
Perspectives in Amyotrophic Lateral Sclerosis: Biomarkers, Omics, and Gene Therapy Informing Disease and Treatment

 ,
,  , ,
, ,  and
and
Abstract
1. Introduction
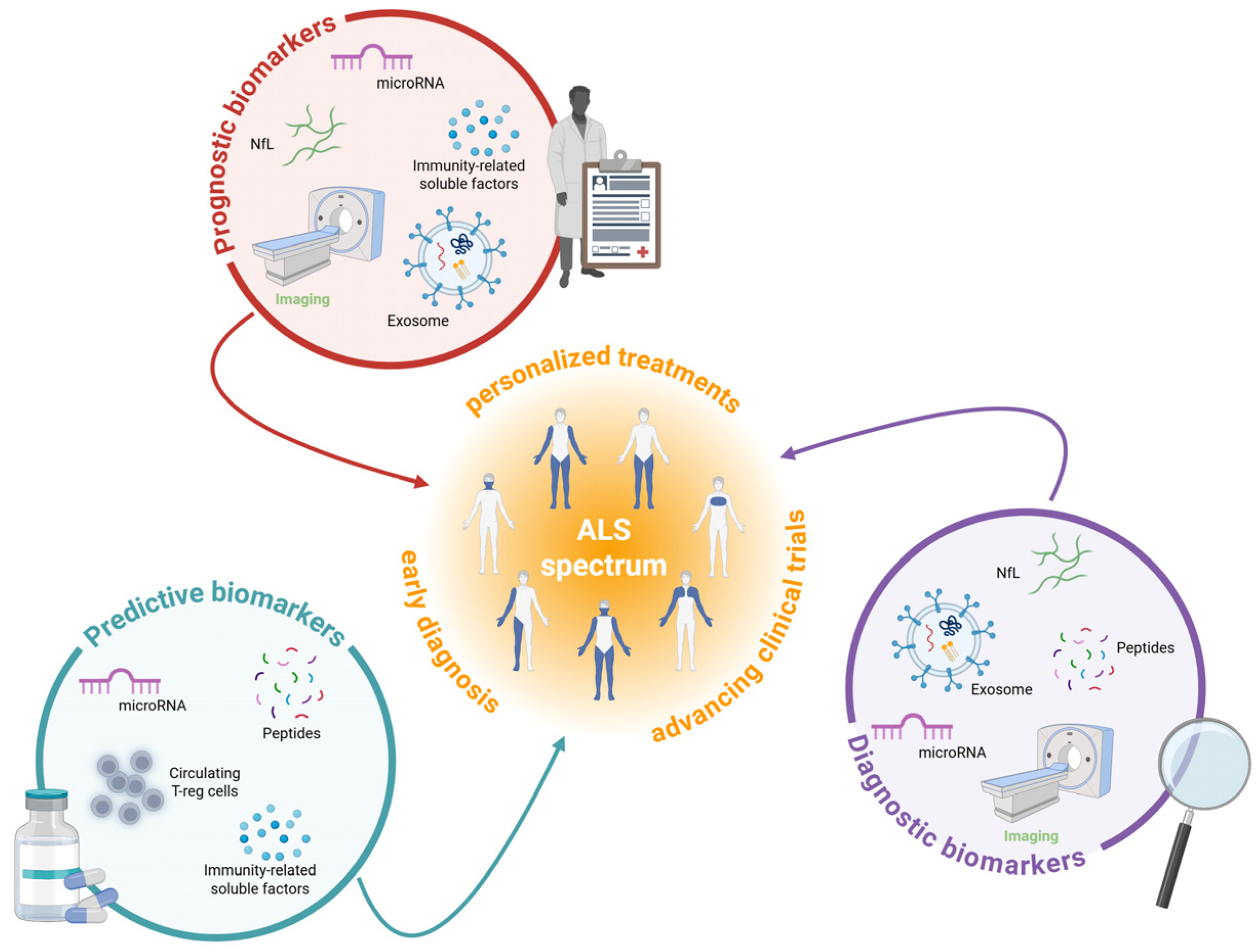
2. Redefining ALS Diagnosis and Treatment Through Biomarker Discovery
2.1. Fluid-Based Biomarkers
2.1.1. Neurofilament Light Chain
2.1.2. Exosomes
2.1.3. Non-Coding RNA
2.1.4. Cryptic Peptides
2.1.5. Neuroinflammation and Metabolism

{kind=link}
{kind=link}
| Details | Roles | References | |
|---|---|---|---|
| Traditional diagnostic and prognostic methods | |||
| Clinical evaluation | El Escorial criteria; Awaji criteria; Gold Coast criteria; The King’s Clinical Staging System and the Milano–Torino (MiToS) Functional Staging System; ALS Functional Rating Scale—Revised (ALSFRS-R). | Diagnostic evaluation | [5] |
| Electrophysiological assessments | Muscle action potential. | Diagnostic evaluation | [6] |
| Neurofilament light chain (NfL) analysis | NfL is a sensitive but non-specific biomarker of neuronal damage whose elevated levels in CSF and blood correlate with ALS progression and severity. | Diagnostic and prognostic fluid biomarkers | [20,21,22] |
| Promising non-invasive diagnostic, prognostic, and predictive methods | |||
| Exosomes analysis | Exosomes are extracellular vesicles capable of crossing the blood–brain barrier and carrying ALS-related biomarkers (e.g., TDP-43, NfL, miRNAs), offering a promising, non-invasive, and cell-specific approach for early diagnosis and disease monitoring. | Prognostic and predictive fluid biomarkers | [23,24,25,26,27,28,29,30,31] |
| Non-coding RNA profiling | miRNAs, small non-coding RNAs involved in gene regulation expressed in CSF, blood, and serum, represent a promising avenue for identifying molecular signatures linked to ALS onset and progression. | Diagnostic, prognostic, and predictive fluid biomarkers | [16,33,34,35,36] |
| Cryptic peptide analysis | Cryptic peptides, closely linked to TDP-43 pathology, are emerging biomarkers expressed in CFS, plasma, and serum, with strong potential for early, pathology-specific ALS diagnosis and patient subtyping. | Diagnostic and predictive fluid biomarkers | [30,31,32,37] |
| Neuroinflammation and metabolism investigation | Chitinase family, specifically CHIT1, CHI3L1, and CHI3L2 expressed in CSF; | Prognostic and predictive fluid biomarkers | [35] |
| S100B expressed in CFS; | [36] | ||
| Treg cells expressed in CSF; | [38] | ||
| p75 and neopterin expressed in urine. | [37,38] | ||
| Neuroimaging and electrophysiological assessment | MRI to measure brain, spinal cord, and muscle volume; | Diagnostic and prognostic non-fluid biomarkers | [39,40,41,42,43,44,45] |
| EMG to measure fasciculations early. | [46,47] | ||
| PET to measure metabolic changes. | [48,49,50,51] |
2.2. Non-Fluid Biomarkers in ALS
2.2.1. Magnetic Resonance Imaging
2.2.2. Electromyography and Positron Emission Tomography
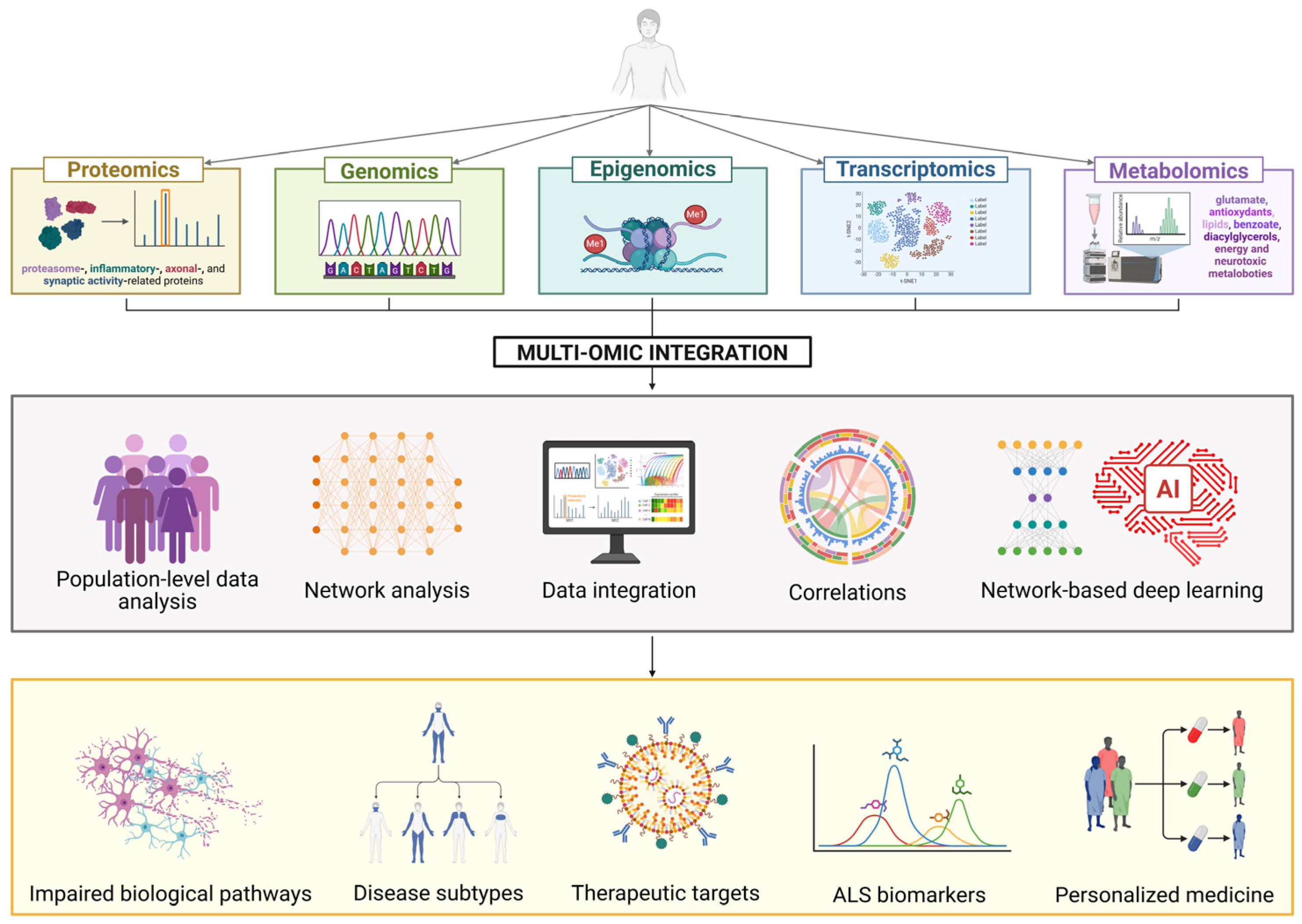
3. Understanding ALS Pathophysiology: Multi-Omics Integration
3.1. Genomics
3.2. Transcriptomics and Epigenomics
3.3. Proteomics
3.4. Metabolomics
3.5. Multi-Omics Integration in ALS
4. The Evolution of ALS Treatment: From Current Options and Innovative Approaches
4.1. Current Treatment Landscape and Limitations
| Name | Drug Type | Target | Delivery System | Phase | Clinical Trial | Developer | Year (Start) | Route of Administration | Key Information |
|---|---|---|---|---|---|---|---|---|---|
| Tofersen (BIIB067) | ASO | SOD1 mutations | Modified oligonucleotide backbone | Phase 3 completed | NCT02623699 | Biogen | 2018 | Intrathecal | Received FDA accelerated approval in 2023. VALOR study showed slowing of disease progression in SOD1-ALS patients. |
| BIIB078 | ASO | C9orf72 mutations | Modified oligonucleotide backbone | Phase 1/2 | NCT03626012 | Biogen | 2019 | Intrathecal | It targets the most common genetic cause of ALS. |
| ION363 (Jacifusen) | ASO | FUS mutations | Modified oligonucleotide backbone | Phase 3 | NCT04768972 | Ionis Pharmaceuticals | 2020 | Intrathecal | Specifically for patients with FUS mutations. Named after Jaci Hermstad, an ALS patient. |
| VM202 (Engensis) | Plasmid DNA | HGF expression | Non-viral plasmid vector | Phase 2 | NCT02427464 | Helixmith | 2018 | Intramuscular | Non-viral plasmid DNA containing the HGF gene, enabling cells to produce this protein to promote nerve regeneration. |
| AAV-GDNF | GDNF gene | Neuroprotection | AAV9 viral vector | Phase 1/2 | NCT01621581 | Various institutions | 2019 | Intrathecal | It delivers the gene for GDNF to promote production of this neuroprotective protein by transduced cells. One-time administration. |
| AL001 | Autologous Treg cell therapy | Immune regulation | AAV9 viral vector | Phase 1/2 | NCT05053035 | Asklepios BioPharmaceutical | 2020 | Intrathecal | One-time AAV delivery of the hepatocyte growth factor gene, allowing transduced cells to express this neuroprotective protein. |
| AMX0035 (Relyvrio) | Small molecule combination (sodium phenylbutyrate and taurursodiol) | Cellular death pathways | - | FDA Approved | NCT03127514 | Amylyx Pharmaceuticals | 2017 | Oral | Combination therapy. FDA approved in 2022. |
| NurOwn | Mesenchymal stem cell therapy | Multiple neuroprotective pathways | Autologous MSCs | Phase 3 completed | NCT03280056 | BrainStorm Cell Therapeutics | 2017 | Intrathecal | Autologous MSCs secreting neurotrophic factors. Mixed results in Phase 3. |
| Masitinib | Small molecule (tyrosine kinase inhibitor) | Mast cells, microglia | - | Phase 3 | NCT03127267 | AB Science | 2017 | Oral | Anti-inflammatory and neuroprotective effects. |
| MN-166 (Ibudilast) | Small molecule (PDE4 inhibitor) | Neuroinflammation | - | Phase 2/3 | NCT02714036 | MediciNova | 2018 | Oral | Anti-inflammatory and neuroprotective. |
| Ravulizumab (Ultomiris) | Monoclonal antibody (C5 complement inhibitor) | Complement system | - | Phase 3 | NCT04248465 | Alexion Pharmaceuticals | 2019 | Intravenous | It targets complement-mediated neuroinflammation. |
| AP-101 | Recombinant human FGF-1 protein | Neuronal survival | - | Phase 3 | NCT05039099 | Artielle Pharmaceuticals | 2019 | Intrathecal | It promotes motor neuron survival. |
| AT-1501 | Monoclonal antibody | CD40L | - | Phase 2 | NCT04322149 | Eledon Pharmaceuticals | 2020 | Intravenous | It targets neuroinflammation. |
| SBT-272 | Small molecule (mitochondria-targeted peptide) | Mitochondrial dysfunction | - | Phase 1 | NCT02297035 | Stealth BioTherapeutics | 2021 | Oral | It improves mitochondrial function in neurons. |
4.2. Emerging Gene Therapies
4.2.1. Gene Replacement and Augmentation
4.2.2. Gene Silencing Approaches
| Gene | Chromosome | Year Discovered | Protein Function | % of fALS | % of sALS | Key Features | References |
|---|---|---|---|---|---|---|---|
| C9orf72 | 9p21.2 | 2011 | Membrane trafficking; autophagy | 30–40% | 5–10% | Hexanucleotide (GGGGCC) repeat expansion; most common genetic cause of ALS/FTD; complex pathomechanisms including RNA toxicity and DPR proteins | [94,129,138] |
| SOD1 | 21q22.11 | 1993 | Antioxidant enzyme | 15–20% | 1–2% | Over 200 mutations identified; most studied ALS gene; first successful gene therapy target (Tofersen) | [93,139,140] |
| TARDBP (TDP-43) | 1p36.22 | 2008 | RNA processing/metabolism | 4–5% | 1–2% | TDP-43 pathology present in ~97% of all ALS cases; primarily missense mutations in C-terminal domain | [136] |
| FUS | 16p11.2 | 2009 | RNA processing/metabolism | 4–5% | <1% | Associated with early-onset and aggressive disease course; primarily C-terminal mutations affecting nuclear localization | [141] |
| OPTN | 10p13 | 2010 | Vesicular transport | <4% | <0.4% | OPTN defects result in mitophagy disorder, protein aggregation, neuroinflammation, vesicular transport, neuronal axonal degeneration, oxidative stress | [142] |
| p62/SQSTM1 | 5q35.3 | 2011 | Autophagy | 2–3% | <1% | Depletion of p62 protein levels inhibits LC3 recruitment to autophagosomes and has been shown to increase cell death induced by mutant huntingtin | [143] |
| ATXN2 | 12q24.12 | 2010 | RNA processing | 1–2% | <1% | Intermediate-length polyQ expansions (27–33 repeats) increase ALS risk; longer expansions cause SCA2 | [144] |
| VCP | 9p13.3 | 2010 | Protein degradation, autophagy | 1–2% | <1% | Also associated with inclusion body myopathy and frontotemporal dementia (IBMPFD) | [145] |
| UBQLN2 | Xp11.21 | 2011 | Protein degradation | 1–2% | <1% | X-linked dominant inheritance; affects protein homeostasis; associated with ALS-FTD | [146] |
| PFN1 | 17p13.2 | 2012 | Cytoskeleton dynamics | 1–2% | <1% | Mutant PFN1 contributes to ALS pathogenesis by altering actin dynamics and inhibiting axon outgrowth | [147,148] |
| TBK1 | 12q14.2 | 2015 | Autophagy, inflammation | 1–2% | <1% | Haploinsufficiency mechanism; involved in autophagy and inflammatory pathways | [149] |
| NEK1 | 4q33 | 2016 | DNA damage response, axonal growth | 1–2% | 1–2% | Loss-of-function variants; identified through large-scale genome-wide association studies | [150] |
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ALS | Amyotrophic lateral sclerosis |
| sALS | Sporadic Amyotrophic lateral sclerosis |
| fALS | Familial Amyotrophic lateral sclerosis |
| C9orf72 | Chromosome 9 Open Reading Frame 72 |
| TARDBP | TAR DNA-binding Protein |
| SOD1 | Superoxide dismutase 1 |
| FUS | Fused in Sarcoma |
| CSF | Cerebrospinal fluid |
| NfL | Neurofilament light chain |
| FTD | Frontotemporal dementia |
| STMN2 | Stathmin-2 |
| UNC13A | Unc-13 Homolog A |
| ncRNA | Noncoding RNA |
| miRNA | Micro RNA |
| CHIT1 | Chitotriosidase |
| CHI3L1 | Chitinase-3-like-1 |
| CHI3L2 | Chitinase-3-like-2 |
| S100B | S100 Calcium Binding Protein B |
| Treg | T regulatory |
| MRI | Magnetic Resonance Imaging |
| PET | Positron Emission Tomography |
| EMG | Electromyography |
| AI | Artificial Intelligence |
| EPHA4 | Ephrin Type-A Receptor 4 |
| CNVs | Copy number variations |
| lncRNA | Long non-coding RNA |
| RNA-seq | High-throughput RNA sequencing |
| hnRNP | Heterogeneous Nuclear Ribonucleoprotein |
| m6A | N6-methyladenosine |
| myomiR | Muscle-specific microRNA |
| iPSC | Induced pluripotent stem cells |
| CNS | Central nervous system |
| ASO | Antisense oligonucleotide |
| siRNA | Small interfering RNA |
| MAPK | Mitogen-activated protein kinase |
| QTL | Quantitative trait loci |
| eQTL | Expression Quantitative trait loci |
| pQTL | Protein Quantitative trait loci |
| sQTL | Splicing Quantitative trait loci |
| meQTL | Methylation Quantitative trait loci |
| haQTL | Histone acetylation Quantitative trait loci |
| AAV | Adeno-associated-virus |
| RNAi | RNA interference |
| HGF | Hepatocyte growth factor |
| GDNF | Glial cell-derived neurotrophic factor |
| LNP | Lipid nanoparticle |
| BBB | Blood–brain barrier |
| DPR | Dipeptide repeat protein |
| ATXN2 | Ataxin-2 |
| siRNA | Small interfering RNA |
| RISC | RNA-induced silencing complex |
| shRNA | Short hairpin RNA |
References
- Riva, N.; Domi, T.; Pozzi, L.; Lunetta, C.; Schito, P.; Spinelli, E.G.; Cabras, S.; Matteoni, E.; Consonni, M.; Bella, E.D.; et al. Update on Recent Advances in Amyotrophic Lateral Sclerosis. J. Neurol. 2024, 271, 4693–4723. [Google Scholar] [CrossRef] [PubMed]
- Chiò, A.; Logroscino, G.; Hardiman, O.; Swingler, R.; Mitchell, D.; Beghi, E.; Traynor, B.G.; On Behalf of the Eurals Consortium. Prognostic Factors in ALS: A Critical Review. Amyotroph. Lateral Scler. 2009, 10, 310–323. [Google Scholar] [CrossRef]
- Boylan, K. Familial Amyotrophic Lateral Sclerosis. Neurol. Clin. 2015, 33, 807–830. [Google Scholar] [CrossRef] [PubMed]
- Bendotti, C.; Bonetto, V.; Pupillo, E.; Logroscino, G.; Al-Chalabi, A.; Lunetta, C.; Riva, N.; Mora, G.; Lauria, G.; Weishaupt, J.H.; et al. Focus on the Heterogeneity of Amyotrophic Lateral Sclerosis. Amyotroph. Lateral Scler. Front. Degener. 2020, 21, 485–495. [Google Scholar] [CrossRef]
- Rizea, R.E.; Corlatescu, A.-D.; Costin, H.P.; Dumitru, A.; Ciurea, A.V. Understanding Amyotrophic Lateral Sclerosis: Pathophysiology, Diagnosis, and Therapeutic Advances. Int. J. Mol. Sci. 2024, 25, 9966. [Google Scholar] [CrossRef]
- Kiani, L. Electrophysiology Test for Early ALS Diagnosis. Nat. Rev. Neurol. 2023, 19, 459. [Google Scholar] [CrossRef]
- Geevasinga, N.; Menon, P.; Scherman, D.B.; Simon, N.; Yiannikas, C.; Henderson, R.D.; Kiernan, M.C.; Vucic, S. Diagnostic Criteria in Amyotrophic Lateral Sclerosis: A Multicenter Prospective Study. Neurology 2016, 87, 684–690. [Google Scholar] [CrossRef]
- Sleutjes, B.T.H.M.; Bystrup Jacobsen, A.; Tankisi, H.; Gorkem Sirin, N.; Emre Oge, A.; Henderson, R.D.; van Doorn, P.A.; van den Berg, L.H.; van Eijk, R.P.A. Advancing Disease Monitoring of Amyotrophic Lateral Sclerosis with the Compound Muscle Action Potential Scan. Clin. Neurophysiol. 2021, 132, 3152–3159. [Google Scholar] [CrossRef] [PubMed]
- Rokade, A.V.; Yelne, P.; Giri, A. Riluzole and Edavarone: The Hope Against Amyotrophic Lateral Sclerosis. Cureus 2022, 14, e30035. [Google Scholar] [CrossRef]
- Prieto Santamaría, L.; García del Valle, E.P.; Zanin, M.; Hernández Chan, G.S.; Pérez Gallardo, Y.; Rodríguez-González, A. Classifying Diseases by Using Biological Features to Identify Potential Nosological Models. Sci. Rep. 2021, 11, 21096. [Google Scholar] [CrossRef]
- Goutman, S.A.; Hardiman, O.; Al-Chalabi, A.; Chió, A.; Savelieff, M.G.; Kiernan, M.C.; Feldman, E.L. Recent Advances in the Diagnosis and Prognosis of Amyotrophic Lateral Sclerosis. Lancet Neurol. 2022, 21, 480–493. [Google Scholar] [CrossRef]
- Irwin, K.E.; Sheth, U.; Wong, P.C.; Gendron, T.F. Fluid Biomarkers for Amyotrophic Lateral Sclerosis: A Review. Mol. Neurodegener. 2024, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Morello, G.; Salomone, S.; D’Agata, V.; Conforti, F.L.; Cavallaro, S. From Multi-Omics Approaches to Precision Medicine in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 577755. [Google Scholar] [CrossRef]
- Caldi Gomes, L.; Hänzelmann, S.; Hausmann, F.; Khatri, R.; Oller, S.; Parvaz, M.; Tzeplaeff, L.; Pasetto, L.; Gebelin, M.; Ebbing, M.; et al. Multiomic ALS Signatures Highlight Subclusters and Sex Differences Suggesting the MAPK Pathway as Therapeutic Target. Nat. Commun. 2024, 15, 4893. [Google Scholar] [CrossRef]
- Witzel, S.; Mayer, K.; Oeckl, P. Biomarkers for Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 2022, 35, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Giagnorio, E.; Malacarne, C.; Cavalcante, P.; Scandiffio, L.; Cattaneo, M.; Pensato, V.; Gellera, C.; Riva, N.; Quattrini, A.; Dalla Bella, E.; et al. MiR-146a in ALS: Contribution to Early Peripheral Nerve Degeneration and Relevance as Disease Biomarker. Int. J. Mol. Sci. 2023, 24, 4610. [Google Scholar] [CrossRef]
- Zhou, J.; Zeng, Q.; Liao, Q.; Niu, Q.; Gu, W.; Su, D.; Li, S.; Xiao, B.; Bi, F. Biomarkers in Cerebrospinal Fluid for Amyotrophic Lateral Sclerosis Phenotypes. Ann. Clin. Transl. Neurol. 2023, 10, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Bonanno, S.; Marcuzzo, S.; Malacarne, C.; Giagnorio, E.; Masson, R.; Zanin, R.; Arnoldi, M.T.; Andreetta, F.; Simoncini, O.; Venerando, A.; et al. Circulating MyomiRs as Potential Biomarkers to Monitor Response to Nusinersen in Pediatric SMA Patients. Biomedicines 2020, 8, 21. [Google Scholar] [CrossRef]
- Vu, L.T.; Bowser, R. Fluid-Based Biomarkers for Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14, 119–134. [Google Scholar] [CrossRef]
- Thompson, A.G.; Gray, E.; Verber, N.; Bobeva, Y.; Lombardi, V.; Shepheard, S.R.; Yildiz, O.; Feneberg, E.; Farrimond, L.; Dharmadasa, T.; et al. Multicentre Appraisal of Amyotrophic Lateral Sclerosis Biofluid Biomarkers Shows Primacy of Blood Neurofilament Light Chain. Brain Commun. 2022, 4, fcac029. [Google Scholar] [CrossRef]
- Alirezaei, Z.; Pourhanifeh, M.H.; Borran, S.; Nejati, M.; Mirzaei, H.; Hamblin, M.R. Neurofilament Light Chain as a Biomarker, and Correlation with Magnetic Resonance Imaging in Diagnosis of CNS-Related Disorders. Mol. Neurobiol. 2020, 57, 469–491. [Google Scholar] [CrossRef] [PubMed]
- Benatar, M.; Wuu, J.; Turner, M.R. Neurofilament Light Chain in Drug Development for Amyotrophic Lateral Sclerosis: A Critical Appraisal. Brain 2023, 146, 2711–2716. [Google Scholar] [CrossRef]
- McCluskey, G.; Morrison, K.E.; Donaghy, C.; Rene, F.; Duddy, W.; Duguez, S. Extracellular Vesicles in Amyotrophic Lateral Sclerosis. Life 2022, 13, 121. [Google Scholar] [CrossRef]
- Darabi, S.; Ariaei, A.; Rustamzadeh, A.; Afshari, D.; Charkhat Gorgich, E.A.; Darabi, L. Cerebrospinal Fluid and Blood Exosomes as Biomarkers for Amyotrophic Lateral Sclerosis; A Systematic Review. Diagn. Pathol. 2024, 19, 47. [Google Scholar] [CrossRef]
- Banack, S.A.; Dunlop, R.A.; Mehta, P.; Mitsumoto, H.; Wood, S.P.; Han, M.; Cox, P.A. A MicroRNA Diagnostic Biomarker for Amyotrophic Lateral Sclerosis. Brain Commun. 2024, 6, fcae268. [Google Scholar] [CrossRef] [PubMed]
- Katsu, M.; Hama, Y.; Utsumi, J.; Takashina, K.; Yasumatsu, H.; Mori, F.; Wakabayashi, K.; Shoji, M.; Sasaki, H. MicroRNA Expression Profiles of Neuron-Derived Extracellular Vesicles in Plasma from Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 2019, 708, 134176. [Google Scholar] [CrossRef]
- Morasso, C.F.; Sproviero, D.; Mimmi, M.C.; Giannini, M.; Gagliardi, S.; Vanna, R.; Diamanti, L.; Bernuzzi, S.; Piccotti, F.; Truffi, M.; et al. Raman Spectroscopy Reveals Biochemical Differences in Plasma Derived Extracellular Vesicles from Sporadic Amyotrophic Lateral Sclerosis Patients. Nanomedicine 2020, 29, 102249. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Veyrat-Durebex, C.; Bocca, C.; Patin, F.; Vourc’h, P.; Kouassi Nzoughet, J.; Lenaers, G.; Andres, C.R.; Simard, G.; Corcia, P.; et al. Lipidomics Reveals Cerebrospinal-Fluid Signatures of ALS. Sci. Rep. 2017, 7, 17652. [Google Scholar] [CrossRef]
- Lai, J.J.; Chau, Z.L.; Chen, S.-Y.; Hill, J.J.; Korpany, K.V.; Liang, N.-W.; Lin, L.-H.; Lin, Y.-H.; Liu, J.K.; Liu, Y.-C.; et al. Exosome Processing and Characterization Approaches for Research and Technology Development. Adv. Sci. 2022, 9, e2103222. [Google Scholar] [CrossRef]
- Chen, Y.; Xia, K.; Chen, L.; Fan, D. Increased Interleukin-6 Levels in the Astrocyte-Derived Exosomes of Sporadic Amyotrophic Lateral Sclerosis Patients. Front. Neurosci. 2019, 13, 574. [Google Scholar] [CrossRef]
- Dehghani, S.; Ocakcı, O.; Hatipoglu, P.T.; Özalp, V.C.; Tevlek, A. Exosomes as Biomarkers and Therapeutic Agents in Neurodegenerative Diseases: Current Insights and Future Directions. Mol. Neurobiol. 2025, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Diaz Duran, R.; Wei, H.; Kim, D.H.; Wu, J.Q. Invited Review: Long Non-coding RNAs: Important Regulators in the Development, Function and Disorders of the Central Nervous System. Neuropathol. Appl. Neurobiol. 2019, 45, 538–556. [Google Scholar] [CrossRef]
- Cao, Y.; Xu, Y.; Cao, M.; Chen, N.; Zeng, Q.; Lai, M.K.P.; Fan, D.; Sethi, G.; Cao, Y. Fluid-Based Biomarkers for Neurodegenerative Diseases. Ageing Res. Rev. 2025, 108, 102739. [Google Scholar] [CrossRef] [PubMed]
- Joilin, G.; Gray, E.; Thompson, A.G.; Bobeva, Y.; Talbot, K.; Weishaupt, J.; Ludolph, A.; Malaspina, A.; Leigh, P.N.; Newbury, S.F.; et al. Identification of a Potential Non-Coding RNA Biomarker Signature for Amyotrophic Lateral Sclerosis. Brain Commun. 2020, 2, fcaa053. [Google Scholar] [CrossRef]
- Waller, R.; Goodall, E.F.; Milo, M.; Cooper-Knock, J.; Da Costa, M.; Hobson, E.; Kazoka, M.; Wollff, H.; Heath, P.R.; Shaw, P.J.; et al. Serum MiRNAs MiR-206, 143-3p and 374b-5p as Potential Biomarkers for Amyotrophic Lateral Sclerosis (ALS). Neurobiol. Aging 2017, 55, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Waller, R.; Wyles, M.; Heath, P.R.; Kazoka, M.; Wollff, H.; Shaw, P.J.; Kirby, J. Small RNA Sequencing of Sporadic Amyotrophic Lateral Sclerosis Cerebrospinal Fluid Reveals Differentially Expressed MiRNAs Related to Neural and Glial Activity. Front. Neurosci. 2018, 11, 731. [Google Scholar] [CrossRef] [PubMed]
- Klim, J.R.; Williams, L.A.; Limone, F.; Guerra San Juan, I.; Davis-Dusenbery, B.N.; Mordes, D.A.; Burberry, A.; Steinbaugh, M.J.; Gamage, K.K.; Kirchner, R.; et al. ALS-Implicated Protein TDP-43 Sustains Levels of STMN2, a Mediator of Motor Neuron Growth and Repair. Nat. Neurosci. 2019, 22, 167–179. [Google Scholar] [CrossRef]
- Reddy, D.S.; Abeygunaratne, H.N. Experimental and Clinical Biomarkers for Progressive Evaluation of Neuropathology and Therapeutic Interventions for Acute and Chronic Neurological Disorders. Int. J. Mol. Sci. 2022, 23, 11734. [Google Scholar] [CrossRef]
- Senda, J.; Atsuta, N.; Watanabe, H.; Bagarinao, E.; Imai, K.; Yokoi, D.; Riku, Y.; Masuda, M.; Nakamura, R.; Watanabe, H.; et al. Structural MRI Correlates of Amyotrophic Lateral Sclerosis Progression. J. Neurol. Neurosurg. Psychiatry 2017, 88, 901–907. [Google Scholar] [CrossRef]
- Ta, D.; Ishaque, A.H.; Elamy, A.; Anand, T.; Wu, A.; Eurich, D.T.; Luk, C.; Yang, Y.H.; Kalra, S. Severity of in Vivo Corticospinal Tract Degeneration Is Associated with Survival in Amyotrophic Lateral Sclerosis: A Longitudinal, Multicohort Study. Eur. J. Neurol. 2023, 30, 1220–1231. [Google Scholar] [CrossRef]
- Müller, H.-P.; Turner, M.R.; Grosskreutz, J.; Abrahams, S.; Bede, P.; Govind, V.; Prudlo, J.; Ludolph, A.C.; Filippi, M.; Kassubek, J.; et al. A Large-Scale Multicentre Cerebral Diffusion Tensor Imaging Study in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2016, 87, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, V.; Pioro, E.P. Graph Theory Network Analysis Provides Brain MRI Evidence of a Partial Continuum of Neurodegeneration in Patients with UMN-Predominant ALS and ALS-FTD. Neuroimage Clin. 2022, 35, 103037. [Google Scholar] [CrossRef]
- Querin, G.; Bede, P.; El Mendili, M.M.; Li, M.; Pélégrini-Issac, M.; Rinaldi, D.; Catala, M.; Saracino, D.; Salachas, F.; Camuzat, A.; et al. Presymptomatic Spinal Cord Pathology in C9orf72 Mutation Carriers: A Longitudinal Neuroimaging Study. Ann. Neurol. 2019, 86, 158–167. [Google Scholar] [CrossRef]
- De Vocht, J.; Van Weehaeghe, D.; Ombelet, F.; Masrori, P.; Lamaire, N.; Devrome, M.; Van Esch, H.; Moisse, M.; Koole, M.; Dupont, P.; et al. Differences in Cerebral Glucose Metabolism in ALS Patients with and without C9orf72 and SOD1 Mutations. Cells 2023, 12, 933. [Google Scholar] [CrossRef]
- Bede, P.; Lulé, D.; Müller, H.-P.; Tan, E.L.; Dorst, J.; Ludolph, A.C.; Kassubek, J. Presymptomatic Grey Matter Alterations in ALS Kindreds: A Computational Neuroimaging Study of Asymptomatic C9orf72 and SOD1 Mutation Carriers. J. Neurol. 2023, 270, 4235–4247. [Google Scholar] [CrossRef] [PubMed]
- Toh, C.; Keslake, A.; Payne, T.; Onwuegbuzie, A.; Harding, J.; Baster, K.; Hoggard, N.; Shaw, P.J.; Wilkinson, I.D.; Jenkins, T.M. Analysis of Brain and Spinal MRI Measures in a Common Domain to Investigate Directional Neurodegeneration in Motor Neuron Disease. J. Neurol. 2023, 270, 1682–1690. [Google Scholar] [CrossRef]
- Kriss, A.; Jenkins, T. Muscle MRI in Motor Neuron Diseases: A Systematic Review. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Jamali, A.M.; Kethamreddy, M.; Burkett, B.J.; Port, J.D.; Pandey, M.K. PET and SPECT Imaging of ALS: An Educational Review. Mol. Imaging 2023, 2023, 5864391. [Google Scholar] [CrossRef]
- Chew, S.; Atassi, N. Positron Emission Tomography Molecular Imaging Biomarkers for Amyotrophic Lateral Sclerosis. Front. Neurol. 2019, 10, 135. [Google Scholar] [CrossRef]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A Long Noncoding RNA Maintains Active Chromatin to Coordinate Homeotic Gene Expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef]
- Kas, A.; Rozenblum, L.; Pyatigorskaya, N. Clinical Value of Hybrid PET/MR Imaging: Brain Imaging Using PET/MR Imaging. Magn. Reson. Imaging Clin. N. Am. 2023, 31, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Menke, R.A.L.; Agosta, F.; Grosskreutz, J.; Filippi, M.; Turner, M.R. Neuroimaging Endpoints in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2017, 14, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Waisberg, E.; Ong, J.; Paladugu, P.; Amiri, D.; Saintyl, J.; Yelamanchi, J.; Nahouraii, R.; Jagadeesan, R.; Tavakkoli, A. Artificial Intelligence-Based Methodologies for Early Diagnostic Precision and Personalized Therapeutic Strategies in Neuro-Ophthalmic and Neurodegenerative Pathologies. Brain Sci. 2024, 14, 1266. [Google Scholar] [CrossRef] [PubMed]
- Nikafshan Rad, H.; Su, Z.; Trinh, A.; Hakim Newton, M.A.; Shamsani, J.; Nygc Als Consortium; Karim, A.; Sattar, A. Amyotrophic Lateral Sclerosis Diagnosis Using Machine Learning and Multi-Omic Data Integration. Heliyon 2024, 10, e38583. [Google Scholar] [CrossRef]
- StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 2025. Available online: https://www.ncbi.nlm.nih.gov/books/NBK430685/ (accessed on 30 May 2025).
- Dunckley, T.; Huentelman, M.J.; Craig, D.W.; Pearson, J.V.; Szelinger, S.; Joshipura, K.; Halperin, R.F.; Stamper, C.; Jensen, K.R.; Letizia, D.; et al. Whole-Genome Analysis of Sporadic Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2007, 357, 775–788. [Google Scholar] [CrossRef] [PubMed]
- van Rheenen, W.; van der Spek, R.A.A.; Bakker, M.K.; van Vugt, J.J.F.A.; Hop, P.J.; Zwamborn, R.A.J.; de Klein, N.; Westra, H.-J.; Bakker, O.B.; Deelen, P.; et al. Common and Rare Variant Association Analyses in Amyotrophic Lateral Sclerosis Identify 15 Risk Loci with Distinct Genetic Architectures and Neuron-Specific Biology. Nat. Genet. 2021, 53, 1636–1648. [Google Scholar] [CrossRef]
- Van Hoecke, A.; Schoonaert, L.; Lemmens, R.; Timmers, M.; Staats, K.A.; Laird, A.S.; Peeters, E.; Philips, T.; Goris, A.; Dubois, B.; et al. EPHA4 Is a Disease Modifier of Amyotrophic Lateral Sclerosis in Animal Models and in Humans. Nat. Med. 2012, 18, 1418–1422. [Google Scholar] [CrossRef]
- Berdyński, M.; Miszta, P.; Safranow, K.; Andersen, P.M.; Morita, M.; Filipek, S.; Żekanowski, C.; Kuźma-Kozakiewicz, M. SOD1 Mutations Associated with Amyotrophic Lateral Sclerosis Analysis of Variant Severity. Sci. Rep. 2022, 12, 103. [Google Scholar] [CrossRef]
- Morello, G.; Guarnaccia, M.; Spampinato, A.G.; La Cognata, V.; D’Agata, V.; Cavallaro, S. Copy Number Variations in Amyotrophic Lateral Sclerosis: Piecing the Mosaic Tiles Together through a Systems Biology Approach. Mol. Neurobiol. 2018, 55, 1299–1322. [Google Scholar] [CrossRef]
- Floriddia, E. Transcriptomics and ALS Outcome. Nat. Neurosci. 2023, 26, 175. [Google Scholar] [CrossRef]
- Gámez-Valero, A.; Guisado-Corcoll, A.; Herrero-Lorenzo, M.; Solaguren-Beascoa, M.; Martí, E. Non-Coding RNAs as Sensors of Oxidative Stress in Neurodegenerative Diseases. Antioxidants 2020, 9, 1095. [Google Scholar] [CrossRef]
- Barmada, S.J. Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 340–351. [Google Scholar] [CrossRef]
- Hop, P.J.; Zwamborn, R.A.J.; Hannon, E.; Shireby, G.L.; Nabais, M.F.; Walker, E.M.; van Rheenen, W.; van Vugt, J.J.F.A.; Dekker, A.M.; Westeneng, H.-J.; et al. Genome-Wide Study of DNA Methylation Shows Alterations in Metabolic, Inflammatory, and Cholesterol Pathways in ALS. Sci. Transl. Med. 2022, 14, eabj0264. [Google Scholar] [CrossRef]
- Balendra, R.; Isaacs, A.M. C9orf72-Mediated ALS and FTD: Multiple Pathways to Disease. Nat. Rev. Neurol. 2018, 14, 544–558. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dou, X.; Liu, J.; Xiao, Y.; Zhang, Z.; Hayes, L.; Wu, R.; Fu, X.; Ye, Y.; Yang, B.; et al. Globally Reduced N6-Methyladenosine (M6A) in C9ORF72-ALS/FTD Dysregulates RNA Metabolism and Contributes to Neurodegeneration. Nat. Neurosci. 2023, 26, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Kovanda, A.; Režen, T.; Rogelj, B. MicroRNA in Skeletal Muscle Development, Growth, Atrophy, and Disease. Wiley Interdiscip. Rev. RNA 2014, 5, 509–525. [Google Scholar] [CrossRef] [PubMed]
- Hoppeler, H. Molecular Networks in Skeletal Muscle Plasticity. J. Exp. Biol. 2016, 219, 205–213. [Google Scholar] [CrossRef]
- Pineda, S.S.; Lee, H.; Ulloa-Navas, M.J.; Linville, R.M.; Garcia, F.J.; Galani, K.; Engelberg-Cook, E.; Castanedes, M.C.; Fitzwalter, B.E.; Pregent, L.J.; et al. Single-Cell Dissection of the Human Motor and Prefrontal Cortices in ALS and FTLD. Cell 2024, 187, 1971–1989. [Google Scholar] [CrossRef]
- Li, J.; Jaiswal, M.K.; Chien, J.-F.; Kozlenkov, A.; Jung, J.; Zhou, P.; Gardashli, M.; Pregent, L.J.; Engelberg-Cook, E.; Dickson, D.W.; et al. Divergent Single Cell Transcriptome and Epigenome Alterations in ALS and FTD Patients with C9orf72 Mutation. Nat. Commun. 2023, 14, 5714. [Google Scholar] [CrossRef]
- Iridoy, M.O.; Zubiri, I.; Zelaya, M.V.; Martinez, L.; Ausín, K.; Lachen-Montes, M.; Santamaría, E.; Fernandez-Irigoyen, J.; Jericó, I. Neuroanatomical Quantitative Proteomics Reveals Common Pathogenic Biological Routes between Amyotrophic Lateral Sclerosis (ALS) and Frontotemporal Dementia (FTD). Int. J. Mol. Sci. 2019, 20, 4. [Google Scholar] [CrossRef]
- Umoh, M.E.; Dammer, E.B.; Dai, J.; Duong, D.M.; Lah, J.J.; Levey, A.I.; Gearing, M.; Glass, J.D.; Seyfried, N.T. A Proteomic Network Approach across the ALS-FTD Disease Spectrum Resolves Clinical Phenotypes and Genetic Vulnerability in Human Brain. EMBO Mol. Med. 2018, 10, 48–62. [Google Scholar] [CrossRef]
- Laferrière, F.; Maniecka, Z.; Pérez-Berlanga, M.; Hruska-Plochan, M.; Gilhespy, L.; Hock, E.-M.; Wagner, U.; Afroz, T.; Boersema, P.J.; Barmettler, G.; et al. TDP-43 Extracted from Frontotemporal Lobar Degeneration Subject Brains Displays Distinct Aggregate Assemblies and Neurotoxic Effects Reflecting Disease Progression Rates. Nat. Neurosci. 2019, 22, 65–77. [Google Scholar] [CrossRef]
- Oeckl, P.; Weydt, P.; Thal, D.R.; Weishaupt, J.H.; Ludolph, A.C.; Otto, M. Proteomics in Cerebrospinal Fluid and Spinal Cord Suggests UCHL1, MAP2 and GPNMB as Biomarkers and Underpins Importance of Transcriptional Pathways in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2020, 139, 119–134. [Google Scholar] [CrossRef]
- Zhu, S.; Wuolikainen, A.; Wu, J.; Öhman, A.; Wingsle, G.; Moritz, T.; Andersen, P.M.; Forsgren, L.; Trupp, M. Targeted Multiple Reaction Monitoring Analysis of CSF Identifies UCHL1 and GPNMB as Candidate Biomarkers for ALS. J. Mol. Neurosci. 2019, 69, 643–657. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.G.; Gray, E.; Mäger, I.; Thézénas, M.-L.; Charles, P.D.; Talbot, K.; Fischer, R.; Kessler, B.M.; Wood, M.; Turner, M.R. CSF Extracellular Vesicle Proteomics Demonstrates Altered Protein Homeostasis in Amyotrophic Lateral Sclerosis. Clin. Proteom. 2020, 17, 31. [Google Scholar] [CrossRef]
- Schumacher-Schuh, A.; Bieger, A.; Borelli, W.V.; Portley, M.K.; Awad, P.S.; Bandres-Ciga, S. Advances in Proteomic and Metabolomic Profiling of Neurodegenerative Diseases. Front. Neurol. 2022, 12, 792227. [Google Scholar] [CrossRef] [PubMed]
- Goutman, S.A.; Boss, J.; Guo, K.; Alakwaa, F.M.; Patterson, A.; Kim, S.; Savelieff, M.G.; Hur, J.; Feldman, E.L. Untargeted Metabolomics Yields Insight into ALS Disease Mechanisms. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1329–1338. [Google Scholar] [CrossRef] [PubMed]
- Marino, C.; Grimaldi, M.; Sommella, E.M.; Ciaglia, T.; Santoro, A.; Buonocore, M.; Salviati, E.; Trojsi, F.; Polverino, A.; Sorrentino, P.; et al. The Metabolomic Profile in Amyotrophic Lateral Sclerosis Changes According to the Progression of the Disease: An Exploratory Study. Metabolites 2022, 12, 837. [Google Scholar] [CrossRef]
- Khanna, R.K.; Catanese, S.; Mortemousque, G.; Dupuy, C.; Lefevre, A.; Emond, P.; Beltran, S.; Gissot, V.; Pisella, P.-J.; Blasco, H.; et al. Metabolomics of Basal Tears in Amyotrophic Lateral Sclerosis: A Cross-Sectional Study. Ocul. Surf. 2024, 34, 363–369. [Google Scholar] [CrossRef]
- Yu, M.; Xu, J.; Dutta, R.; Trapp, B.; Pieper, A.A.; Cheng, F. Network Medicine Informed Multiomics Integration Identifies Drug Targets and Repurposable Medicines for Amyotrophic Lateral Sclerosis. npj Syst. Biol. Appl. 2024, 10, 128. [Google Scholar] [CrossRef]
- Baxi, E.G.; Thompson, T.; Li, J.; Kaye, J.A.; Lim, R.G.; Wu, J.; Ramamoorthy, D.; Lima, L.; Vaibhav, V.; Matlock, A.; et al. Answer ALS, a Large-Scale Resource for Sporadic and Familial ALS Combining Clinical and Multi-Omics Data from Induced Pluripotent Cell Lines. Nat. Neurosci. 2022, 25, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Stavrou, M.; Elliott, E.; Gregory, J.M.; Leigh, N.; Pinto, A.A.; Williams, T.L.; Chataway, J.; Swingler, R.; Parmar, M.K.B.; et al. Clinical Trials in Amyotrophic Lateral Sclerosis: A Systematic Review and Perspective. Brain Commun. 2021, 3, fcab242. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.; Palomo Ruiz, M.D.V.; Perez, D.I.; Gil, C. Drugs in Clinical Development for the Treatment of Amyotrophic Lateral Sclerosis. Expert Opin. Investig. Drugs 2017, 26, 403–414. [Google Scholar] [CrossRef]
- Cook, S.F.; Rhodes, T.; Schlusser, C.; Han, S.; Chen, C.; Zach, N.; Murthy, V.; Davé, S. A Descriptive Review of Global Real World Evidence Efforts to Advance Drug Discovery and Clinical Development in Amyotrophic Lateral Sclerosis. Front. Neurol. 2021, 12, 770001. [Google Scholar] [CrossRef] [PubMed]
- Ruffo, P.; Traynor, B.J.; Conforti, F.L. Advancements in Genetic Research and RNA Therapy Strategies for Amyotrophic Lateral Sclerosis (ALS): Current Progress and Future Prospects. J. Neurol. 2025, 272, 233. [Google Scholar] [CrossRef]
- Ediriweera, G.R.; Chen, L.; Yerbury, J.J.; Thurecht, K.J.; Vine, K.L. Non-Viral Vector-Mediated Gene Therapy for ALS: Challenges and Future Perspectives. Mol. Pharm. 2021, 18, 2142–2160. [Google Scholar] [CrossRef]
- Liepert, J.; Schwenkreis, P.; Tegenthoff, M.; Malin, J.-P. The Glutamate Antagonist Riluzole Suppresses Intracortical Facilitation. J. Neural Transm. 1997, 104, 1207–1214. [Google Scholar] [CrossRef]
- Cruz, M.P. Edaravone (Radicava): A Novel Neuroprotective Agent for the Treatment of Amyotrophic Lateral Sclerosis. Pharm. Ther. 2018, 43, 25–28. [Google Scholar]
- Nikitin, D.; Makam, A.N.; Suh, K.; McKenna, A.; Carlson, J.J.; Richardson, M.; Rind, D.M.; Pearson, S.D. The Effectiveness and Value of AMX0035 and Oral Edaravone for Amyotrophic Lateral Sclerosis: A Summary from the Institute for Clinical and Economic Review’s Midwest Comparative Effectiveness Public Advisory Council. J. Manag. Care Spec. Pharm. 2023, 29, 216–221. [Google Scholar] [CrossRef]
- Devinsky, O.; Coller, J.; Ahrens-Nicklas, R.; Liu, X.S.; Ahituv, N.; Davidson, B.L.; Bishop, K.M.; Weiss, Y.; Mingorance, A. Gene Therapies for Neurogenetic Disorders. Trends Mol. Med. 2025. [Google Scholar] [CrossRef]
- Cappella, M.; Ciotti, C.; Cohen-Tannoudji, M.; Biferi, M.G. Gene Therapy for ALS—A Perspective. Int. J. Mol. Sci. 2019, 20, 4388. [Google Scholar] [CrossRef]
- van Es, M.A.; Dahlberg, C.; Birve, A.; Veldink, J.H.; van den Berg, L.H.; Andersen, P.M. Large-Scale SOD1 Mutation Screening Provides Evidence for Genetic Heterogeneity in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 562–566. [Google Scholar] [CrossRef]
- Majounie, E.; Renton, A.E.; Mok, K.; Dopper, E.G.; Waite, A.; Rollinson, S.; Chiò, A.; Restagno, G.; Nicolaou, N.; Simon-Sanchez, J.; et al. Frequency of the C9orf72 Hexanucleotide Repeat Expansion in Patients with Amyotrophic Lateral Sclerosis and Frontotemporal Dementia: A Cross-Sectional Study. Lancet Neurol. 2012, 11, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Katyal, N.; Govindarajan, R. Shortcomings in the Current Amyotrophic Lateral Sclerosis Trials and Potential Solutions for Improvement. Front. Neurol. 2017, 8, 521. [Google Scholar] [CrossRef] [PubMed]
- Su, X.W.; Broach, J.R.; Connor, J.R.; Gerhard, G.S.; Simmons, Z. Genetic Heterogeneity of Amyotrophic Lateral Sclerosis: Implications for Clinical Practice and Research. Muscle Nerve 2014, 49, 786–803. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel Genes Associated with Amyotrophic Lateral Sclerosis: Diagnostic and Clinical Implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Brenner, D.; Freischmidt, A. Update on Genetics of Amyotrophic Lateral Sclerosis. Curr. Opin. Neurol. 2022, 35, 672–677. [Google Scholar] [CrossRef]
- Brenner, D.; Weishaupt, J.H. Update on Amyotrophic Lateral Sclerosis Genetics. Curr. Opin. Neurol. 2019, 32, 735–739. [Google Scholar] [CrossRef]
- Kaur, S.J.; McKeown, S.R.; Rashid, S. Mutant SOD1 Mediated Pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [Google Scholar] [CrossRef]
- Zhou, L.; Wang, Y.; Xu, Y.; Zhang, Y.; Zhu, C. Advances in AAV-Mediated Gene Replacement Therapy for Pediatric Monogenic Neurological Disorders. Mol. Ther. Methods Clin. Dev. 2024, 32, 101357. [Google Scholar] [CrossRef]
- Dwivedi, J.; Kaushal, S.; Jeslin, D.; Karpagavalli, L.; Kumar, R.; Dev, D.; Wal, P. Gene Augmentation Techniques to Stimulate Wound Healing Process: Progress and Prospects. Curr. Gene Ther. 2024, 25, 1–23. [Google Scholar] [CrossRef]
- Goga, A.; Stoffel, M. Therapeutic RNA-Silencing Oligonucleotides in Metabolic Diseases. Nat. Rev. Drug Discov. 2022, 21, 417–439. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Brown, R.H., Jr. Finding a Treatment for ALS—Will Gene Editing Cut It? N. Engl. J. Med. 2018, 378, 1454–1456. [Google Scholar] [CrossRef] [PubMed]
- Amado, D.A.; Davidson, B.L. Gene Therapy for ALS: A Review. Mol. Ther. 2021, 29, 3345–3358. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cao, Y.; Xie, W.; Guo, Y.; Cai, J.; Huang, T.; Li, P. Advances in Clinical Translation of Stem Cell-Based Therapy in Neurological Diseases. J. Cereb. Blood Flow Metab. 2025, 45, 600–616. [Google Scholar] [CrossRef]
- Bono, N.; Ponti, F.; Mantovani, D.; Candiani, G. Non-Viral in Vitro Gene Delivery: It Is Now Time to Set the Bar! Pharmaceutics 2020, 12, 183. [Google Scholar] [CrossRef] [PubMed]
- Blessing, D.; Déglon, N. Adeno-Associated Virus and Lentivirus Vectors: A Refined Toolkit for the Central Nervous System. Curr. Opin. Virol. 2016, 21, 61–66. [Google Scholar] [CrossRef]
- Kumagai, S.; Nakajima, T.; Muramatsu, S.-I. Intraparenchymal Delivery of Adeno-Associated Virus Vectors for the Gene Therapy of Neurological Diseases. Expert Opin. Biol. Ther. 2024, 24, 773–785. [Google Scholar] [CrossRef]
- Raoul, C.; Abbas-Terki, T.; Bensadoun, J.-C.; Guillot, S.; Haase, G.; Szulc, J.; Henderson, C.E.; Aebischer, P. Lentiviral-Mediated Silencing of SOD1 through RNA Interference Retards Disease Onset and Progression in a Mouse Model of ALS. Nat. Med. 2005, 11, 423–428. [Google Scholar] [CrossRef]
- Lentz, T.B.; Gray, S.J.; Samulski, R.J. Viral Vectors for Gene Delivery to the Central Nervous System. Neurobiol. Dis. 2012, 48, 179–188. [Google Scholar] [CrossRef]
- Ozceylan, O.; Sezgin-Bayindir, Z. Current Overview on the Use of Nanosized Drug Delivery Systems in the Treatment of Neurodegenerative Diseases. ACS Omega 2024, 9, 35223–35242. [Google Scholar] [CrossRef] [PubMed]
- Ponti, F.; Campolungo, M.; Melchiori, C.; Bono, N.; Candiani, G. Cationic Lipids for Gene Delivery: Many Players, One Goal. Chem. Phys. Lipids 2021, 235, 105032. [Google Scholar] [CrossRef] [PubMed]
- Tosolini, A.P.; Sleigh, J.N. Motor Neuron Gene Therapy: Lessons from Spinal Muscular Atrophy for Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2017, 10, 405. [Google Scholar] [CrossRef] [PubMed]
- Kole, R.; Krainer, A.R.; Altman, S. RNA Therapeutics: Beyond RNA Interference and Antisense Oligonucleotides. Nat. Rev. Drug Discov. 2012, 11, 125–140. [Google Scholar] [CrossRef]
- Brillante, S.; Volpe, M.; Indrieri, A. Advances in MicroRNA Therapeutics: From Preclinical to Clinical Studies. Hum. Gene Ther. 2024, 35, 628–648. [Google Scholar] [CrossRef]
- Fang, T.; Je, G.; Pacut, P.; Keyhanian, K.; Gao, J.; Ghasemi, M. Gene Therapy in Amyotrophic Lateral Sclerosis. Cells 2022, 11, 2066. [Google Scholar] [CrossRef]
- Miller, T.; Cudkowicz, M.; Shaw, P.J.; Andersen, P.M.; Atassi, N.; Bucelli, R.C.; Genge, A.; Glass, J.; Ladha, S.; Ludolph, A.L.; et al. Phase 1–2 Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2020, 383, 109–119. [Google Scholar] [CrossRef]
- Geng, Y.; Cai, Q. Role of C9orf72 Hexanucleotide Repeat Expansions in ALS/FTD Pathogenesis. Front. Mol. Neurosci. 2024, 17, 1322720. [Google Scholar] [CrossRef]
- Liu, Y.; Pattamatta, A.; Zu, T.; Reid, T.; Bardhi, O.; Borchelt, D.R.; Yachnis, A.T.; Ranum, L.P.W. C9orf72 BAC Mouse Model with Motor Deficits and Neurodegenerative Features of ALS/FTD. Neuron 2016, 90, 521–534. [Google Scholar] [CrossRef]
- Martier, R.; Konstantinova, P. Gene Therapy for Neurodegenerative Diseases: Slowing Down the Ticking Clock. Front. Neurosci. 2020, 14, 580179. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, L. MicroRNAs in Amyotrophic Lateral Sclerosis: From Pathogenetic Involvement to Diagnostic Biomarker and Therapeutic Agent Development. Neurol. Sci. 2020, 41, 3569–3577. [Google Scholar] [CrossRef] [PubMed]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for Widespread MicroRNA-155 Inhibition Prolongs Survival in ALS-Model Mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown, R.H., Jr.; Sena-Esteves, M. Adeno-associated Virus–Delivered Artificial MicroRNA Extends Survival and Delays Paralysis in an Amyotrophic Lateral Sclerosis Mouse Model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Xu, X.; Shen, D.; Gao, Y.; Zhou, Q.; Ni, Y.; Meng, H.; Shi, H.; Le, W.; Chen, S.; Chen, S. A Perspective on Therapies for Amyotrophic Lateral Sclerosis: Can Disease Progression Be Curbed? Transl. Neurodegener. 2021, 10, 29. [Google Scholar] [CrossRef]
- Mueller, C.; Berry, J.D.; McKenna-Yasek, D.M.; Gernoux, G.; Owegi, M.A.; Pothier, L.M.; Douthwright, C.L.; Gelevski, D.; Luppino, S.D.; Blackwood, M.; et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. N. Engl. J. Med. 2020, 383, 151–158. [Google Scholar] [CrossRef]
- Zeng, Y.; Yi, R.; Cullen, B.R. MicroRNAs and Small Interfering RNAs Can Inhibit MRNA Expression by Similar Mechanisms. Proc. Natl. Acad. Sci. USA 2003, 100, 9779–9784. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Zhao, Y.; Lu, L.; Gao, Q.; Yu, D.; Sun, M. CRISPR/Cas9: Implication for Modeling and Therapy of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2023, 17, 1223777. [Google Scholar] [CrossRef]
- Krishnan, G.; Zhang, Y.; Gu, Y.; Kankel, M.W.; Gao, F.-B.; Almeida, S. CRISPR Deletion of the C9ORF72 Promoter in ALS/FTD Patient Motor Neurons Abolishes Production of Dipeptide Repeat Proteins and Rescues Neurodegeneration. Acta Neuropathol. 2020, 140, 81–84. [Google Scholar] [CrossRef]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D. V In Vivo Genome Editing Improves Motor Function and Extends Survival in a Mouse Model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef]
- Chen, Y.A.; Kankel, M.W.; Hana, S.; Lau, S.K.; Zavodszky, M.I.; McKissick, O.; Mastrangelo, N.; Dion, J.; Wang, B.; Ferretti, D.; et al. In Vivo Genome Editing Using Novel AAV-PHP Variants Rescues Motor Function Deficits and Extends Survival in a SOD1-ALS Mouse Model. Gene Ther. 2023, 30, 443–454. [Google Scholar] [CrossRef]
- Meijboom, K.E.; Abdallah, A.; Fordham, N.P.; Nagase, H.; Rodriguez, T.; Kraus, C.; Gendron, T.F.; Krishnan, G.; Esanov, R.; Andrade, N.S.; et al. CRISPR/Cas9-Mediated Excision of ALS/FTD-Causing Hexanucleotide Repeat Expansion in C9ORF72 Rescues Major Disease Mechanisms in Vivo and in Vitro. Nat. Commun. 2022, 13, 6286. [Google Scholar] [CrossRef]
- Lopez-Gonzalez, R.; Lu, Y.; Gendron, T.F.; Karydas, A.; Tran, H.; Yang, D.; Petrucelli, L.; Miller, B.L.; Almeida, S.; Gao, F.-B. Poly(GR) in C9ORF72-Related ALS/FTD Compromises Mitochondrial Function and Increases Oxidative Stress and DNA Damage in IPSC-Derived Motor Neurons. Neuron 2016, 92, 383–391. [Google Scholar] [CrossRef]
- Bhinge, A.; Namboori, S.C.; Zhang, X.; VanDongen, A.M.J.; Stanton, L.W. Genetic Correction of SOD1 Mutant IPSCs Reveals ERK and JNK Activated AP1 as a Driver of Neurodegeneration in Amyotrophic Lateral Sclerosis. Stem Cell Rep. 2017, 8, 856–869. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Naujock, M.; Fumagalli, L.; Vandoorne, T.; Baatsen, P.; Boon, R.; Ordovás, L.; Patel, A.; Welters, M.; Vanwelden, T.; et al. HDAC6 Inhibition Reverses Axonal Transport Defects in Motor Neurons Derived from FUS-ALS Patients. Nat. Commun. 2017, 8, 861. [Google Scholar] [CrossRef] [PubMed]
- Tann, J.Y.; Wong, L.-W.; Sajikumar, S.; Ibáñez, C.F. Abnormal TDP-43 Function Impairs Activity-Dependent BDNF Secretion, Synaptic Plasticity, and Cognitive Behavior through Altered Sortilin Splicing. EMBO J. 2019, 38, e100989. [Google Scholar] [CrossRef]
- Dafinca, R.; Barbagallo, P.; Farrimond, L.; Candalija, A.; Scaber, J.; Ababneh, N.A.; Sathyaprakash, C.; Vowles, J.; Cowley, S.A.; Talbot, K. Impairment of Mitochondrial Calcium Buffering Links Mutations in C9ORF72 and TARDBP in IPS-Derived Motor Neurons from Patients with ALS/FTD. Stem Cell Rep. 2020, 14, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Vahsen, B.F.; Nalluru, S.; Morgan, G.R.; Farrimond, L.; Carroll, E.; Xu, Y.; Cramb, K.M.L.; Amein, B.; Scaber, J.; Katsikoudi, A.; et al. C9orf72-ALS Human IPSC Microglia Are pro-Inflammatory and Toxic to Co-Cultured Motor Neurons via MMP9. Nat. Commun. 2023, 14, 5898. [Google Scholar] [CrossRef]
- Kiskinis, E.; Sandoe, J.; Williams, L.A.; Boulting, G.L.; Moccia, R.; Wainger, B.J.; Han, S.; Peng, T.; Thams, S.; Mikkilineni, S.; et al. Pathways Disrupted in Human ALS Motor Neurons Identified through Genetic Correction of Mutant SOD1. Cell Stem Cell 2014, 14, 781–795. [Google Scholar] [CrossRef]
- Caron, I.; Micotti, E.; Paladini, A.; Merlino, G.; Plebani, L.; Forloni, G.; Modo, M.; Bendotti, C. Comparative Magnetic Resonance Imaging and Histopathological Correlates in Two SOD1 Transgenic Mouse Models of Amyotrophic Lateral Sclerosis. PLoS ONE 2015, 10, e0132159. [Google Scholar] [CrossRef]
- Blair, I.P.; Williams, K.L.; Warraich, S.T.; Durnall, J.C.; Thoeng, A.D.; Manavis, J.; Blumbergs, P.C.; Vucic, S.; Kiernan, M.C.; Nicholson, G.A. FUS Mutations in Amyotrophic Lateral Sclerosis: Clinical, Pathological, Neurophysiological and Genetic Analysis. J. Neurol. Neurosurg. Psychiatry 2010, 81, 639–645. [Google Scholar] [CrossRef]
- Zhao, S.; Chen, R.; Gao, Y.; Lu, Y.; Bai, X.; Zhang, J. Fundamental Roles of the Optineurin Gene in the Molecular Pathology of Amyotrophic Lateral Sclerosis. Front. Neurosci. 2023, 17, 1319706. [Google Scholar] [CrossRef] [PubMed]
- Kwok, C.T.; Morris, A.; de Belleroche, J.S. Sequestosome-1 (SQSTM1) Sequence Variants in ALS Cases in the UK: Prevalence and Coexistence of SQSTM1 Mutations in ALS Kindred with PDB. Eur. J. Hum. Genet. 2014, 22, 492–496. [Google Scholar] [CrossRef]
- Tan, R.H.; Kril, J.J.; McGinley, C.; Hassani, M.; Masuda-Suzukake, M.; Hasegawa, M.; Mito, R.; Kiernan, M.C.; Halliday, G.M. Cerebellar Neuronal Loss in Amyotrophic Lateral Sclerosis Cases with ATXN 2 Intermediate Repeat Expansions. Ann. Neurol. 2016, 79, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [PubMed]
- Renaud, L.; Picher-Martel, V.; Codron, P.; Julien, J.-P. Key Role of UBQLN2 in Pathogenesis of Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. Acta Neuropathol. Commun. 2019, 7, 103. [Google Scholar] [CrossRef]
- Wu, C.-H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the Profilin 1 Gene Cause Familial Amyotrophic Lateral Sclerosis. Nature 2012, 488, 499–503. [Google Scholar] [CrossRef]
- Daoud, H.; Dobrzeniecka, S.; Camu, W.; Meininger, V.; Dupré, N.; Dion, P.A.; Rouleau, G.A. Mutation Analysis of PFN1 in Familial Amyotrophic Lateral Sclerosis Patients. Neurobiol. Aging 2013, 34, 1311.e1–1311.e2. [Google Scholar] [CrossRef] [PubMed]
- Freischmidt, A.; Wieland, T.; Richter, B.; Ruf, W.; Schaeffer, V.; Müller, K.; Marroquin, N.; Nordin, F.; Hübers, A.; Weydt, P.; et al. Haploinsufficiency of TBK1 Causes Familial ALS and Fronto-Temporal Dementia. Nat. Neurosci. 2015, 18, 631–636. [Google Scholar] [CrossRef]
- Kenna, K.P.; van Doormaal, P.T.C.; Dekker, A.M.; Ticozzi, N.; Kenna, B.J.; Diekstra, F.P.; van Rheenen, W.; van Eijk, K.R.; Jones, A.R.; Keagle, P.; et al. NEK1 Variants Confer Susceptibility to Amyotrophic Lateral Sclerosis. Nat. Genet. 2016, 48, 1037–1042. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bono, N.; Fruzzetti, F.; Farinazzo, G.; Candiani, G.; Marcuzzo, S. Perspectives in Amyotrophic Lateral Sclerosis: Biomarkers, Omics, and Gene Therapy Informing Disease and Treatment. Int. J. Mol. Sci. 2025, 26, 5671. https://doi.org/10.3390/ijms26125671
Bono N, Fruzzetti F, Farinazzo G, Candiani G, Marcuzzo S. Perspectives in Amyotrophic Lateral Sclerosis: Biomarkers, Omics, and Gene Therapy Informing Disease and Treatment. International Journal of Molecular Sciences. 2025; 26(12):5671. https://doi.org/10.3390/ijms26125671
Chicago/Turabian StyleBono, Nina, Flaminia Fruzzetti, Giorgia Farinazzo, Gabriele Candiani, and Stefania Marcuzzo. 2025. "Perspectives in Amyotrophic Lateral Sclerosis: Biomarkers, Omics, and Gene Therapy Informing Disease and Treatment" International Journal of Molecular Sciences 26, no. 12: 5671. https://doi.org/10.3390/ijms26125671
APA StyleBono, N., Fruzzetti, F., Farinazzo, G., Candiani, G., & Marcuzzo, S. (2025). Perspectives in Amyotrophic Lateral Sclerosis: Biomarkers, Omics, and Gene Therapy Informing Disease and Treatment. International Journal of Molecular Sciences, 26(12), 5671. https://doi.org/10.3390/ijms26125671