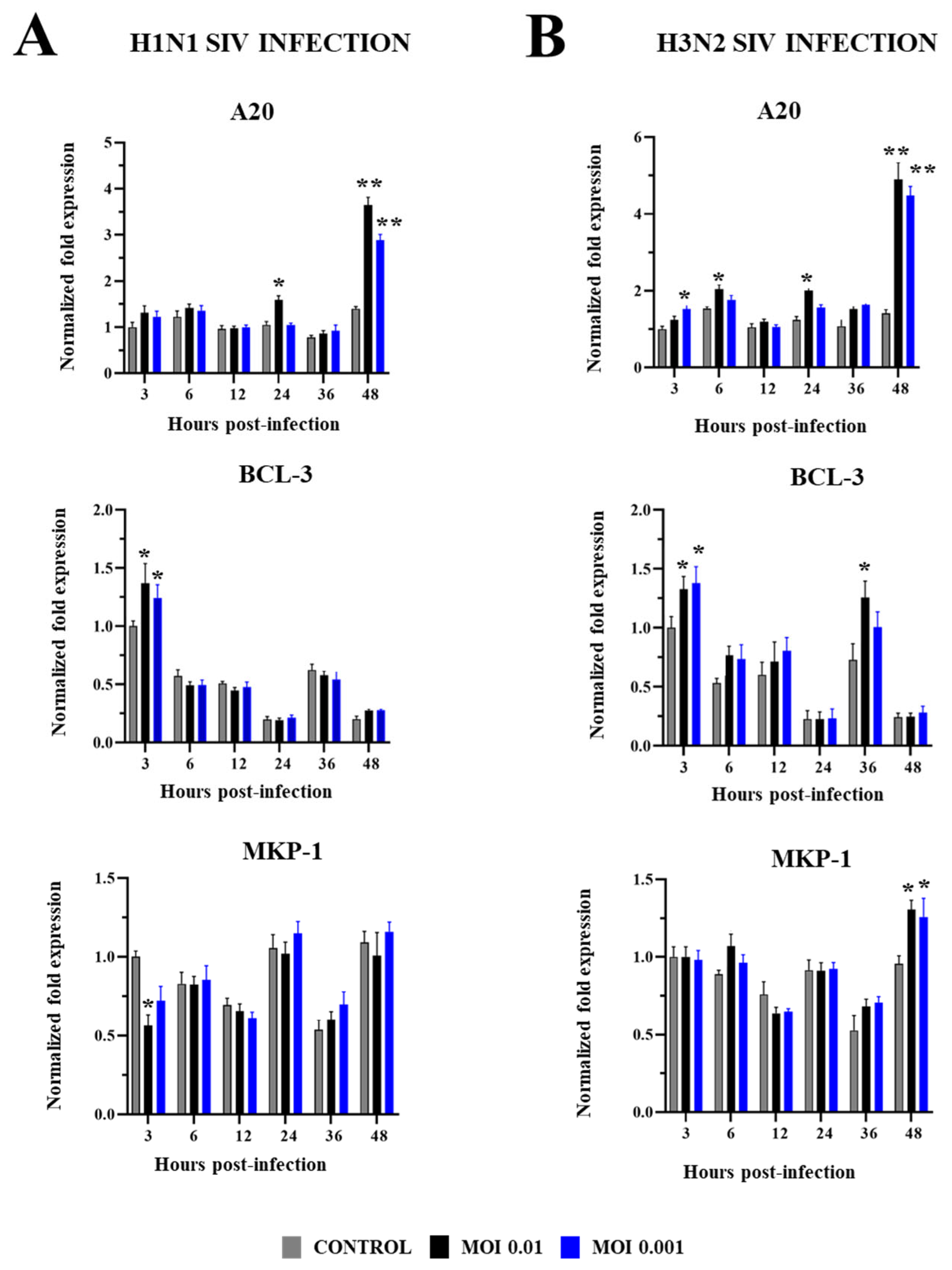
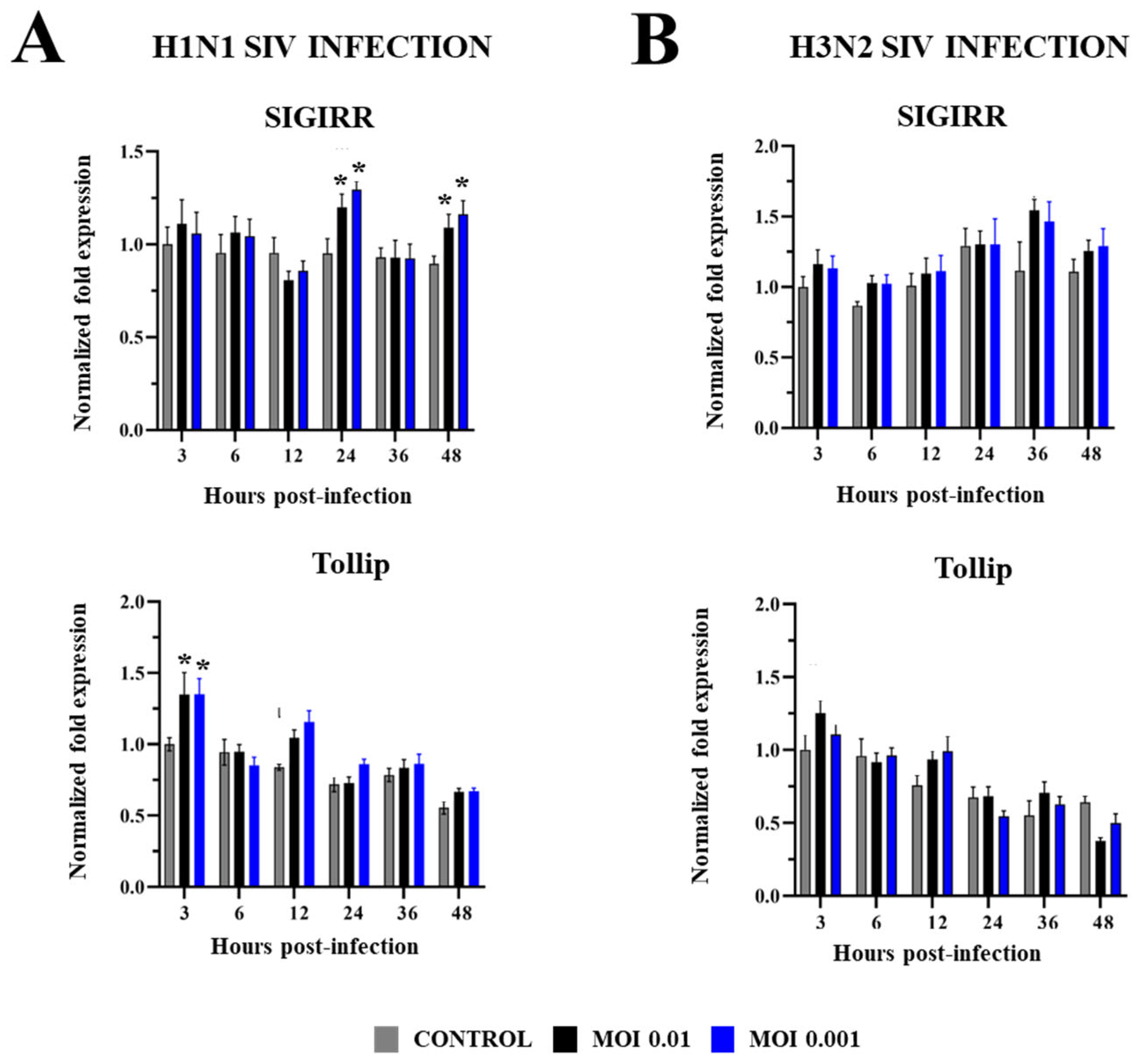
2.2. Innate Immune Response in PBE Cells Triggered by SIV Infection
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Treatments
4.2. Virus and Titration
4.3. Immunofluorescence Staining for SIV
4.4. Transepithelial Electrical Resistance (TEER)
4.5. Real-Time Quantitative PCR
4.6. Scanning Electron Microscopy (SEM)
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Crisci, E.; Mussa, T.; Fraile, L.; Montoya, M. Review: Influenza virus in pigs. Mol. Immunol. 2013, 55, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Sarli, G.; D’Annunzio, G.; Gobbo, F.; Benazzi, C.; Ostanello, F. The role of pathology in the diagnosis of swine respiratory disease. Vet. Sci. 2021, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Baker, R.E.; Mahmud, A.S.; Miller, I.F.; Rajeev, M.; Rasambainarivo, F.; Rice, B.L.; Takahashi, S.; Tatem, A.J.; Wagner, C.E.; Wang, L.F.; et al. Infectious disease in an era of global change. Nat. Rev. Microbiol. 2022, 20, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Ma, W. Swine influenza virus: Current status and challenge. Virus Res. 2020, 288, 198118. [Google Scholar] [CrossRef]
- Roach, S.N.; Shepherd, F.K.; Mickelson, C.K.; Fiege, J.K.; Thielen, B.K.; Pross, L.M.; Sanders, A.E.; Mitchell, J.S.; Robertson, M.; Fife, B.T.; et al. Tropism for ciliated cells is the dominant driver of influenza viral burst size in the human airway. Proc. Natl. Acad. Sci. USA 2024, 121, e2320303121. [Google Scholar] [CrossRef]
- Punyadarsaniya, D.; Liang, C.H.; Winter, C.; Petersen, H.; Rautenschlein, S.; Hennig-Pauka, I.; Schwegmann-Wessels, C.; Wu, C.Y.; Wong, C.H.; Herrler, G. Infection of differentiated porcine airway epithelial cells by influenza virus: Differential susceptibility to infection by porcine and avian viruses. PLoS ONE 2011, 6, e28429. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Ching, R.H.H.; Chan, S.K.H.; Nicholls, J.M.; Sachs, N.; Clevers, H.; Peiris, J.S.M.; Chan, M.C.W. Tropism, replication competence, and innate immune responses of influenza virus: An analysis of human airway organoids and ex-vivo bronchus cultures. Lancet Respir. Med. 2018, 6, 846–854. [Google Scholar] [CrossRef]
- Ibricevic, A.; Pekosz, A.; Walter, M.J.; Newby, C.; Battaile, J.T.; Brown, E.G.; Holtzman, M.J.; Brody, S.L. Influenza virus receptor specificity and cell tropism in mouse and human airway epithelial cells. J. Virol. 2006, 80, 7469–7480. [Google Scholar] [CrossRef]
- Sreenivasan, C.C.; Thomas, M.; Antony, L.; Wormstadt, T.; Hildreth, M.B.; Wang, D.; Hause, B.; Francis, D.H.; Li, F.; Kaushik, R.S. Development and characterization of swine primary respiratory epithelial cells and their susceptibility to infection by four influenza virus types. Virology 2019, 528, 152–163. [Google Scholar] [CrossRef]
- Krunkosky, M.; Krunkosky, T.M.; Meliopoulos, V.; Kyriakis, C.S.; Schultz-Cherry, S.; Tompkins, S.M. Establishment of Swine Primary Nasal, Tracheal, and Bronchial Epithelial Cell Culture Models for the Study of Influenza Virus Infection. J. Virol. Methods 2024, 327, 114943. [Google Scholar] [CrossRef]
- Reeves, S.R.; Barrow, K.A.; White, M.P.; Rich, L.M.; Naushab, M.; Debley, J.S. Stability of gene expression by primary bronchial epithelial cells over increasing passage number. BMC Pulm. Med. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Meliopoulos, V.; Cherry, S.; Wohlgemuth, N.; Honce, R.; Barnard, K.; Gauger, P. Primary swine respiratory epithelial cell lines for the efficient isolation and propagation of influenza a viruses. J. Virol. 2020, 94, e01091-20. [Google Scholar] [CrossRef] [PubMed]
- Rijsbergen, L.C.; van Dijk, L.L.A.; Engel, M.F.M.; de Vries, R.D.; de Swart, R.L. In vitro modelling of respiratory virus infections in human airway epithelial cells—A systematic review. Front. Immunol. 2021, 12, 683002. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Pierson, M.; Uprety, T.; Zhu, L.; Ran, Z.; Sreenivasan, C.C.; Wang, D.; Hause, B.; Francis, D.H.; Li, F.; et al. Comparison of Porcine Airway and Intestinal Epithelial Cell Lines for the Susceptibility and Expression of Pattern Recognition Receptors upon Influenza Virus Infection. Viruses 2018, 10, 312. [Google Scholar] [CrossRef]
- Ferrari, M.; Scalvini, A.; Losio, M.N.; Corradi, A.; Soncini, M.; Bignotti, E.; Milanesi, E.; Ajmone-Marsan, P.; Barlati, S.; Bellotti, D.; et al. Establishment and characterization of two new pig cell lines for use in virological diagnostic laboratories. J. Virol. Methods 2003, 107, 205–212. [Google Scholar] [CrossRef]
- Xie, X.; Gan, Y.; Pang, M.; Shao, G.; Zhang, L.; Liu, B.; Xu, Q.; Wang, H.; Feng, Y.; Yu, Y.; et al. Establishment and characterization of a telomerase-immortalized porcine bronchial epithelial cell line. J. Cell Physiol. 2018, 233, 9763–9776. [Google Scholar] [CrossRef]
- Fukuyama, K.; Zhuang, T.; Toyoshi, E.; Raya Tonetti, F.; Saha, S.; Zhou, B.; Ikeda-Ohtsubo, W.; Nishiyama, K.; Aso, H.; Villena, J.; et al. Establishment of a porcine bronchial epithelial cell line and its application to study innate immunity in the respiratory epithelium. Front. Immunol. 2023, 14, 1117102. [Google Scholar] [CrossRef]
- Delgado-Ortega, M.; Melo, S.; Punyadarsaniya, D.; Ramé, C.; Olivier, M.; Soubieux, D.; Marc, D.; Simon, G.; Herrler, G.; Berri, M.; et al. Innate immune response to a H3N2 subtype swine influenza virus in newborn porcine trachea cells, alveolar macrophages, and precision-cut lung slices. Vet. Res. 2014, 45, 42. [Google Scholar] [CrossRef]
- Wu, N.H.; Yang, W.; Beineke, A.; Dijkman, R.; Matrosovich, M.; Baumgärtner, W.; Thiel, V.; Valentin-Weigand, P.; Meng, F.; Herrler, G. The differentiated airway epithelium infected by influenza viruses maintains the bar rier function despite a dramatic loss of ciliated cells. Sci. Rep. 2016, 6, 39668. [Google Scholar] [CrossRef]
- Yu, W.C.; Chan, R.W.; Wang, J.; Travanty, E.A.; Nicholls, J.M.; Peiris, J.S.; Mason, R.J.; Chan, M.C. Viral replication and innate host responses in primary human alveolar epithelial cells and alveolar macrophages infected with influenza H5N1 and H1N1 viruses. J. Virol. 2011, 85, 6844–6855. [Google Scholar] [CrossRef]
- Weinheimer, V.K.; Becher, A.; Tönnies, M.; Holland, G.; Knepper, J.; Bauer, T.T.; Schneider, P.; Neudecker, J.; Rückert, J.C.; Szymanski, K.; et al. Influenza A viruses target type II pneumocytes in the human lung. J. Infect. Dis. 2012, 206, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Dürrwald, R.; Meng, F.; Tong, J.; Wu, N.H.; Su, A.; Yin, X.; Haas, L.; Schmidtke, M.; Zell, R.; et al. Infection Studies in Pigs and Porcine Airway Epithelial Cells Reveal an Evolution of A(H1N1)pdm09 Influenza A Viruses Toward Lower Virulence. J. Infect. Dis. 2019, 219, 1596–1604. [Google Scholar] [CrossRef]
- Villalón-Letelier, F.; Brooks, A.G.; Saunders, P.M.; Londrigan, S.L.; Reading, P.C. Host Cell Restriction Factors That Limit Influenza A Infection. Viruses 2017, 9, 376. [Google Scholar] [CrossRef]
- Fields, B.N. Fields’ Virology; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; Volume 1. [Google Scholar]
- Haller, O.; Staeheli, P.; Kochs, G. Interferon-induced Mx proteins in antiviral host defense. Biochimie 2007, 89, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Haller, O.; Frese, M.; Kochs, G. Mx proteins: Mediators of innate resistance to RNA viruses. Rev. Sci. Tech. 1998, 17, 220–230. [Google Scholar] [CrossRef]
- Drappier, M.; Michiels, T. Inhibition of the OAS/RNase L pathway by viruses. Curr. Opin. Virol. 2015, 15, 19–26. [Google Scholar] [CrossRef]
- Chang, Y.; Kang, J.S.; Jung, K.; Chung, D.H.; Ha, S.J.; Kim, Y.J.; Kim, H.Y. OASL1-Mediated Inhibition of Type I IFN Reduces Influenza A Infection-Induced Airway Inflammation by Regulating ILC2s. Allergy Asthma Immunol. Res. 2022, 14, 99–116. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Correll, K.; Zemans, R.L.; Leslie, C.C.; Murphy, R.C.; Mason, R.J. Influenza induces IL-8 and GM-CSF secretion by human alveolar epithelial cells through HGF/c-Met and TGF-α/EGFR signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2015, 308, L1178–L1188. [Google Scholar] [CrossRef]
- Farzin, H.; Toroghi, R.; Haghparast, A. Up-Regulation of Pro-Inflammatory Cytokines and Chemokine Production in Avian Influenza H9N2 Virus-Infected Human Lung Epithelial Cell Line (A549). Immunol. Investig. 2016, 45, 116–129. [Google Scholar] [CrossRef]
- Song, B.M.; Kang, Y.M.; Kim, H.S.; Seo, S.H. Induction of inflammatory cytokines and toll-like receptors in human normal respiratory epithelial cells infected with seasonal H1N1, 2009 pandemic H1N1, seasonal H3N2, and highly pathogenic H5N1 influenza virus. Viral Immunol. 2011, 24, 179–187. [Google Scholar] [CrossRef]
- Alagarasu, K.; Kaushal, H.; Shinde, P.; Kakade, M.; Chaudhary, U.; Padbidri, V.; Sangle, S.A.; Salvi, S.; Bavdekar, A.R.; D’costa, P.; et al. TNFA and IL10 Polymorphisms and IL-6 and IL-10 Levels Influence Disease Severity in Influenza A(H1N1)pdm09 Virus Infected Patients. Genes 2021, 12, 1914. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, J.; Gong, Y.; Gu, Y.; Xiang, Q.; Tang, L.L. Interleukin-6 and granulocyte colony-stimulating factor as predictors of the prognosis of influenza-associated pneumonia. BMC Infect. Dis. 2022, 22, 343. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yan, R.; Chen, B.; Pan, Q.; Chen, Y.; Hong, J.; Zhang, L.; Liu, W.; Wang, S.; Chen, J.L. Influenza Virus-Induced Robust Expression of SOCS3 Contributes to Excessive Production of IL-6. Front. Immunol. 2019, 10, 1843. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.L.; Wang, C.T.; Yang, S.J.; Leu, C.H.; Chen, S.H.; Wu, C.L.; Shiau, A.L. IL-6 ameliorates acute lung injury in influenza virus infection. Sci. Rep. 2017, 7, 43829. [Google Scholar] [CrossRef]
- Bakre, A.A.; Jones, L.P.; Murray, J.; Reneer, Z.B.; Meliopoulos, V.A.; Cherry, S.; Schultz-Cherry, S.; Tripp, R.A. Innate Antiviral Cytokine Response to Swine Influenza Virus by Swine Respiratory Epithelial Cells. J. Virol. 2021, 95, e0069221. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Diamond, M.S.; Suthar, M.S. Negative regulators of the RIG-I-like receptor signaling pathway. Eur. J. Immunol. 2017, 47, 615–628. [Google Scholar] [CrossRef]
- Maelfait, J.; Roose, K.; Vereecke, L.; Mc Guire, C.; Sze, M.; Schuijs, M.J.; Willart, M.; Ibañez, L.I.; Hammad, H.; Lambrecht, B.N.; et al. A20 Deficiency in Lung Epithelial Cells Protects against Influenza A Virus Infection. PLoS Pathog. 2016, 12, e1005410. [Google Scholar] [CrossRef]
- Franzoso, G.; Carlson, L.; Scharton-Kersten, T.; Shores, E.W.; Epstein, S.; Grinberg, A.; Tran, T.; Shacter, E.; Leonardi, A.; Anver, M.; et al. Critical roles for the Bcl-3 oncoprotein in T cell-mediated immunity, splenic microarchitecture, and germinal center reactions. Immunity 1997, 6, 479–490. [Google Scholar] [CrossRef]
- Wancket, L.; de Oliveira, S.; Liu, Y. MAP kinase phosphatase (MKP)-1 in the innate immune response to influenza infection (42.4). J. Immunol. 2010, 1, 184. [Google Scholar] [CrossRef]
- Feng, W.; Sun, X.; Shi, N.; Zhang, M.; Guan, Z.; Duan, M. Influenza a virus NS1 protein induced A20 contributes to viral replication by suppressing interferon-induced antiviral response. Biochem. Biophys. Res. Commun. 2017, 482, 1107–1113. [Google Scholar] [CrossRef]
- Onose, A.; Hashimoto, S.; Hayashi, S.; Maruoka, S.; Kumasawa, F.; Mizumura, K.; Jibiki, I.; Matsumoto, K.; Gon, Y.; Kobayashi, T.; et al. An inhibitory effect of A20 on NF-kappaB activation in airway epithelium upon influenza virus infection. Eur. J. Pharmacol. 2006, 541, 198–204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Dong, C.; Sun, X.; Li, Z.; Zhang, M.; Guan, Z.; Duan, M. Induction of the cellular miR-29c by influenza virus inhibits the innate immune response through protection of A20 mRNA. Biochem. Biophys. Res. Commun. 2014, 450, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Hsu, A.C.; Zuo, X.; Guo, X.; Zhou, Z.; Jiang, S.; Ouyang, Z.; Wang, F. Chronic exposure to low-level lipopolysaccharide dampens influenza-mediated inflammatory response via A20 and PPAR network. Front. Immunol. 2023, 14, 1119473. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, J.; Huang, Y.; Song, X.; Niu, Z.; Gao, Z.; Wang, H. Bcl-3 suppresses Tax-induced NF-κB activation through p65 nuclear translocation blockage in HTLV-1-infected cells. Int. J. Oncol. 2013, 42, 269–276. [Google Scholar] [CrossRef]
- Jamaluddin, M.; Choudhary, S.; Wang, S.; Casola, A.; Huda, R.; Garofalo, R.P.; Ray, S.; Brasier, A.R. Respiratory syncytial virus-inducible BCL-3 expression antagonizes the STAT/IRF and NF-kappaB signaling pathways by inducing histone deacetylase 1 recruitment to the interleukin-8 promoter. J. Virol. 2005, 79, 15302–15313. [Google Scholar] [CrossRef]
- Nencioni, L.; De Chiara, G.; Sgarbanti, R.; Amatore, D.; Aquilano, K.; Marcocci, M.E.; Serafino, A.; Torcia, M.; Cozzolino, F.; Ciriolo, M.R.; et al. Bcl-2 expression and p38MAPK activity in cells infected with influenza A virus: Impact on virally induced apoptosis and viral replication. J. Biol. Chem. 2009, 284, 16004–16015. [Google Scholar] [CrossRef]
- Ye, Y.; Shi, Y.; Wei, Z.; Liu, H.; Li, W. SIGIRR suppresses hepatitis B virus X protein-induced chronic inflammation in hepatocytes. Gene 2024, 928, 148768. [Google Scholar] [CrossRef]
- Zhang, B.; Xu, S.; Liu, M.; Wei, Y.; Wang, Q.; Shen, W.; Lei, C.Q.; Zhu, Q. The nucleoprotein of influenza A virus inhibits the innate immune response by inducing mitophagy. Autophagy 2023, 19, 1916–1933. [Google Scholar] [CrossRef]
- Huang, C.; Jiang, D.; Francisco, D.; Berman, R.; Wu, Q.; Ledford, J.G.; Moore, C.M.; Ito, Y.; Stevenson, C.; Munson, D.; et al. Tollip SNP rs5743899 modulates human airway epithelial responses to rhinovirus infection. Clin. Exp. Allergy 2016, 46, 1549–1563. [Google Scholar] [CrossRef]
- Dakhama, A.; Al Mubarak, R.; Pavelka, N.; Voelker, D.; Seibold, M.; Ledford, J.G.; Kraft, M.; Li, L.; Chu, H.W. Tollip inhibits ST2 signaling in airway epithelial cells exposed to type 2 cytokines and rhinovirus. J. Innate Immun. 2020, 12, 103–115. [Google Scholar] [CrossRef]
- Schaunaman, N.; Dimasuay, K.G.; Cervantes, D.; Li, L.; Numata, M.; Kraft, M.; Chu, H.W. Tollip Inhibits IL-33 Release and Inflammation in Influenza A Virus-Infected Mouse Airways. J. Innate Immun. 2023, 15, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Agraval, H.; Gao, J.; Schaunaman, N.; Hua, H.; Vandivier, R.W.; Numata, M.; Day, B.J.; Chu, H.W. TOLLIP downregulation by cigarette smoke exposure impairs human lung defense against Influenza A virus infection. Am. J. Pathol. 2025, 195, 1124–1140. [Google Scholar]
- Zhang, C.; Wu, X.; Zhao, Y.; Deng, Z.; Qian, G. SIGIRR inhibits toll-like receptor 4, 5, 9-mediated immune responses in human airway epithelial cells. Mol. Biol. Rep. 2011, 38, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Ueno-Shuto, K.; Kamei, S.; Hayashi, M.; Fukuyama, A.; Uchida, Y.; Tokutomi, N.; Suico, M.A.; Kai, H.; Shuto, T. A Splice Switch in SIGIRR Causes a Defect of IL-37-Dependent Anti-Inflammatory Activity in Cystic Fibrosis Airway Epithelial Cells. Int. J. Mol. Sci. 2022, 23, 7748. [Google Scholar] [CrossRef]
- Cumplido-Laso, G.; Benitez, D.A.; Mulero-Navarro, S.; Carvajal-Gonzalez, J.M. Transcriptional Regulation of Airway Epithelial Cell Differentiation: Insights into the Notch Pathway and Beyond. Int. J. Mol. Sci. 2023, 24, 14789. [Google Scholar] [CrossRef]
- Kuek, L.E.; Lee, R.J. First contact: The role of respiratory cilia in host-pathogen interactions in the airways. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L603–L619. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, X.-C.; Fukuyama, K.; Albarracin, L.; Imamura, Y.; Namai, F.; Gong, W.; Ikeda-Ohtsubo, W.; Nishiyama, K.; Villena, J.; Kitazawa, H. The Originally Established PBE Cell Line as a Reliable In Vitro Model for Investigating SIV Infection and Immunity. Int. J. Mol. Sci. 2025, 26, 5764. https://doi.org/10.3390/ijms26125764
Bai X-C, Fukuyama K, Albarracin L, Imamura Y, Namai F, Gong W, Ikeda-Ohtsubo W, Nishiyama K, Villena J, Kitazawa H. The Originally Established PBE Cell Line as a Reliable In Vitro Model for Investigating SIV Infection and Immunity. International Journal of Molecular Sciences. 2025; 26(12):5764. https://doi.org/10.3390/ijms26125764
Chicago/Turabian StyleBai, Xi-Chen, Kohtaro Fukuyama, Leonardo Albarracin, Yoshiya Imamura, Fu Namai, Weichen Gong, Wakako Ikeda-Ohtsubo, Keita Nishiyama, Julio Villena, and Haruki Kitazawa. 2025. "The Originally Established PBE Cell Line as a Reliable In Vitro Model for Investigating SIV Infection and Immunity" International Journal of Molecular Sciences 26, no. 12: 5764. https://doi.org/10.3390/ijms26125764
APA StyleBai, X.-C., Fukuyama, K., Albarracin, L., Imamura, Y., Namai, F., Gong, W., Ikeda-Ohtsubo, W., Nishiyama, K., Villena, J., & Kitazawa, H. (2025). The Originally Established PBE Cell Line as a Reliable In Vitro Model for Investigating SIV Infection and Immunity. International Journal of Molecular Sciences, 26(12), 5764. https://doi.org/10.3390/ijms26125764