
Exploring the Clinical Implication of S100A9 in Ulcerative Colitis and Its Progression to Cancer: A Journey from Inflammation to Cancer
Abstract
1. Introduction
2. Results
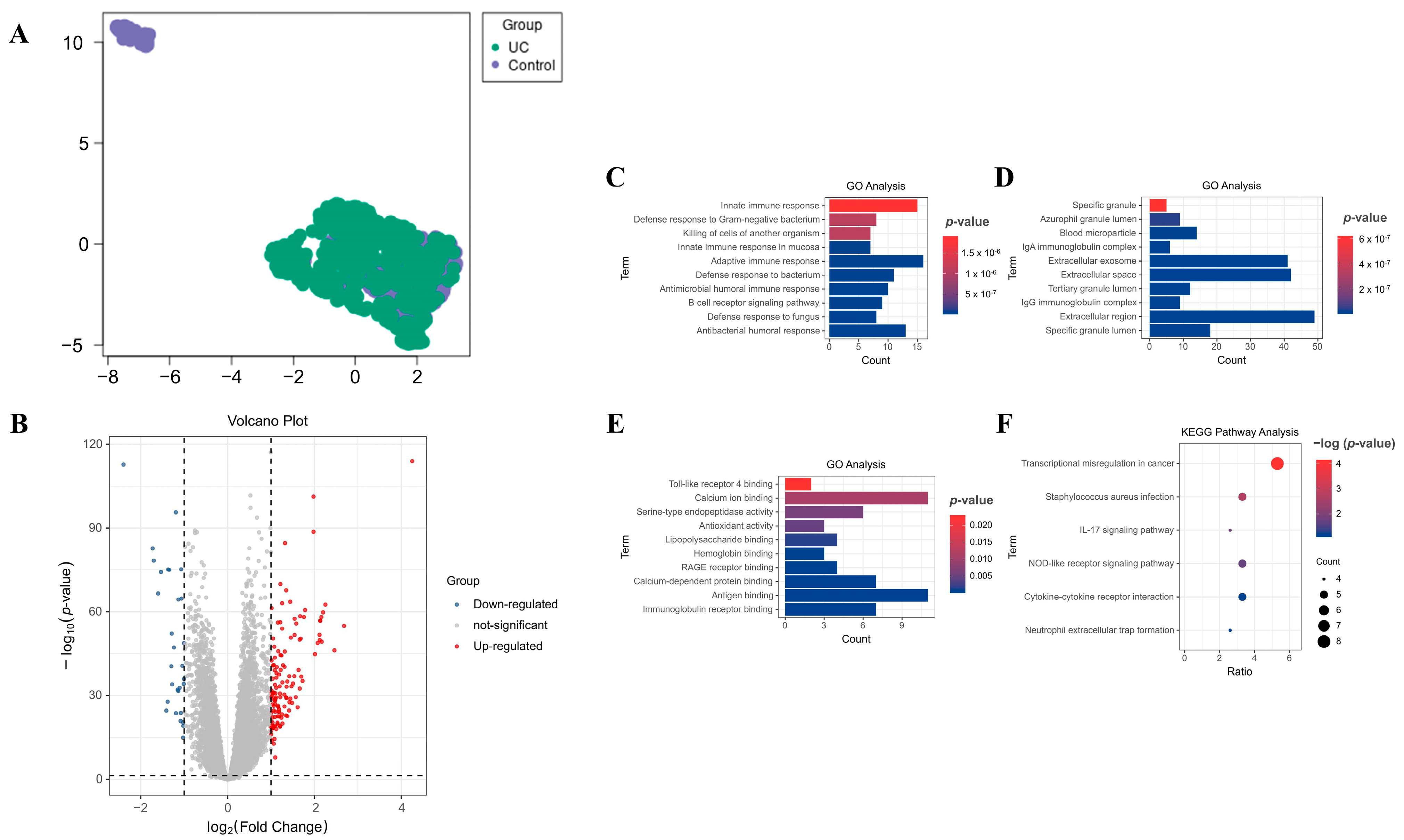
2.1. Discovery of Differentially Expressed Genes (DEGs) in Blood Samples from Patients with UC
2.2. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Pathway Analyses on DEGs
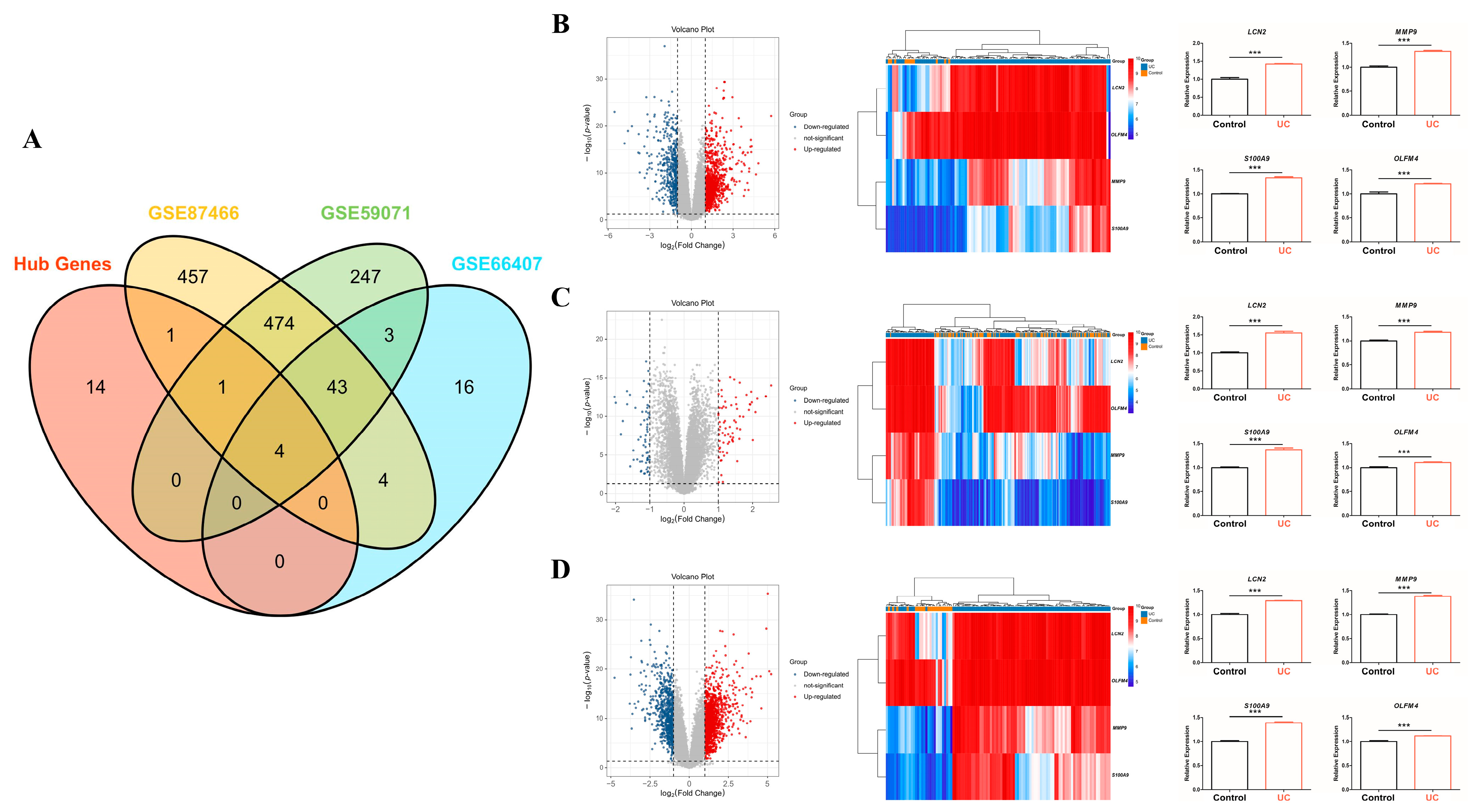
2.3. Protein–Protein Interaction (PPI) Network Construction for Hub Genes Detection
2.4. Key Hub Genes Selection and Validation
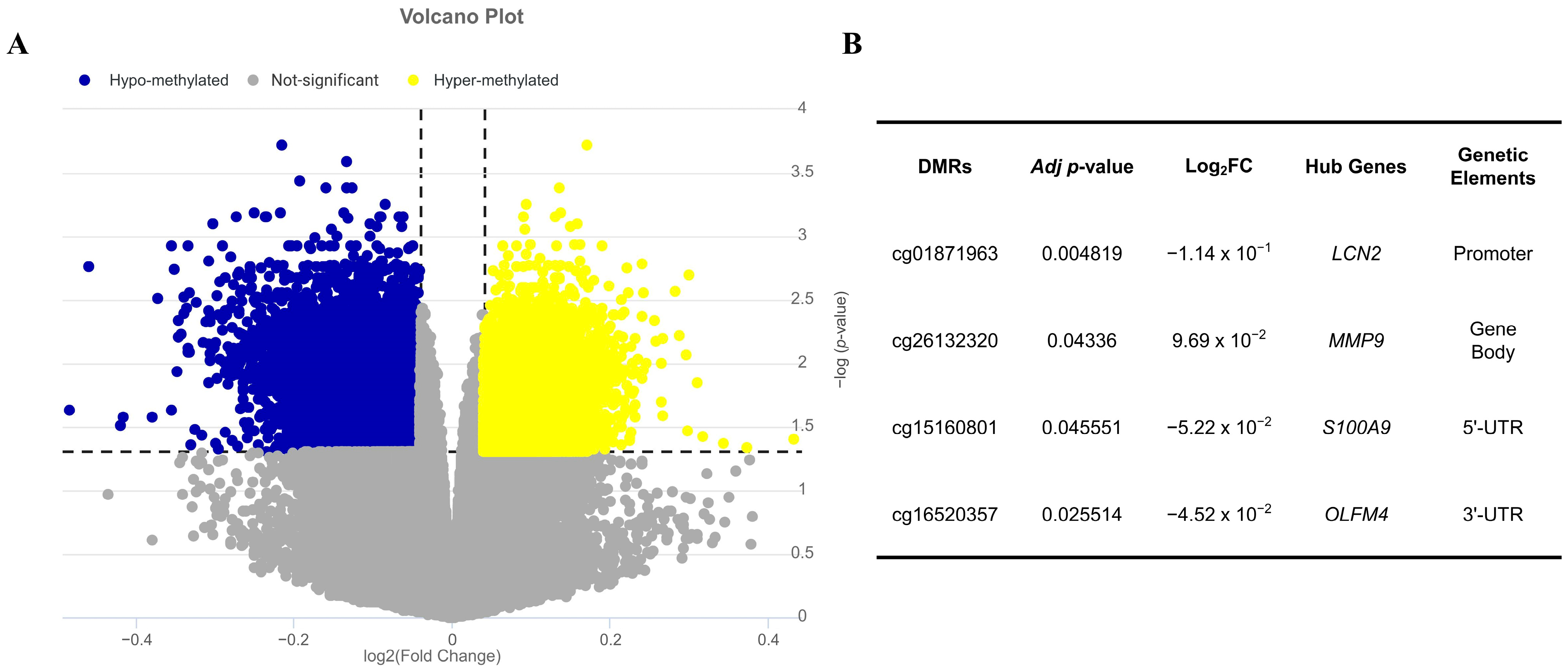
2.5. Validation of Key Hub Genes Expression Through Epigenetic Factors
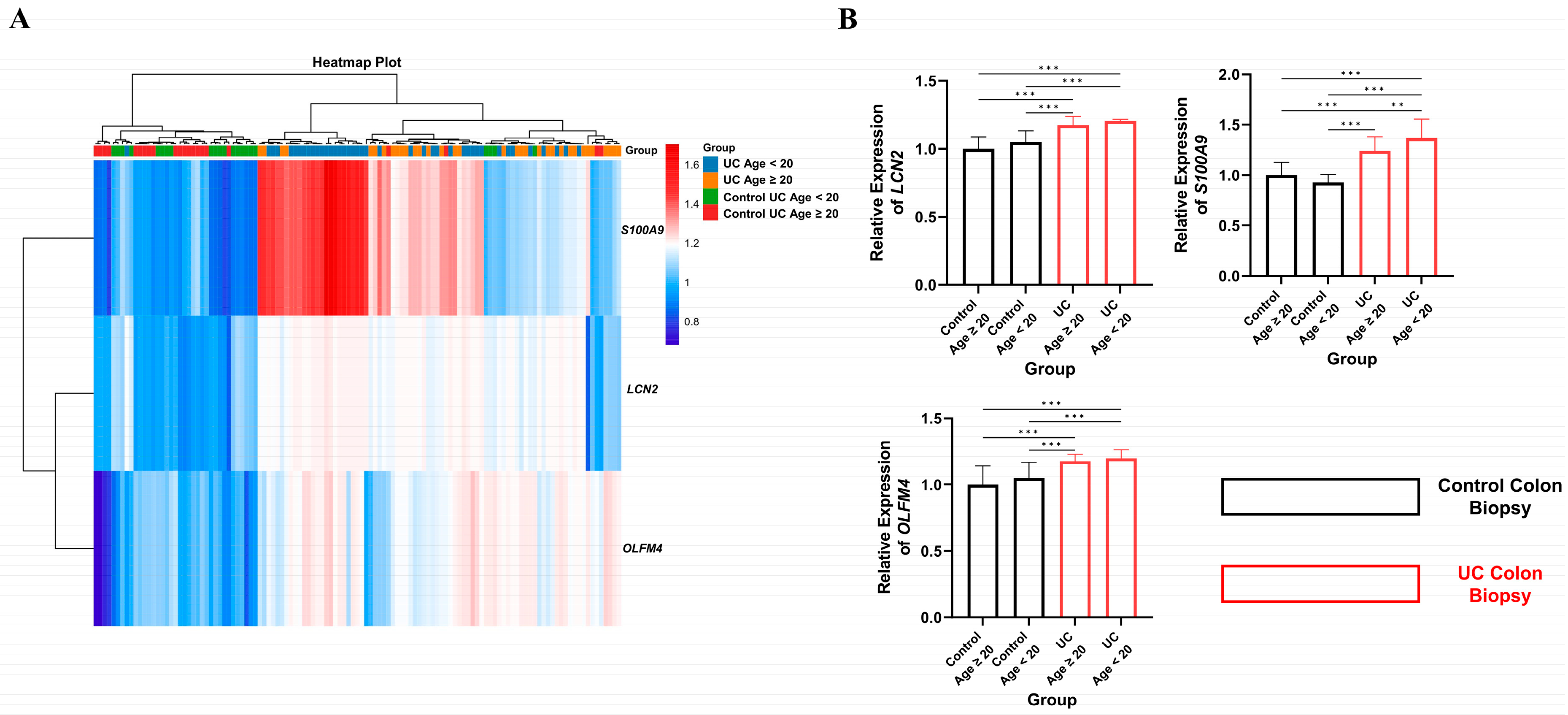
2.6. Age-Dependent Expression Analysis of Key Hub Genes in UC
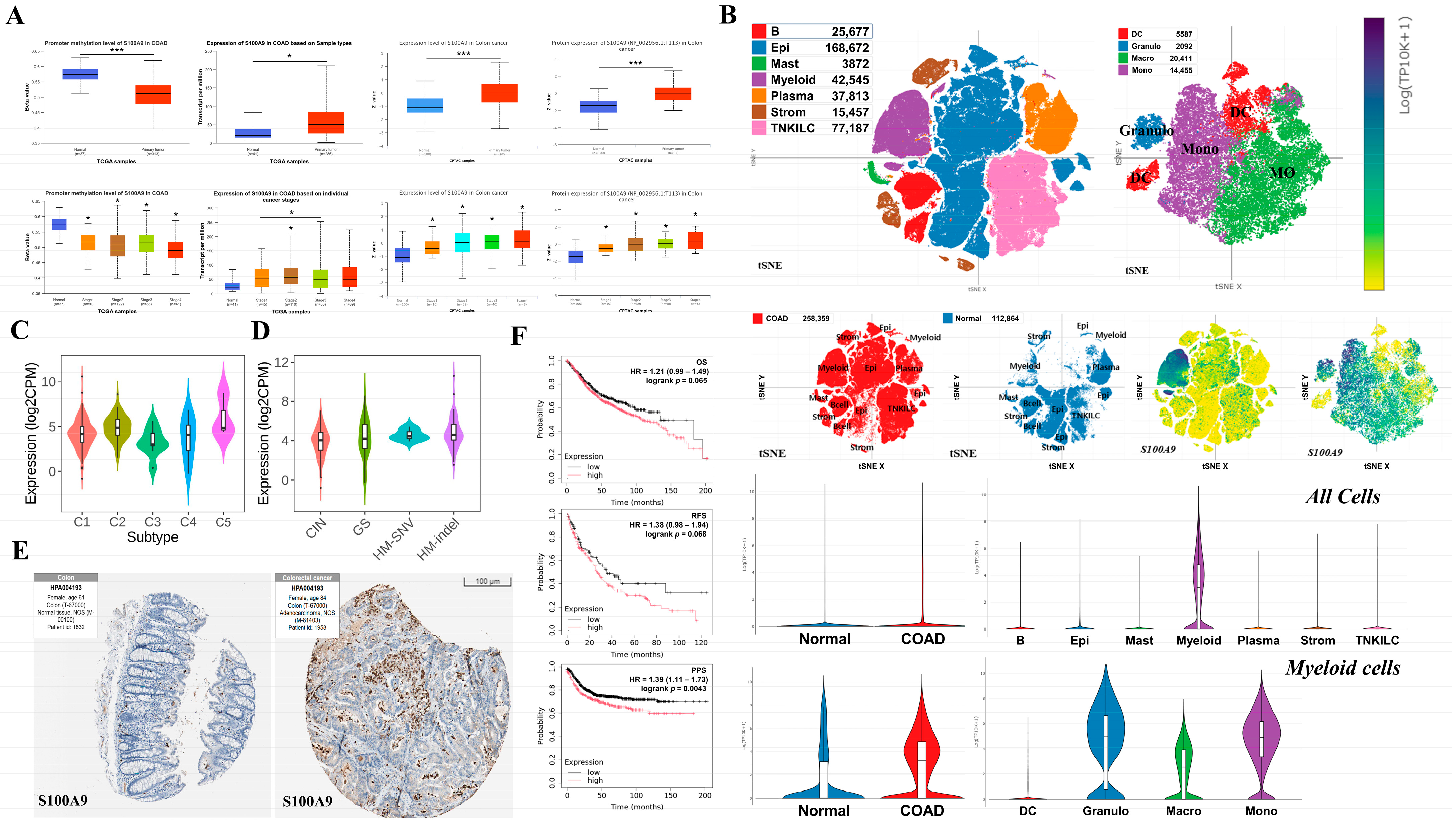
2.7. Analysis of the Effects of Key Hub Gene Expression in Cancer
3. Discussion
4. Materials and Methods
4.1. Next-Generation Sequencing (NGS) Dataset
4.2. Identification of DEGs
4.3. GO and KEGG Pathway Analysis of DEGs
4.4. PPI Network Build Up for Hub Genes Detection
4.5. Validation Using Colon Mucosa Samples of Patients with UC
4.6. Variable Selection via Using LASSO
4.7. Analysis of DMRs of Key Hub Genes
4.8. Analysis of Age-Related Expression Patterns of Key Hub Genes in Patients with UC
4.9. Analysis of the Effects of Key Hub Gene Expression on Cancer
4.10. Data Visualization and Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kobayashi, T.; Siegmund, B.; Le Berre, C.; Wei, S.C.; Ferrante, M.; Shen, B.; Bernstein, C.N.; Danese, S.; Peyrin-Biroulet, L.; Hibi, T. Ulcerative colitis. Nat. Rev. Dis. Primers 2020, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Magro, F.; Gionchetti, P.; Eliakim, R.; Ardizzone, S.; Armuzzi, A.; Barreiro-de Acosta, M.; Burisch, J.; Gecse, K.B.; Hart, A.L.; Hindryckx, P.; et al. Third European evidence-based consensus on diagnosis and management of ulcerative colitis. Part 1: Definitions, diagnosis, extra-intestinal manifestations, pregnancy, cancer surveillance, surgery, and ileo-anal pouch disorders. J. Crohns Colitis 2017, 11, 649–670. [Google Scholar] [CrossRef] [PubMed]
- Feuerstein, J.D.; Cheifetz, A.S. Ulcerative colitis: Epidemiology, diagnosis, and management. Mayo Clin. Proc. 2014, 89, 1553–1563. [Google Scholar] [CrossRef]
- Keller, R.; Mazurak, N.; Fantasia, L.; Fusco, S.; Malek, N.P.; Wehkamp, J.; Enck, P.; Klag, T. Quality of life in inflammatory bowel diseases: It is not all about the bowel. Intest. Res. 2021, 19, 45–52. [Google Scholar] [CrossRef] [PubMed]
- van Gils, T.; Törnblom, H.; Hreinsson, J.P.; Jonefjäll, B.; Strid, H.; Simrén, M. Factors associated with abdominal pain in patients with active and quiescent ulcerative colitis: A multicohort study. Aliment. Pharmacol. Ther. 2024, 61, 268–277. [Google Scholar] [CrossRef]
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e065186. [Google Scholar] [CrossRef]
- Zhou, J.L.; Bao, J.C.; Liao, X.Y.; Chen, Y.J.; Wang, L.W.; Fan, Y.Y.; Xu, Q.Y.; Hao, L.X.; Li, K.J.; Liang, M.X.; et al. Trends and projections of inflammatory bowel disease at the global, regional and national levels, 1990–2050: A bayesian age-period-cohort modeling study. BMC Public Health 2023, 23, 2507. [Google Scholar] [CrossRef]
- Park, J.; Jeong, G.H.; Song, M.; Yon, D.K.; Lee, S.W.; Koyanagi, A.; Jacob, L.; Kostev, K.; Dragioti, E.; Radua, J.; et al. The global, regional, and national burden of inflammatory bowel diseases, 1990–2019: A systematic analysis for the global burden of disease study 2019. Dig. Liver Dis. 2023, 55, 1352–1359. [Google Scholar] [CrossRef]
- Lin, D.; Jin, Y.; Shao, X.; Xu, Y.; Ma, G.; Jiang, Y.; Xu, Y.; Jiang, Y.; Hu, D. Global, regional, and national burden of inflammatory bowel disease, 1990–2021: Insights from the global burden of disease 2021. Int. J. Colorectal Dis. 2024, 39, 139. [Google Scholar] [CrossRef]
- Qing, F.; Tian, H.; Wang, B.; Xie, B.; Sui, L.; Xie, X.; He, W.; He, T.; Li, Y.; He, L.; et al. Interferon regulatory factor 7 alleviates the experimental colitis through enhancing IL-28A-mediated intestinal epithelial integrity. J. Transl. Med. 2024, 22, 905. [Google Scholar] [CrossRef]
- Liang, Y.; Li, Y.; Lee, C.; Yu, Z.; Chen, C.; Liang, C. Ulcerative colitis: Molecular insights and intervention therapy. Mol. biomed 2024, 5, 42. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Liu, C.; Jiang, S.; Qian, D.; Duan, J. Cross talk between gut microbiota and intestinal mucosal immunity in the development of ulcerative colitis. Infect. Immun. 2021, 89, e0001421. [Google Scholar] [CrossRef]
- Danne, C.; Lamas, B.; Lavelle, A.; Michel, M.L.; Da Costa, G.; Pham, H.P.; Lefevre, A.; Bridonneau, C.; Bredon, M.; Planchais, J.; et al. Dissecting the respective roles of microbiota and host genetics in the susceptibility of Card9−/− mice to colitis. Microbiome 2024, 12, 76. [Google Scholar] [CrossRef]
- Lim, J.G.; Kang, B.; Oh, S.H.; Ryoo, E.; Kim, Y.B.; Choe, Y.H.; Lee, Y.J.; Shin, M.; Yang, H.R.; Kim, S.C.; et al. Characteristics of pediatric ulcerative colitis at diagnosis in Korea: Results from a multicenter, registry-based, inception cohort study. J. Korean Med. Sci. 2024, 39, e303. [Google Scholar] [CrossRef]
- Weidner, J.; Kern, I.; Reinecke, I.; Bathelt, F.; Manuwald, U.; Henke, E.; Zoch, M.; Rothe, U.; Kugler, J. A systematic review and meta-regression on international trends in the incidence of ulcerative colitis in children and adolescents associated with socioeconomic and geographic factors. Eur. J. Pediatr. 2024, 183, 1723–1732. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, L.; Huang, S.; Yang, L.; Wang, L.; Yang, F.; Huang, J.; Ding, X. Predicting novel biomarkers for early diagnosis and dynamic severity monitoring of human ulcerative colitis. Front. Genet. 2024, 15, 1429482. [Google Scholar] [CrossRef]
- Kuwahara, R.; Ikeuchi, H.; Kusunoki, K.; Minagawa, T.; Horio, Y.; Kimura, K.; Kataoka, K.; Beppu, N.; Ikeda, M.; Uchino, M. Clinical features and surgical selection in colitis-associated colorectal cancer with ulcerative colitis. BMC Surg. 2023, 23, 256. [Google Scholar] [CrossRef] [PubMed]
- Wetwittayakhlang, P.; Golovics, P.A.; Gonczi, L.; Lakatos, L.; Lakatos, P.L.; Kurti, Z.; David, G.; Erdelyi, Z.; Szita, I.; Pandur, T.; et al. Stable incidence and risk factors of colorectal cancer in ulcerative colitis: A population-based cohort between 1977–2020. Clin. Gastroenterol. Hepatol. 2024, 22, 191–193.e3. [Google Scholar] [CrossRef]
- Kanters, A.; Liska, D. The impact of ulcerative colitis on colorectal cancer prognosis: The jury is still out. Ann. Surg. Oncol. 2022, 29, 2761–2762. [Google Scholar] [CrossRef]
- Yin, Y.; Yang, X.; Cheng, Z.; Wang, H.; Lei, J.; Wang, D.; Wang, P.; Li, B.; Mi, J.; Yuan, Q. Identification of extracellular matrix-related biomarkers in colon adenocarcinoma by bioinformatics and experimental validation. Front. Immunol. 2024, 15, 1371584. [Google Scholar] [CrossRef]
- Cheon, J.; Jung, H.; Kang, B.Y.; Kim, M. Impact of potential biomarkers, SNRPE, COX7C, and RPS27, on idiopathic Parkinson’s disease. Genes Genomics 2025, 47, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Cheon, J.; Kim, B.; Park, J.; Shin, J.; Kim, T.H. Unveiling biomarkers in head and neck squamous cell carcinoma through bioinformatics: The role of SPP1 and KRT78. Int. J. Mol. Sci. 2024, 25, 12062. [Google Scholar] [CrossRef]
- Zhao, M.; Gönczi, L.; Lakatos, P.L.; Burisch, J. The burden of inflammatory bowel disease in Europe in 2020. J. Crohns Colitis 2021, 15, 1573–1587. [Google Scholar] [CrossRef]
- Hind, D.; Sheldon, E.; Lillington, G.; Greig, N.; Buckley, V.; D’Afflitto, M.; Ezaydi, N.; Ali, B.; Haamed, H.; Alhashemi, T.; et al. Sources of diagnostic delay for people with Crohn’s disease and ulcerative colitis: Qualitative research study. PLoS ONE 2024, 19, e0301672. [Google Scholar]
- Rosen, M.J.; Dhawan, A.; Saeed, S.A. Inflammatory bowel disease in children and adolescents. JAMA Pediatr. 2015, 169, 1053–1060. [Google Scholar] [CrossRef]
- Turner, D.; Levine, A.; Escher, J.C.; Griffiths, A.M.; Russell, R.K.; Dignass, A.; Dias, J.A.; Bronsky, J.; Braegger, C.P.; Cucchiara, S.; et al. Management of pediatric ulcerative colitis: Joint ECCO and ESPGHAN evidence-based consensus guidelines. J. Pediatr. Gastroenterol. Nutr. 2012, 55, 340–361. [Google Scholar] [CrossRef]
- Yang, H.; Mirsepasi-Lauridsen, H.C.; Struve, C.; Allaire, J.M.; Sivignon, A.; Vogl, W.; Bosman, E.S.; Ma, C.; Fotovati, A.; Reid, G.S.; et al. Ulcerative colitis-associated E. coli pathobionts potentiate colitis in susceptible hosts. Gut Microbes 2020, 12, 1847976. [Google Scholar] [CrossRef] [PubMed]
- Tahara, T.; Shibata, T.; Kawamura, T.; Okubo, M.; Ichikawa, Y.; Sumi, K.; Miyata, M.; Ishizuka, T.; Nakamura, M.; Nagasaka, M.; et al. Fusobacterium detected in colonic biopsy and clinicopathological features of ulcerative colitis in Japan. Dig. Dis. Sci. 2015, 60, 205–210. [Google Scholar] [CrossRef]
- Liu, F.; Chen, S.; Luu, L.D.W.; Lee, S.A.; Tay, A.C.Y.; Wu, R.; Riordan, S.M.; Lan, R.; Liu, L.; Zhang, L. Analysis of complete Campylobacter concisus genomes identifies genomospecies features, secretion systems and novel plasmids and their association with severe ulcerative colitis. Microb. Genom. 2020, 6, mgen000457. [Google Scholar] [CrossRef]
- Castro-Dopico, T.; Dennison, T.W.; Ferdinand, J.R.; Mathews, R.J.; Fleming, A.; Clift, D.; Stewart, B.J.; Jing, C.; Strongili, K.; Labzin, L.I.; et al. Anti-commensal IgG drives intestinal inflammation and Type 17 immunity in ulcerative colitis. Immunity 2019, 50, 1099–1114.e10. [Google Scholar] [CrossRef]
- Uzzan, M.; Martin, J.C.; Mesin, L.; Livanos, A.E.; Castro-Dopico, T.; Huang, R.; Petralia, F.; Magri, G.; Kumar, S.; Zhao, Q.; et al. Ulcerative colitis is characterized by a plasmablast-skewed humoral response associated with disease activity. Nat. Med. 2022, 28, 766–779. [Google Scholar] [CrossRef] [PubMed]
- Rydell, N.; Ekoff, H.; Hellström, P.M.; Movérare, R. Measurement of serum IgG anti-integrin αvβ6 autoantibodies is a promising tool in the diagnosis of ulcerative colitis. J. Clin. Med. 2022, 11, 1881. [Google Scholar] [CrossRef] [PubMed]
- Uo, M.; Hisamatsu, T.; Miyoshi, J.; Kaito, D.; Yoneno, K.; Kitazume, M.T.; Mori, M.; Sugita, A.; Koganei, K.; Matsuoka, K.; et al. Mucosal CXCR4+ IgG plasma cells contribute to the pathogenesis of human ulcerative colitis through FcγR-mediated CD14 macrophage activation. Gut 2013, 62, 1734–1744. [Google Scholar] [CrossRef]
- García, M.J.; Pascual, M.; Del Pozo, C.; Díaz-González, A.; Castro, B.; Rasines, L.; Crespo, J.; Rivero, M. Impact of immune-mediated diseases in inflammatory bowel disease and implications in therapeutic approach. Sci. Rep. 2020, 10, 10731. [Google Scholar] [CrossRef]
- Wen, Z.; Fiocchi, C. Inflammatory bowel disease: Autoimmune or immune-mediated pathogenesis? Clin. Dev. Immunol. 2004, 11, 195–204. [Google Scholar] [CrossRef]
- Subudhi, R.N.; Poonia, N.; Singh, D.; Arora, V. Natural approaches for the management of ulcerative colitis: Evidence of preclinical and clinical investigations. Nat. Prod. Bioprospect. 2024, 14, 42. [Google Scholar] [CrossRef] [PubMed]
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979. [Google Scholar] [CrossRef]
- Casalegno Garduño, R.; Däbritz, J. New insights on CD8+ T cells in inflammatory bowel disease and therapeutic approaches. Front. Immunol. 2021, 12, 738762. [Google Scholar] [CrossRef]
- Monteleone, I.; Sarra, M.; Pallone, F.; Monteleone, G. Th17-related cytokines in inflammatory bowel diseases: Friends or foes? Curr. Mol. Med. 2012, 12, 592–597. [Google Scholar] [CrossRef]
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019, 178, 714–730.e22. [Google Scholar] [CrossRef]
- Kim, H.; Kim, H.K.; Hong, D.; Kim, M.; Jang, S.; Yang, C.S.; Yoon, S. Identification of ulcerative colitis-specific immune cell signatures from public single-cell RNA-seq data. Genes Genomics 2023, 45, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Fesneau, O.; Thevin, V.; Pinet, V.; Goldsmith, C.; Vieille, B.; M’Homa Soudja, S.; Lattanzio, R.; Hahne, M.; Dardalhon, V.; Hernandez-Vargas, H.; et al. An intestinal TH17 cell-derived subset can initiate cancer. Nat. Immunol. 2024, 25, 1637–1649. [Google Scholar] [CrossRef]
- Eaden, J.A.; Abrams, K.R.; Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: A meta-analysis. Gut 2001, 48, 526–535. [Google Scholar] [CrossRef]
- Taman, H.; Fenton, C.G.; Hensel, I.V.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. Genome-wide DNA methylation in treatment-naïve ulcerative colitis. J. Crohns Colitis 2018, 12, 1338–1347. [Google Scholar] [CrossRef]
- Xu, H.; Xu, J.; Yang, M.; Liang, Y.; Peng, Q.; Zhang, Y.; Tian, C.; Nie, Y.; Wang, L.; Yao, J.; et al. Epigenetic DNA methylation of Zbtb7b regulates the population of double-positive CD4+CD8+ T cells in ulcerative colitis. J. Transl. Med. 2022, 20, 289. [Google Scholar] [CrossRef] [PubMed]
- Taman, H.; Fenton, C.G.; Anderssen, E.; Florholmen, J.; Paulssen, R.H. DNA hypo-methylation facilitates anti-inflammatory responses in severe ulcerative colitis. PLoS ONE 2021, 16, e0248905. [Google Scholar] [CrossRef] [PubMed]
- Cowland, J.B.; Borregaard, N. Molecular characterization and pattern of tissue expression of the gene for neutrophil gelatinase-associated lipocalin from humans. Genomics 1997, 45, 17–23. [Google Scholar] [CrossRef]
- Friedl, A.; Stoesz, S.P.; Buckley, P.; Gould, M.N. Neutrophil gelatinase-associated lipocalin in normal and neoplastic human tissues. Cell type-specific pattern of expression. Histochem. J. 1999, 31, 433–441. [Google Scholar] [CrossRef]
- Yang, J.; Goetz, D.; Li, J.Y.; Wang, W.; Mori, K.; Setlik, D.; Du, T.; Erdjument-Bromage, H.; Tempst, P.; Strong, R.; et al. An Iron Delivery Pathway Mediated by a lipocalin. Mol. Cell 2002, 10, 1045–1056. [Google Scholar] [CrossRef]
- Bakke, I.; Walaas, G.A.; Bruland, T.; Røyset, E.S.; van Beelen Granlund, A.; Escudero-Hernández, C.; Thorsvik, S.; Münch, A.; Sandvik, A.K.; Østvik, A.E. Mucosal and faecal neutrophil gelatinase-associated lipocalin as potential biomarkers for collagenous colitis. J. Gastroenterol. 2021, 56, 914–927. [Google Scholar] [CrossRef]
- Oikonomou, K.A.; Kapsoritakis, A.N.; Theodoridou, C.; Karangelis, D.; Germenis, A.; Stefanidis, I.; Potamianos, S.P. Neutrophil gelatinase-associated lipocalin (NGAL) in inflammatory bowel disease: Association with pathophysiology of inflammation, established markers, and disease activity. J. Gastroenterol. 2012, 47, 519–530. [Google Scholar] [CrossRef]
- Abdulganieva, D.I.; Mukhametova, D.D.; Valeeva, I.K.; Blatt, N.; Zinkevich, O.D.; Safina, N.A.; Odintsova, A.K.; Korovina, M.O. Faecal neutrophil gelatinase-associated lipocalin as a non-invasive biomarker in inflammatory bowel disease. BioNanoScience 2022, 12, 1381–1387. [Google Scholar] [CrossRef]
- Kim, S.L.; Shin, M.W.; Seo, S.Y.; Kim, S.W. Lipocalin 2 potentially contributes to tumorigenesis from colitis via IL-6/STAT3/NF-κB signaling pathway. Biosci. Rep. 2022, 42, BSR20212418. [Google Scholar] [CrossRef]
- Yang, Y.H.; Yan, F.; Shi, P.S.; Yang, L.C.; Cui, D.J. HIF-1α pathway orchestration by LCN2: A key player in hypoxia-mediated colitis exacerbation. Inflammation 2024, 47, 1491–1519. [Google Scholar] [CrossRef]
- Luo, L. IDDF2024-ABS-0273 LCN2 promotes ferroptosis and induces impairment of intestinal barrier function in ulcerative colitis by regulating ALOX15-mediated lipid peroxidation. Gut 2024, 73, A173. [Google Scholar]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Vogl, T.; Tenbrock, K.; Ludwig, S.; Leukert, N.; Ehrhardt, C.; Van Zoelen, M.A.D.; Nacken, W.; Foell, D.; Van Der Poll, T.; Sorg, C.; et al. Mrp8 and Mrp14 are endogenous activators of toll-like receptor 4, promoting lethal, endotoxin-induced shock. Nat. Med. 2007, 13, 1042–1049. [Google Scholar] [CrossRef] [PubMed]
- Boyd, J.H.; Kan, B.; Roberts, H.; Wang, Y.; Walley, K.R. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ. Res. 2008, 102, 1239–1246. [Google Scholar] [CrossRef]
- Schenten, V.; Plançon, S.; Jung, N.; Hann, J.; Bueb, J.L.; Bréchard, S.; Tschirhart, E.J.; Tolle, F. Secretion of the phosphorylated form of S100A9 from neutrophils is essential for the proinflammatory functions of extracellular S100A8/A9. Front. Immunol. 2018, 9, 447. [Google Scholar] [CrossRef]
- Croce, K.; Gao, H.; Wang, Y.; Mooroka, T.; Sakuma, M.; Shi, C.; Sukhova, G.K.; Packard, R.R.S.; Hogg, N.; Libby, P.; et al. Myeloid-related protein-8/14 is critical for the biological response to vascular injury. Circulation 2009, 120, 427–436. [Google Scholar] [CrossRef]
- Azramezani Kopi, T.; Amini Kadijani, A.; Parsian, H.; Shahrokh, S.; Asadzadeh Aghdaei, H.; Mirzaei, A.; Balaii, H.; Zali, M.R. The value of mRNA expression of S100A8 and S100A9 as blood-based biomarkers of inflammatory bowel disease. Arab. J. Gastroenterol. 2019, 20, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Itoh, H.; Ikemoto, M. Circulating S100A8/A9 is potentially a biomarker that could reflect the severity of experimental colitis in rats. Heliyon 2020, 6, e03470. [Google Scholar] [CrossRef]
- Li, Z.; Zhang, X.; Liu, C.; Peng, Q.; Wu, Y.; Wen, Y.; Zheng, R.; Yan, Q.; Ma, J. Macrophage-biomimetic nanoparticles ameliorate ulcerative colitis through reducing inflammatory factors expression. J. Innate Immun. 2022, 14, 380–392. [Google Scholar] [CrossRef] [PubMed]
- Zha, Z.X.; Lin, Y.; Wang, K.X.; Zhang, Y.L.; Li, D.; Xu, G.Q.; Xu, Q.M.; Liu, Y.L. Hederacoside C ameliorates colitis via restoring impaired intestinal barrier through moderating S100A9/MAPK and neutrophil recruitment inactivation. Acta Pharmacol. Sin. 2023, 44, 105–119. [Google Scholar] [CrossRef] [PubMed]
- Kahn, S.A. The transition from pediatric to adult inflammatory bowel disease care. Gastroenterol. Hepatol. 2016, 12, 403–406. [Google Scholar]
- Jalanka, J.; Cheng, J.; Hiippala, K.; Ritari, J.; Salojärvi, J.; Ruuska, T.; Kalliomäki, M.; Satokari, R. Colonic Mucosal Microbiota and Association of Bacterial Taxa with the Expression of Host Antimicrobial Peptides in Pediatric Ulcerative Colitis. Int. J. Mol. Sci. 2020, 21, 6044. [Google Scholar] [CrossRef]
- Yoshida, M.; Worlock, K.B.; Huang, N.; Lindeboom, R.G.; Butler, C.R.; Kumasaka, N.; Conde, C.D.; Mamanova, L.; Bolt, L.; Richardson, L.; et al. Local and systemic responses to SARS-CoV-2 infection in children and adults. Nature 2022, 602, 321–327. [Google Scholar] [CrossRef]
- Barsness, K.A.; Partrick, D.A.; Bensard, D.D.; Calkins, C.M.; Hendrickson, R.J.; McIntyre, R.C. Endotoxin induces an exaggerated pro-inflammatory response in peritoneal macrophages of children compared to adults. J. Surg. Res. 2003, 114, 261. [Google Scholar] [CrossRef]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Durán, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C.; et al. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-κB activity during disease progression. Blood 2017, 130, 777–788. [Google Scholar] [CrossRef]
- Zhang, X.; Ai, F.; Li, X.; She, X.; Li, N.; Tang, A.; Qin, Z.; Ye, Q.; Tian, L.; Li, G.; et al. Inflammation-induced S100A8 activates I d3 and promotes colorectal tumorigenesis. Int. J. Cancer 2015, 137, 2803–2814. [Google Scholar] [CrossRef]
- Tang, A.; Li, N.; Li, X.; Yang, H.; Wang, W.; Zhang, L.; Li, G.; Xiong, W.; Ma, J.; Shen, S. Dynamic activation of the key pathways: Linking colitis to colorectal cancer in a mouse model. Carcinogenesis 2012, 33, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Wu, Z.; Yi, X. Elucidating the pan-oncologic landscape of S100A9: Prognostic and therapeutic corollaries from an integrative bioinformatics and Mendelian randomization analysis. Sci. Rep. 2024, 14, 19071. [Google Scholar] [CrossRef]
- Bao, X.; Wang, D.; Dai, X.; Liu, C.; Zhang, H.; Jin, Y.; Tong, Z.; Li, B.; Tong, C.; Xin, S.; et al. An immunometabolism subtyping system identifies S100A9+ macrophage as an immune therapeutic target in colorectal cancer based on multiomics analysis. Cell Rep. Med. 2023, 4, 100987. [Google Scholar] [CrossRef] [PubMed]
- Cho, E.; Mun, S.-J.; Kim, H.K.; Ham, Y.S.; Gil, W.J.; Yang, C.-S. Colon-targeted S100A8/A9-specific peptide systems ameliorate colitis and colitis-associated colorectal cancer in mouse models. Acta Pharmacol. Sin. 2024, 45, 581–593. [Google Scholar] [CrossRef]
- Zhang, X.; Wei, L.; Wang, J.; Qin, Z.; Wang, J.; Lu, Y.; Zheng, X.; Peng, Q.; Ye, Q.; Ai, F.; et al. Suppression colitis and colitis-associated colon cancer by anti-S100a9 antibody in mice. Front. Immunol. 2017, 8, 1774. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, W.L.; Tang, D.C.; Chen, L.; Wang, M.; Pack, S.D.; Zhuang, Z.; Rodgers, G.P. Identification and characterization of a novel member of olfactomedin-related protein family, hGC-1, expressed during myeloid lineage development. Gene 2002, 283, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki, S.; Nakamura, T.; Iimura, M.; Kato, Y.; Iizuka, B.; Kobayashi, M.; Hayashi, N. Upregulation of Reg 1α and GW112 in the epithelium of inflamed colonic mucosa. Gut 2001, 48, 623–629. [Google Scholar] [CrossRef]
- Gersemann, M.; Becker, S.; Nuding, S.; Antoni, L.; Ott, G.; Fritz, P.; Oue, N.; Yasui, W.; Wehkamp, J.; Stange, E.F. Olfactomedin-4 is a glycoprotein secreted into mucus in active IBD. J. Crohns Colitis 2012, 6, 425–434. [Google Scholar] [CrossRef]
- Xing, Z.; Li, X.; He, J.; Chen, Y.; Zhu, L.; Zhang, X.; Huang, Z.; Tang, J.; Guo, Y.; He, Y. OLFM4 modulates intestinal inflammation by promoting IL-22+ILC3 in the gut. Commun. Biol. 2024, 7, 914. [Google Scholar] [CrossRef]
- Besson, D.; Pavageau, A.H.; Valo, I.; Bourreau, A.; Bélanger, A.; Eymerit-Morin, C.; Moulière, A.; Chassevent, A.; Boisdron-Celle, M.; Morel, A.; et al. A quantitative proteomic approach of the different stages of colorectal cancer establishes OLFM4 as a new nonmetastatic tumor marker. Mol. Cell Proteom. 2011, 10, M111.009712. [Google Scholar] [CrossRef]
- Wirries, A.; Breyer, S.; Quint, K.; Schobert, R.; Ocker, M. Olfactomedin 4 (GW112, hGC-1) is an independent prognostic marker for survival in patients with colorectal cancer. Exp. Ther. Med. 2010, 1, 73–78. [Google Scholar]
- Chen, Z.; Zhang, X.; Xing, Z.; Lv, S.; Huang, L.; Liu, J.; Ye, S.; Li, X.; Chen, M.; Zuo, S.; et al. OLFM4 deficiency delays the progression of colitis to colorectal cancer by abrogating PMN-MDSCs recruitment. Oncogene 2022, 41, 3131–3150. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Meltzer, P.S. GE Oquery: A bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23, 1846–1847. [Google Scholar] [CrossRef] [PubMed]
- Sherman, B.T.; Huang, D.W.; Tan, Q.; Guo, Y.; Bour, S.; Liu, D.; Stephens, R.; Baseler, M.W.; Lane, C.H.; Lempicki, R.A. David KnowledgeBase: A gene-centered database integrating heterogeneous gene annotation resources to facilitate high-throughput gene functional analysis. BMC Bioinform. 2007, 8, 426. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Egger, G.; Aparicio, A.; Jones, P.A.; Liang, G. Epigenetics in human diseases and prospects of epigenetic therapy. Nature 2004, 429, 457–463. [Google Scholar] [CrossRef]
- Chandrashekar, D.S.; Bashel, B.; Balasubramanya, S.A.H.; Creighton, C.J.; Ponce-Rodriguez, I.; Chakravarthi, B.V.S.K.; Varambally, S. UALCAN: A portal for facilitating tumor subgroup gene expression and survival analyses. Neoplasia 2017, 19, 649–658. [Google Scholar] [CrossRef]
- Tarhan, L.; Bistline, J.; Chang, J.; Galloway, B.; Hanna, E.; Weitz, E. Single Cell Portal: An interactive home for single-cell genomics data. bioRxiv 2023, 2023.07.13.548886. [Google Scholar]
- Ru, B.; Wong, C.N.; Tong, Y.; Zhong, J.Y.; Zhong, S.S.W.; Wu, W.C.; Chu, K.C.; Wong, C.Y.; Lau, C.Y.; Chen, I.; et al. TISIDB: An integrated repository portal for tumor-immune system interactions. Bioinformatics 2019, 35, 4200–4202. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Győrffy, B. Transcriptome-level discovery of survival-associated biomarkers and therapy targets in non-small-cell lung cancer. Br. J. Pharmacol. 2024, 181, 362–374. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Miao, B.; Wang, S.; Dong, W.; Xu, H.; Si, C.; Wang, W.; Duan, S.; Lou, J.; Bao, Z.; et al. Hiplot: A comprehensive and easy-to-use web service for boosting publication-ready biomedical data visualization. Brief. Bioinform. 2022, 23, bbac261. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genes | LASSO Coefficient | log2FC | Adj. p-Value | Rank (of 113) |
|---|---|---|---|---|
| LCN2 | −3.40228 | 1.42065 | 1.45 × 10−37 | 2 |
| MMP9 | −1.38405 | 1.45637 | 2.02 × 10−51 | 25 |
| S100A9 | −0.32635 | 1.32343 | 2.59 × 10−85 | 86 |
| OLFM4 | −0.05912 | 2.68168 | 1.26 × 10−55 | 110 |
| Characteristics | Ulcerative Colitis | Healthy Controls | p-Value |
|---|---|---|---|
| No. of patients | n = 481 | n = 311 | - |
| % of men/women | Men: 34.21% Women: 65.79% | Men: 67.11% Women: 32.89% | 0.4538 |
| Age (y), mean (SD) | 41.09 ± 14.22 | 48.02 ± 12.14 | < 0.001 |
| Characteristics | UC (Pediatric and Adolescent) | UC (Adult) | Control (Pediatric and Adolescent) | Control (Adult) |
|---|---|---|---|---|
| No. of patients | n = 41 | n = 34 | n = 22 | n = 22 |
| % of men/women | Men: 58.54% Women: 41.46% | Men: 47.06% Women: 52.94% | Men: 72.73% Women: 27.27% | Men: 54.55% Women: 45.45% |
| Age(y), mean (SD) | 17.68 ± 2.27 | 37.29 ± 12.94 | 17.28 ± 2.25 | 37.09 ± 13.19 |
| Datasets | Sample Type | UC Samples | Control Samples | Utilization |
|---|---|---|---|---|
| GSE177044 | Whole blood | n = 481 | n = 311 | DEGs and hub gene selection |
| GSE59071 | Colonic tissue | n = 97 | n = 11 | Hub genes validation based on shared expression patterns in both blood and tissue |
| GSE66407 | n = 161 | n = 99 | ||
| GSE87466 | n = 87 | n = 21 | ||
| GSE32149 | Colonic tissue | n = 4 | n = 10 | Key hub genes selection |
| GSE107597 | Colonic tissue | n = 75 | n = 44 | Age-related expression validation |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheon, J.; Kim, S.H.; Park, J.; Kim, T.H. Exploring the Clinical Implication of S100A9 in Ulcerative Colitis and Its Progression to Cancer: A Journey from Inflammation to Cancer. Int. J. Mol. Sci. 2025, 26, 5693. https://doi.org/10.3390/ijms26125693
Cheon J, Kim SH, Park J, Kim TH. Exploring the Clinical Implication of S100A9 in Ulcerative Colitis and Its Progression to Cancer: A Journey from Inflammation to Cancer. International Journal of Molecular Sciences. 2025; 26(12):5693. https://doi.org/10.3390/ijms26125693
Chicago/Turabian StyleCheon, Jaehwan, Sang Hyun Kim, Jaehyung Park, and Tae Hoon Kim. 2025. "Exploring the Clinical Implication of S100A9 in Ulcerative Colitis and Its Progression to Cancer: A Journey from Inflammation to Cancer" International Journal of Molecular Sciences 26, no. 12: 5693. https://doi.org/10.3390/ijms26125693
APA StyleCheon, J., Kim, S. H., Park, J., & Kim, T. H. (2025). Exploring the Clinical Implication of S100A9 in Ulcerative Colitis and Its Progression to Cancer: A Journey from Inflammation to Cancer. International Journal of Molecular Sciences, 26(12), 5693. https://doi.org/10.3390/ijms26125693