
Evaluating Genetic Regulators of MicroRNAs Using Machine Learning Models
 ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
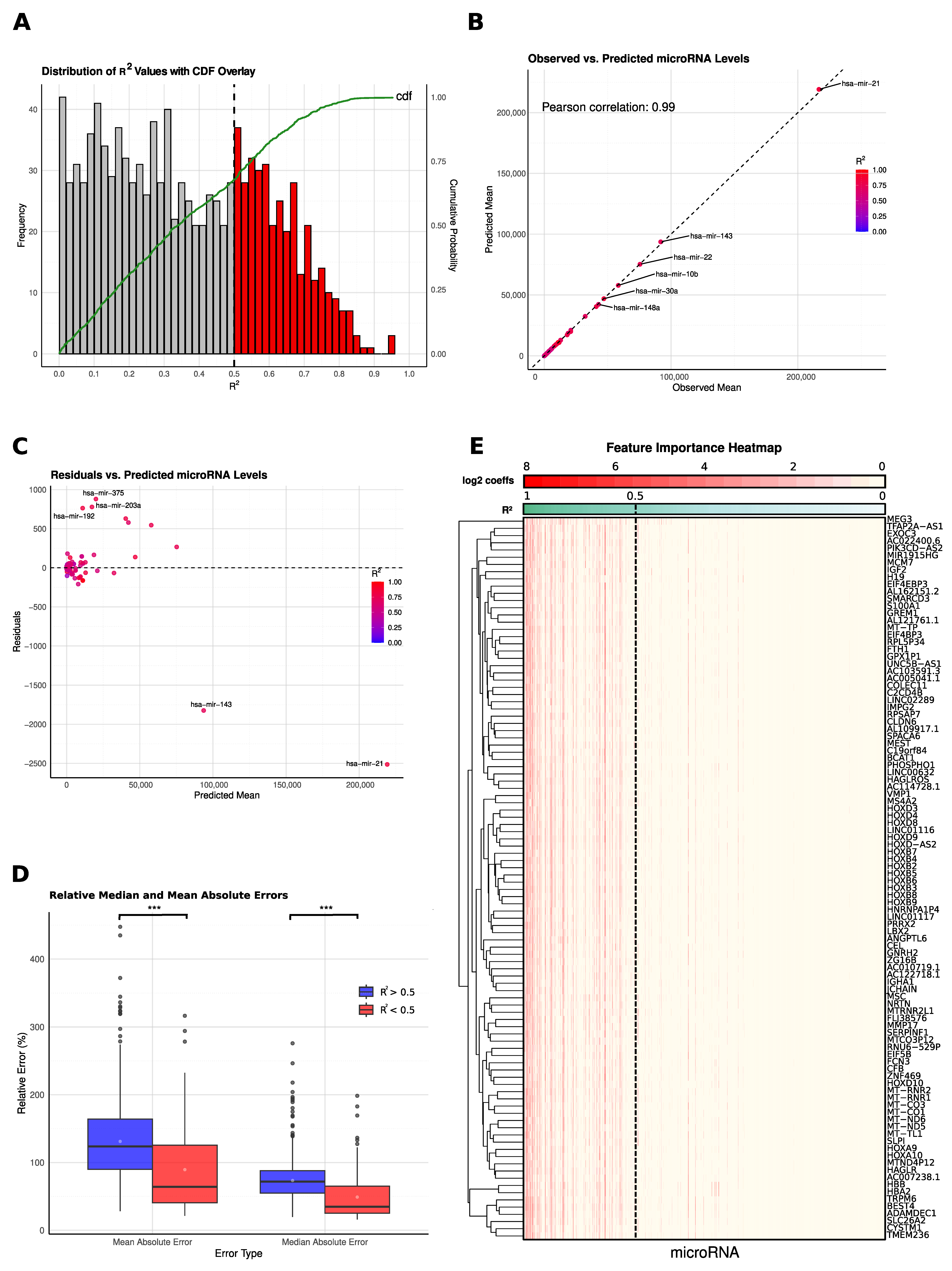
2.1. Model Development and Performance Assessment
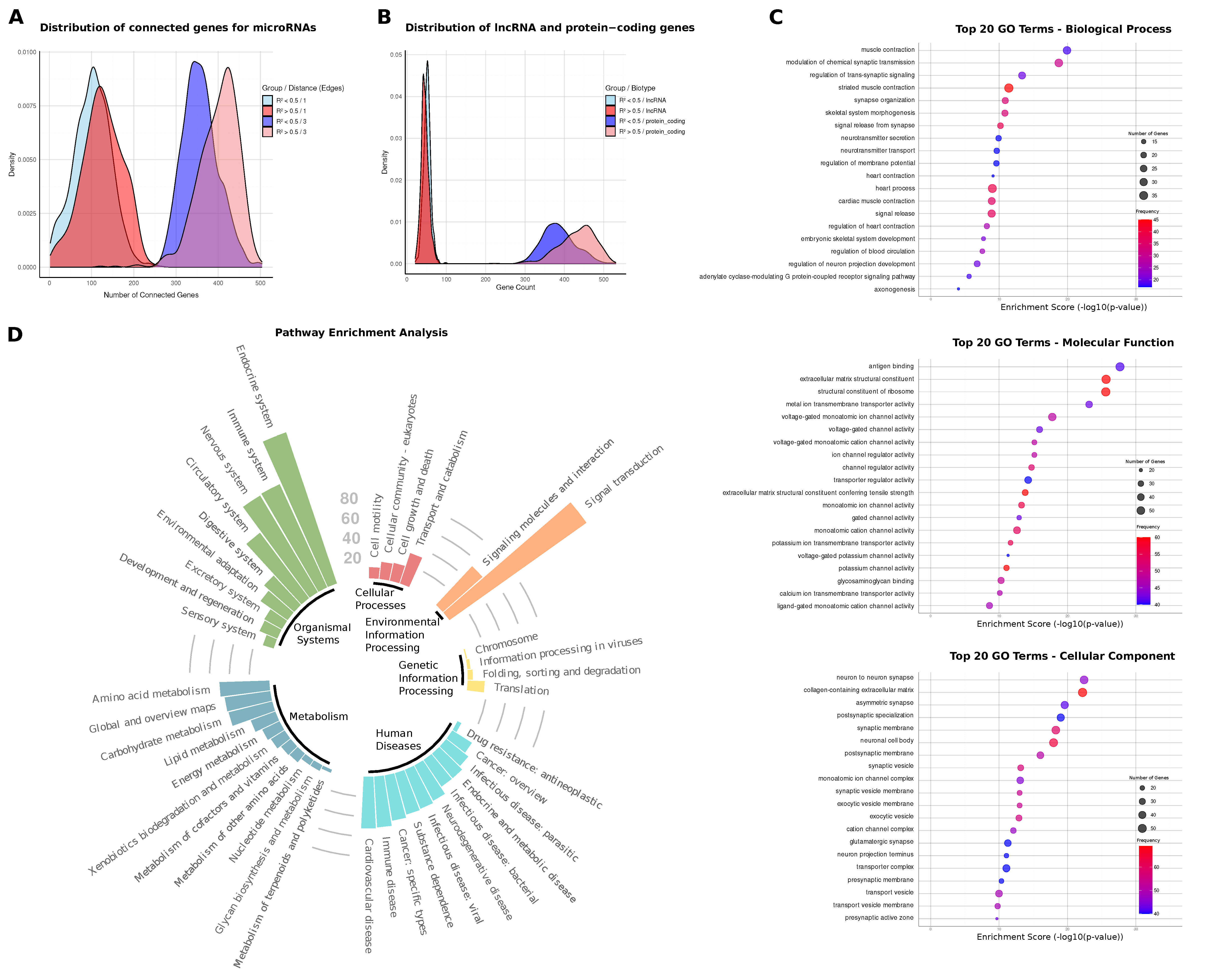
2.2. MiRNA–Gene Network Connectivity and Centrality Analysis
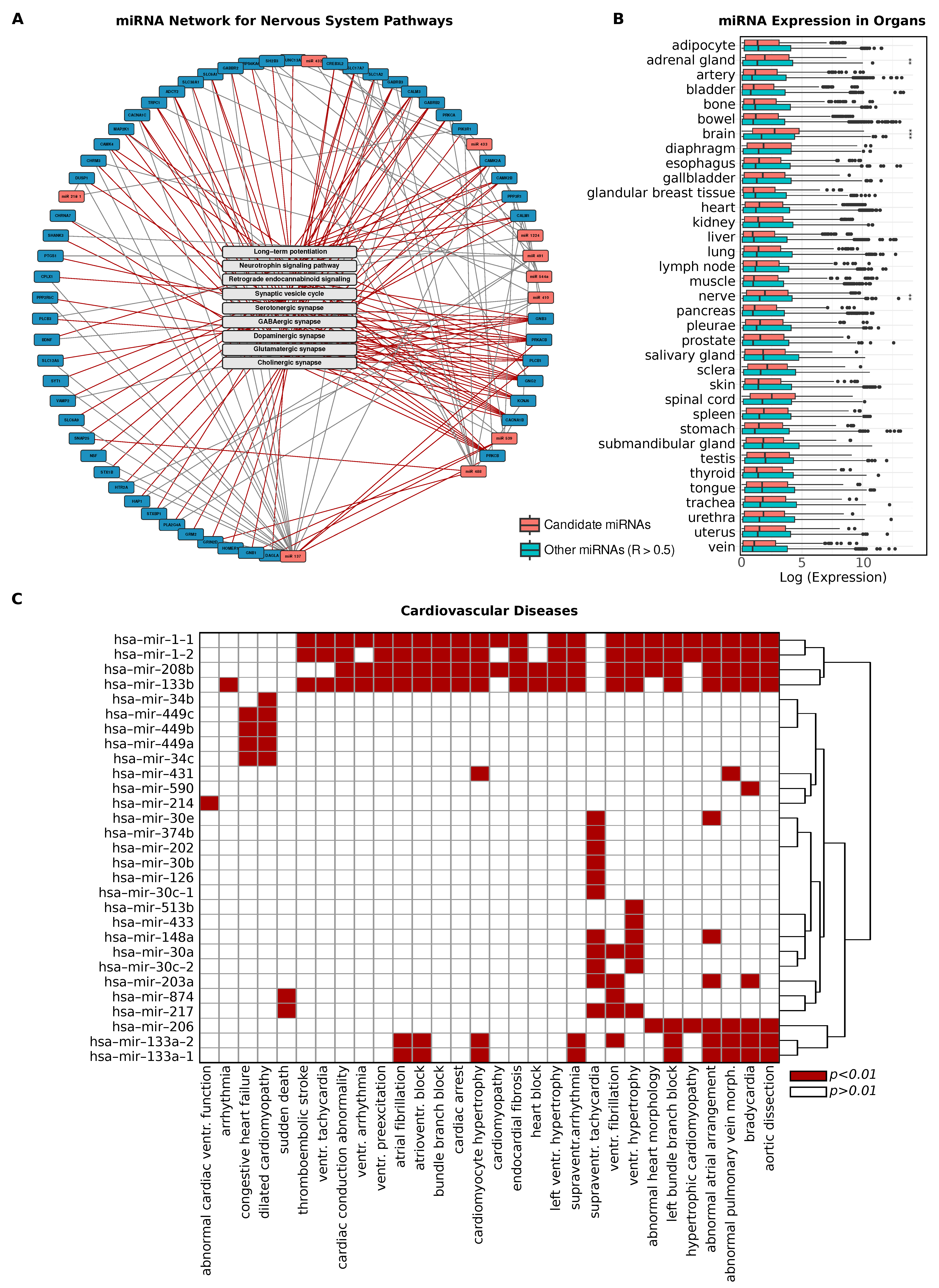
2.3. Biological Signatures of Predictive miRNA Regulators
2.4. miRNA-Linked Pathway Enrichments
2.5. Cardiovascular Disease Associations in Predictive Gene Networks
3. Discussion
4. Methods
4.1. Data Collection
4.2. Data Preparation
4.3. miRNA Expression Modeling
4.4. Model Performance Evaluation
4.5. Network Construction and Subsetting
4.6. Connectivity Metrics
4.7. Gene Ontology (GO) Term Analysis
4.8. Pathway Analysis
4.9. Disease Enrichment
4.10. Organ-Specific miRNA Expression
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in Cancer: Biomarkers, Functions and Therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating High Confidence microRNAs Using Deep Sequencing Data. Nucleic Acids Res. 2014, 42, D68–D73. [Google Scholar] [CrossRef]
- Stempor, P.A.; Cauchi, M.; Wilson, P. MMpred: Functional miRNA—mRNA Interaction Analyses by miRNA Expression Prediction. BMC Genom. 2012, 13, 620. [Google Scholar] [CrossRef]
- Huang, J.C.; Babak, T.; Corson, T.W.; Chua, G.; Khan, S.; Gallie, B.L.; Hughes, T.R.; Blencowe, B.J.; Frey, B.J.; Morris, Q.D. Using Expression Profiling Data to Identify Human microRNA Targets. Nat. Methods 2007, 4, 1045–1049. [Google Scholar] [CrossRef] [PubMed]
- Ruike, Y.; Ichimura, A.; Tsuchiya, S.; Shimizu, K.; Kunimoto, R.; Okuno, Y.; Tsujimoto, G. Global Correlation Analysis for Micro-RNA and mRNA Expression Profiles in Human Cell Lines. J. Hum. Genet. 2008, 53, 515–523. [Google Scholar] [CrossRef]
- Alevizos, I.; Illei, G.G. MicroRNAs as Biomarkers in Rheumatic Diseases. Nat. Rev. Rheumatol. 2010, 6, 391–398. [Google Scholar] [CrossRef]
- Reda El Sayed, S.; Cristante, J.; Guyon, L.; Denis, J.; Chabre, O.; Cherradi, N. MicroRNA Therapeutics in Cancer: Current Advances and Challenges. Cancers 2021, 13, 2680. [Google Scholar] [CrossRef]
- McGeary, S.E.; Lin, K.S.; Shi, C.Y.; Pham, T.M.; Bisaria, N.; Kelley, G.M.; Bartel, D.P. The Biochemical Basis of microRNA Targeting Efficacy. Science 2019, 366, eaav1741. [Google Scholar] [CrossRef]
- Jin, S.; Zeng, X.; Fang, J.; Lin, J.; Chan, S.Y.; Erzurum, S.C.; Cheng, F. A Network-Based Approach to Uncover microRNA-Mediated Disease Comorbidities and Potential Pathobiological Implications. npj Syst. Biol. Appl. 2019, 5, 41. [Google Scholar] [CrossRef]
- Cihan, M.; Andrade-Navarro, M.A. Detection of Features Predictive of microRNA Targets by Integration of Network Data. PLoS ONE 2022, 17, e0269731. [Google Scholar] [CrossRef] [PubMed]
- van Iterson, M.; Bervoets, S.; de Meijer, E.J.; Buermans, H.P.; ‘t Hoen, P.A.C.; Menezes, R.X.; Boer, J.M. Integrated Analysis of microRNA and mRNA Expression: Adding Biological Significance to microRNA Target Predictions. Nucleic Acids Res. 2013, 41, e146. [Google Scholar] [CrossRef]
- Xuan, J.; Shi, L.; Guo, L. microRNA Profiling: Strategies and Challenges. In microRNAs in Toxicology and Medicine; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2013; pp. 437–455. ISBN 978-1-118-69599-9. [Google Scholar]
- Wright, C.; Rajpurohit, A.; Burke, E.E.; Williams, C.; Collado-Torres, L.; Kimos, M.; Brandon, N.J.; Cross, A.J.; Jaffe, A.E.; Weinberger, D.R.; et al. Comprehensive Assessment of Multiple Biases in Small RNA Sequencing Reveals Significant Differences in the Performance of Widely Used Methods. BMC Genom. 2019, 20, 513. [Google Scholar] [CrossRef]
- Benesova, S.; Kubista, M.; Valihrach, L. Small RNA-Sequencing: Approaches and Considerations for miRNA Analysis. Diagnostics 2021, 11, 964. [Google Scholar] [CrossRef]
- Matullo, G.; Naccarati, A.; Pardini, B. MicroRNA Expression Profiling in Bladder Cancer: The Challenge of next-Generation Sequencing in Tissues and Biofluids. Int. J. Cancer 2016, 138, 2334–2345. [Google Scholar] [CrossRef] [PubMed]
- Backes, C.; Sedaghat-Hamedani, F.; Frese, K.; Hart, M.; Ludwig, N.; Meder, B.; Meese, E.; Keller, A. Bias in High-Throughput Analysis of miRNAs and Implications for Biomarker Studies. Anal. Chem. 2016, 88, 2088–2095. [Google Scholar] [CrossRef]
- Madadi, S.; Schwarzenbach, H.; Lorenzen, J.; Soleimani, M. MicroRNA Expression Studies: Challenge of Selecting Reliable Reference Controls for Data Normalization. Cell. Mol. Life Sci. 2019, 76, 3497–3514. [Google Scholar] [CrossRef] [PubMed]
- Schwarzenbach, H.; da Silva, A.M.; Calin, G.; Pantel, K. Data Normalization Strategies for MicroRNA Quantification. Clin. Chem. 2015, 61, 1333–1342. [Google Scholar] [CrossRef]
- Webber, J.W.; Elias, K.M. Fast and Robust Imputation for miRNA Expression Data Using Constrained Least Squares. BMC Bioinform. 2022, 23, 145. [Google Scholar] [CrossRef]
- Nielsen, M.M.; Pedersen, J.S. miRNA Activity Inferred from Single Cell mRNA Expression. Sci. Rep. 2021, 11, 9170. [Google Scholar] [CrossRef]
- Olgun, G.; Gopalan, V.; Hannenhalli, S. miRSCAPE—Inferring miRNA Expression from scRNA-Seq Data. iScience 2022, 25, 104962. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Li, L.M. Inferring MicroRNA Activities by Combining Gene Expression with MicroRNA Target Prediction. PLoS ONE 2008, 3, e1989. [Google Scholar] [CrossRef] [PubMed]
- Le, T.D.; Liu, L.; Tsykin, A.; Goodall, G.J.; Liu, B.; Sun, B.-Y.; Li, J. Inferring microRNA–mRNA Causal Regulatory Relationships from Expression Data. Bioinformatics 2013, 29, 765–771. [Google Scholar] [CrossRef]
- Tan, H.; Huang, S.; Zhang, Z.; Qian, X.; Sun, P.; Zhou, X. Pan-Cancer Analysis on microRNA-Associated Gene Activation. EBioMedicine 2019, 43, 82–97. [Google Scholar] [CrossRef]
- Monteys, A.M.; Spengler, R.M.; Wan, J.; Tecedor, L.; Lennox, K.A.; Xing, Y.; Davidson, B.L. Structure and Activity of Putative Intronic miRNA Promoters. RNA 2010, 16, 495–505. [Google Scholar] [CrossRef]
- Hoerl, A.E.; Kennard, R.W. Ridge Regression: Biased Estimation for Nonorthogonal Problems. Technometrics 1970, 12, 55–67. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Li, J.; Han, X.; Wan, Y.; Zhang, S.; Zhao, Y.; Fan, R.; Cui, Q.; Zhou, Y. TAM 2.0: Tool for MicroRNA Set Analysis. Nucleic Acids Res. 2018, 46, W180–W185. [Google Scholar] [CrossRef] [PubMed]
- Frouin, A.; Dandine-Roulland, C.; Pierre-Jean, M.; Deleuze, J.-F.; Ambroise, C.; Le Floch, E. Exploring the Link Between Additive Heritability and Prediction Accuracy from a Ridge Regression Perspective. Front. Genet. 2020, 11, 581594. [Google Scholar] [CrossRef]
- Novianti, P.W.; Snoek, B.C.; Wilting, S.M.; van de Wiel, M.A. Better Diagnostic Signatures from RNAseq Data through Use of Auxiliary Co-Data. Bioinformatics 2017, 33, 1572–1574. [Google Scholar] [CrossRef]
- Mbatchou, J.; Barnard, L.; Backman, J.; Marcketta, A.; Kosmicki, J.A.; Ziyatdinov, A.; Benner, C.; O’Dushlaine, C.; Barber, M.; Boutkov, B.; et al. Computationally Efficient Whole-Genome Regression for Quantitative and Binary Traits. Nat. Genet. 2021, 53, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wei, D.; Xiang, J.; Ren, F.; Huang, L.; Lang, J.; Tian, G.; Li, Y.; Yang, J. An Improved Anticancer Drug-Response Prediction Based on an Ensemble Method Integrating Matrix Completion and Ridge Regression. Mol. Ther.—Nucleic Acids 2020, 21, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.-Y.; Lin, Y.-C.-D.; Cui, S.; Huang, Y.; Tang, Y.; Xu, J.; Bao, J.; Li, Y.; Wen, J.; Zuo, H.; et al. miRTarBase Update 2022: An Informative Resource for Experimentally Validated miRNA-Target Interactions. Nucleic Acids Res. 2022, 50, D222–D230. [Google Scholar] [CrossRef]
- Karagkouni, D.; Paraskevopoulou, M.D.; Chatzopoulos, S.; Vlachos, I.S.; Tastsoglou, S.; Kanellos, I.; Papadimitriou, D.; Kavakiotis, I.; Maniou, S.; Skoufos, G.; et al. DIANA-TarBase v8: A Decade-Long Collection of Experimentally Supported miRNA-Gene Interactions. Nucleic Acids Res. 2018, 46, D239–D245. [Google Scholar] [CrossRef] [PubMed]
- McDonald, G.C. Ridge Regression. WIREs Comput. Stat. 2009, 1, 93–100. [Google Scholar] [CrossRef]
- Cule, E.; De Iorio, M. Ridge Regression in Prediction Problems: Automatic Choice of the Ridge Parameter. Genet. Epidemiol. 2013, 37, 704–714. [Google Scholar] [CrossRef]
- Zhang, R.; McDonald, G.C. Characterization of Ridge Trace Behavior. Commun. Stat.—Theory Methods 2005, 34, 1487–1501. [Google Scholar] [CrossRef]
- Yang, Y.; Xu, Z.; Song, D. Missing Value Imputation for microRNA Expression Data by Using a GO-Based Similarity Measure. BMC Bioinform. 2016, 17, 10. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Cardiac-Specific miRNA in Cardiogenesis, Heart Function, and Cardiac Pathology (with Focus on Myocardial Infarction). J. Mol. Cell. Cardiol. 2016, 94, 107–121. [Google Scholar] [CrossRef]
- Navickas, R.; Gal, D.; Laucevičius, A.; Taparauskaitė, A.; Zdanytė, M.; Holvoet, P. Identifying Circulating microRNAs as Biomarkers of Cardiovascular Disease: A Systematic Review. Cardiovasc. Res. 2016, 111, 322–337. [Google Scholar] [CrossRef]
- Kaur, A.; Mackin, S.T.; Schlosser, K.; Wong, F.L.; Elharram, M.; Delles, C.; Stewart, D.J.; Dayan, N.; Landry, T.; Pilote, L. Systematic Review of microRNA Biomarkers in Acute Coronary Syndrome and Stable Coronary Artery Disease. Cardiovasc. Res. 2020, 116, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Widera, C.; Gupta, S.K.; Lorenzen, J.M.; Bang, C.; Bauersachs, J.; Bethmann, K.; Kempf, T.; Wollert, K.C.; Thum, T. Diagnostic and Prognostic Impact of Six Circulating microRNAs in Acute Coronary Syndrome. J. Mol. Cell. Cardiol. 2011, 51, 872–875. [Google Scholar] [CrossRef]
- Small, E.M.; Olson, E.N. Pervasive Roles of microRNAs in Cardiovascular Biology. Nature 2011, 469, 336–342. [Google Scholar] [CrossRef]
- Kalozoumi, G.; Yacoub, M.; Sanoudou, D. MicroRNAs in Heart Failure: Small Molecules with Major Impact. Glob. Cardiol. Sci. Pract. 2014, 2014, 79–102. [Google Scholar] [CrossRef] [PubMed]
- Sambandan, S.; Akbalik, G.; Kochen, L.; Rinne, J.; Kahlstatt, J.; Glock, C.; Tushev, G.; Alvarez-Castelao, B.; Heckel, A.; Schuman, E.M. Activity-Dependent Spatially Localized miRNA Maturation in Neuronal Dendrites. Science 2017, 355, 634–637. [Google Scholar] [CrossRef]
- Liu, X.; Xie, H.; Liu, W.; Zuo, J.; Li, S.; Tian, Y.; Zhao, J.; Bai, M.; Li, J.; Bao, L.; et al. Dynamic Regulation of Alternative Polyadenylation by PQBP1 during Neurogenesis. Cell Rep. 2024, 43, 114525. [Google Scholar] [CrossRef]
- Cihan, M.; Schmauck, G.; Sprang, M.; Andrade-Navarro, M.A. Unveiling cell-type-specific microRNA networks through alternative polyadenylation in glioblastoma. BMC Biol. 2025, 23, 15. [Google Scholar] [CrossRef]
- Lagos-Quintana, M.; Rauhut, R.; Yalcin, A.; Meyer, J.; Lendeckel, W.; Tuschl, T. Identification of Tissue-Specific MicroRNAs from Mouse. Curr. Biol. 2002, 12, 735–739. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Leidinger, P.; Becker, K.; Backes, C.; Fehlmann, T.; Pallasch, C.; Rheinheimer, S.; Meder, B.; Stähler, C.; Meese, E.; et al. Distribution of miRNA Expression across Human Tissues. Nucleic Acids Res. 2016, 44, 3865–3877. [Google Scholar] [CrossRef]
- Zhao, Y.; Ransom, J.F.; Li, A.; Vedantham, V.; von Drehle, M.; Muth, A.N.; Tsuchihashi, T.; McManus, M.T.; Schwartz, R.J.; Srivastava, D. Dysregulation of Cardiogenesis, Cardiac Conduction, and Cell Cycle in Mice Lacking miRNA-1-2. Cell 2007, 129, 303–317. [Google Scholar] [CrossRef]
- Yang, B.; Lin, H.; Xiao, J.; Lu, Y.; Luo, X.; Li, B.; Zhang, Y.; Xu, C.; Bai, Y.; Wang, H.; et al. The Muscle-Specific microRNA miR-1 Regulates Cardiac Arrhythmogenic Potential by Targeting GJA1 and KCNJ2. Nat. Med. 2007, 13, 486–491. [Google Scholar] [CrossRef] [PubMed]
- van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of Stress-Dependent Cardiac Growth and Gene Expression by a MicroRNA. Science 2007, 316, 575–579. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudi, E.; Cairns, M.J. MiR-137: An Important Player in Neural Development and Neoplastic Transformation. Mol. Psychiatry 2017, 22, 44–55. [Google Scholar] [CrossRef]
- Sun, J.; Sun, J.; Ming, G.; Song, H. Epigenetic Regulation of Neurogenesis in the Adult Mammalian Brain. Eur. J. Neurosci. 2011, 33, 1087–1093. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wang, Y.-C.; Wang, B.; Kuo, C.-C.J. Graph Representation Learning: A Survey. APSIPA Trans. Signal Inf. Process. 2020, 9, e15. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]
- Matys, V.; Kel-Margoulis, O.V.; Fricke, E.; Liebich, I.; Land, S.; Barre-Dirrie, A.; Reuter, I.; Chekmenev, D.; Krull, M.; Hornischer, K.; et al. TRANSFAC and Its Module TRANSCompel: Transcriptional Gene Regulation in Eukaryotes. Nucleic Acids Res. 2006, 34, D108–D110. [Google Scholar] [CrossRef]
- Harrison, P.W.; Amode, M.R.; Austine-Orimoloye, O.; Azov, A.G.; Barba, M.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2024. Nucleic Acids Res. 2024, 52, D891–D899. [Google Scholar] [CrossRef]
- Csárdi, G.; Nepusz, T. The Igraph Software Package for Complex Network Research. InterJournal—Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a Reference Resource for Gene and Protein Annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Gargano, M.A.; Matentzoglu, N.; Coleman, B.; Addo-Lartey, E.B.; Anagnostopoulos, A.V.; Anderton, J.; Avillach, P.; Bagley, A.M.; Bakštein, E.; Balhoff, J.P.; et al. The Human Phenotype Ontology in 2024: Phenotypes around the World. Nucleic Acids Res. 2024, 52, D1333–D1346. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, L.; Raudvere, U.; Kuzmin, I.; Adler, P.; Vilo, J.; Peterson, H. G:Profiler—Interoperable Web Service for Functional Enrichment Analysis and Gene Identifier Mapping (2023 Update). Nucleic Acids Res. 2023, 51, W207–W212. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cihan, M.; Anyaegbunam, U.A.; Albrecht, S.; Andrade-Navarro, M.A.; Sprang, M. Evaluating Genetic Regulators of MicroRNAs Using Machine Learning Models. Int. J. Mol. Sci. 2025, 26, 5757. https://doi.org/10.3390/ijms26125757
Cihan M, Anyaegbunam UA, Albrecht S, Andrade-Navarro MA, Sprang M. Evaluating Genetic Regulators of MicroRNAs Using Machine Learning Models. International Journal of Molecular Sciences. 2025; 26(12):5757. https://doi.org/10.3390/ijms26125757
Chicago/Turabian StyleCihan, Mert, Uchenna Alex Anyaegbunam, Steffen Albrecht, Miguel A. Andrade-Navarro, and Maximilian Sprang. 2025. "Evaluating Genetic Regulators of MicroRNAs Using Machine Learning Models" International Journal of Molecular Sciences 26, no. 12: 5757. https://doi.org/10.3390/ijms26125757
APA StyleCihan, M., Anyaegbunam, U. A., Albrecht, S., Andrade-Navarro, M. A., & Sprang, M. (2025). Evaluating Genetic Regulators of MicroRNAs Using Machine Learning Models. International Journal of Molecular Sciences, 26(12), 5757. https://doi.org/10.3390/ijms26125757